版權信息: ?四川大學華西醫院華西期刊社《中華眼底病雜志》版權所有,未經授權不得轉載、改編
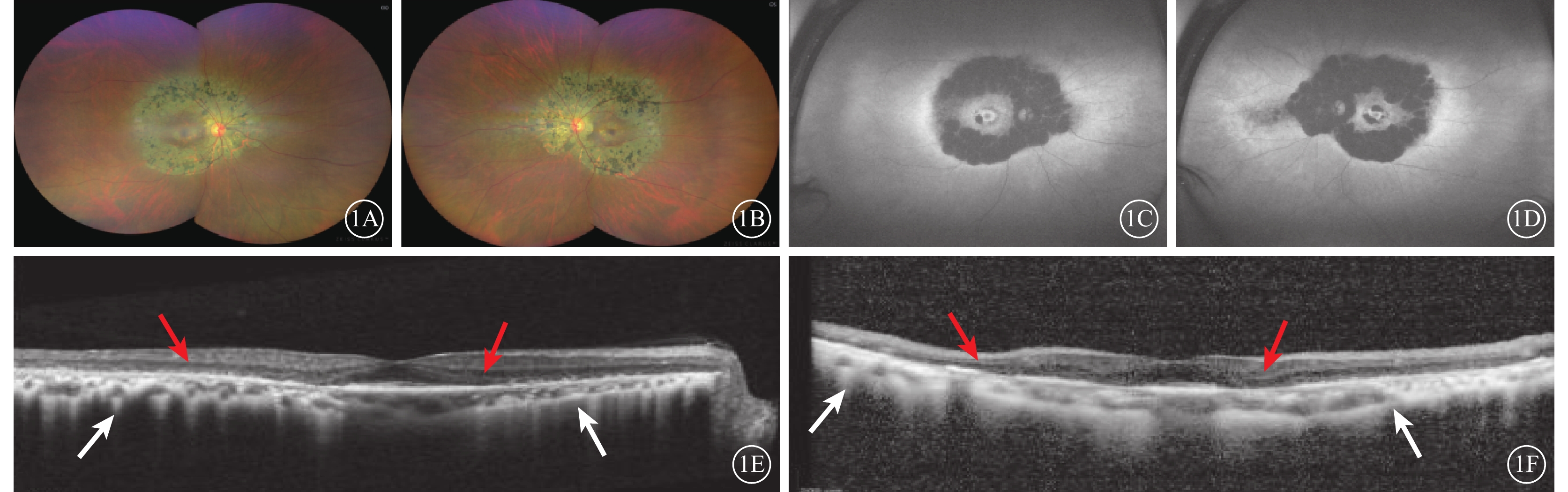
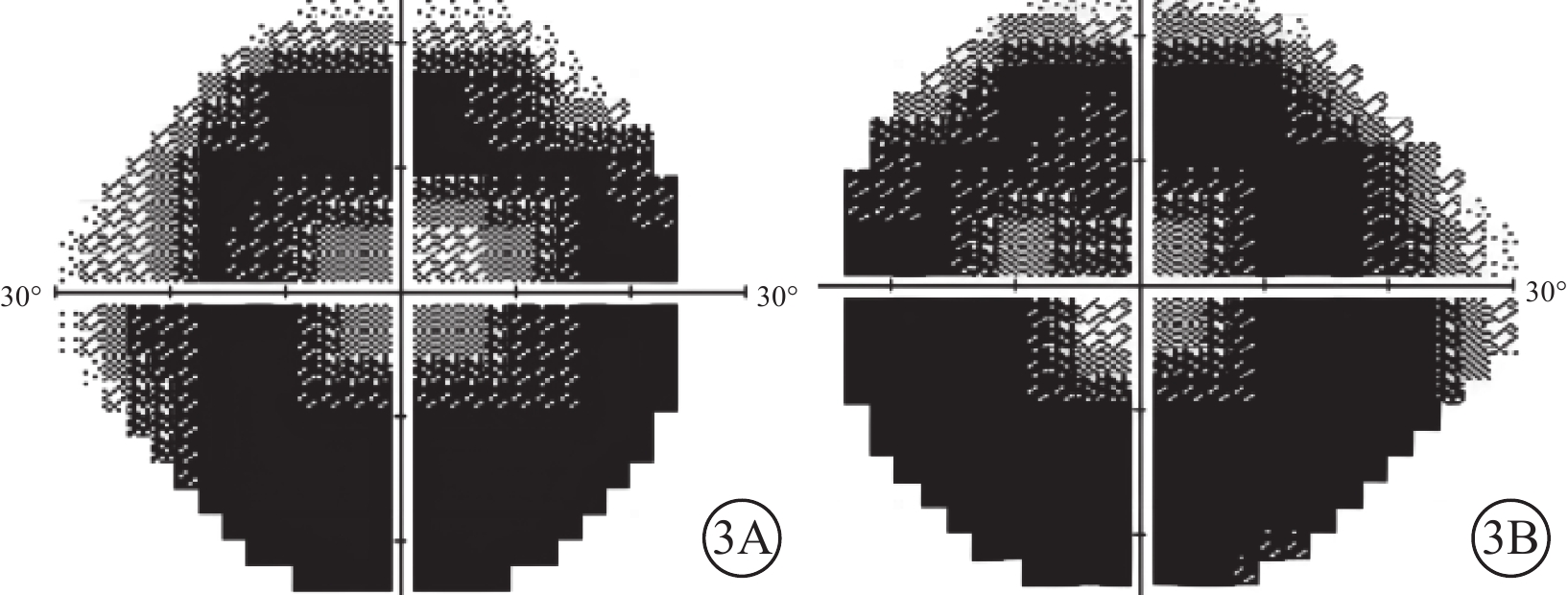
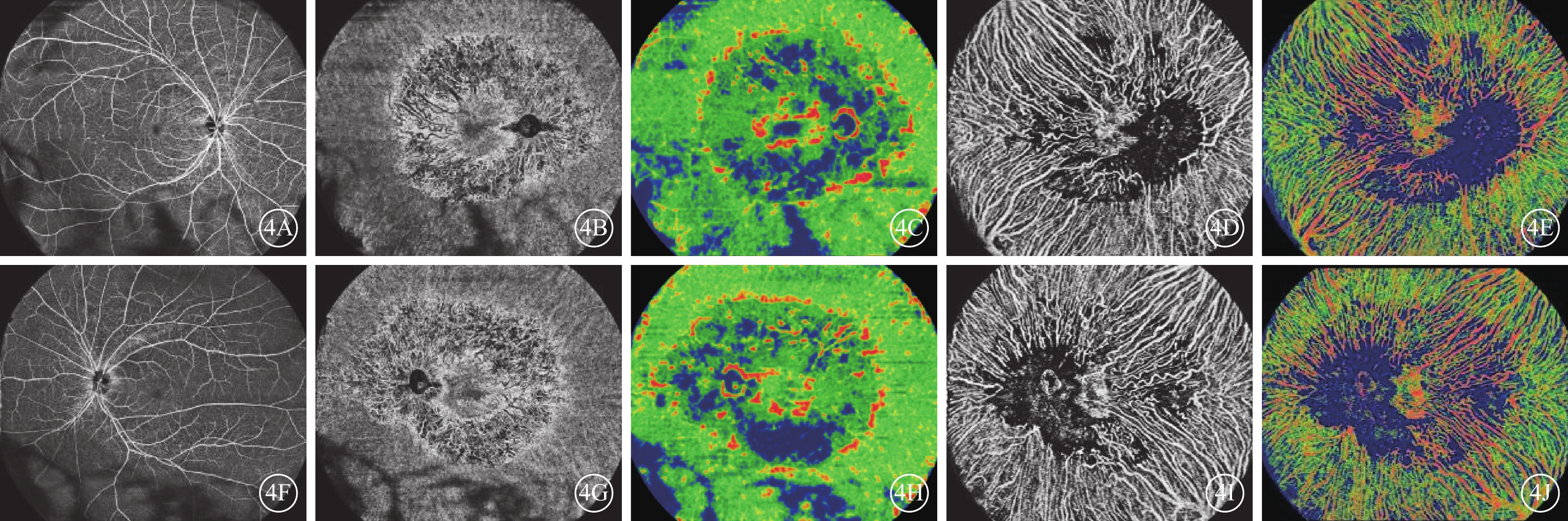
患者女,54歲。因雙眼晚上視物模糊4年、加重2年(白天視物無異常),于2024年4月25日到成都愛迪眼科醫院就診。既往身體健康。家族中無類似疾病史。眼部檢查:雙眼祼眼視力1.0;右眼、左眼眼壓分別為10、13 mm Hg(1 mm Hg=0.133 kPa)。雙眼眼前節未見明顯異常。眼底檢查,雙眼視盤、視網膜色澤紅潤,黃斑區顏色、形態基本正常;后極部以視盤、黃斑為中心,邊界清楚呈對稱性環形萎縮區,色澤晦暗,其間可見骨細胞樣色素沉著,萎縮區外視網膜色澤、形態未見明顯異常(圖1A,1B)。眼底自發熒光(FAF)檢查,雙眼病灶呈邊界清晰的弱熒光區(圖1C,1D)。光相干斷層掃描(OCT)檢查,雙眼環形萎縮區橢圓體帶(EZ)連續性中斷,視網膜色素上皮(RPE)層萎縮、變薄和不規則,脈絡膜層厚度明顯變薄(圖1E,1F);周邊視網膜EZ正常。全視野視網膜電圖(ERG)檢查,各波形態正常,僅部分波形振幅不同程度降低(圖2)。30°視野檢查,雙眼呈與萎縮區相吻合的對稱性巨大環形暗點(圖3)。OCT血管成像(OCTA)檢查,雙眼萎縮區視網膜全層血流密度無明顯改變;脈絡膜中小毛細血管層可見邊界清楚的萎縮區域,血管床密度、血流量降低;脈絡膜大血管層受累相對較輕,血管床密度、血流量中至重度降低(圖4)。患者母親、兄妹、女兒均否認夜盲史。其中母親眼底檢查正常。女兒眼前節、眼底視網膜色澤、脈絡膜全層結構和血流量未見異常。臨床診斷:色素性視網膜炎(RP)。
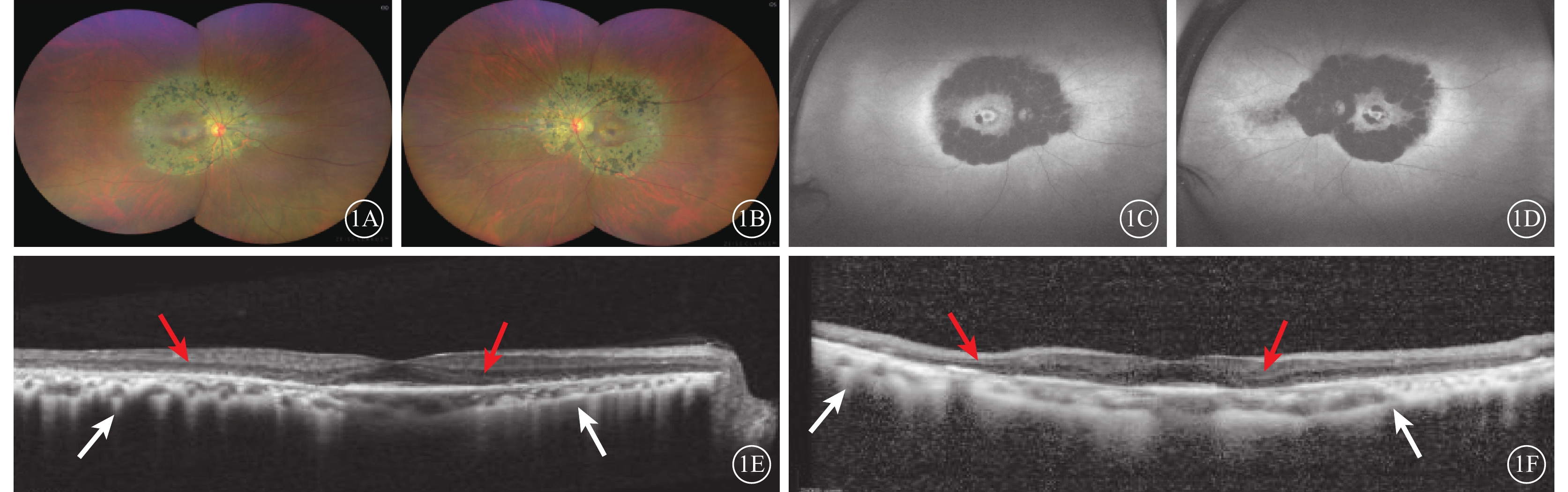 圖1
色素性視網膜炎患者雙眼多模式影像圖 1A、1B分別示右眼、左眼彩色眼底像,雙眼后極部對稱性、邊界清楚的環形萎縮病灶,其間骨細胞樣色素沉著;黃斑區顏色相對正常。1C、1D分別示右眼、左眼眼底自發熒光像,雙眼對稱性環形弱熒光。1E、1F分別示右眼、左眼光相干斷層掃描像,中心凹外橢圓體帶、視網膜色素上皮層部分缺失、斷裂和變薄(紅箭),脈絡膜中小毛細血管層萎縮、厚度明顯變薄(白箭)
圖1
色素性視網膜炎患者雙眼多模式影像圖 1A、1B分別示右眼、左眼彩色眼底像,雙眼后極部對稱性、邊界清楚的環形萎縮病灶,其間骨細胞樣色素沉著;黃斑區顏色相對正常。1C、1D分別示右眼、左眼眼底自發熒光像,雙眼對稱性環形弱熒光。1E、1F分別示右眼、左眼光相干斷層掃描像,中心凹外橢圓體帶、視網膜色素上皮層部分缺失、斷裂和變薄(紅箭),脈絡膜中小毛細血管層萎縮、厚度明顯變薄(白箭)
 圖2
色素性視網膜炎患者全視野視網膜電圖像各波形態基本正常,暗適應10.0 a、b波和明適應3.0 a波振幅正常;其余檢查指標b波不同程度降低
圖2
色素性視網膜炎患者全視野視網膜電圖像各波形態基本正常,暗適應10.0 a、b波和明適應3.0 a波振幅正常;其余檢查指標b波不同程度降低
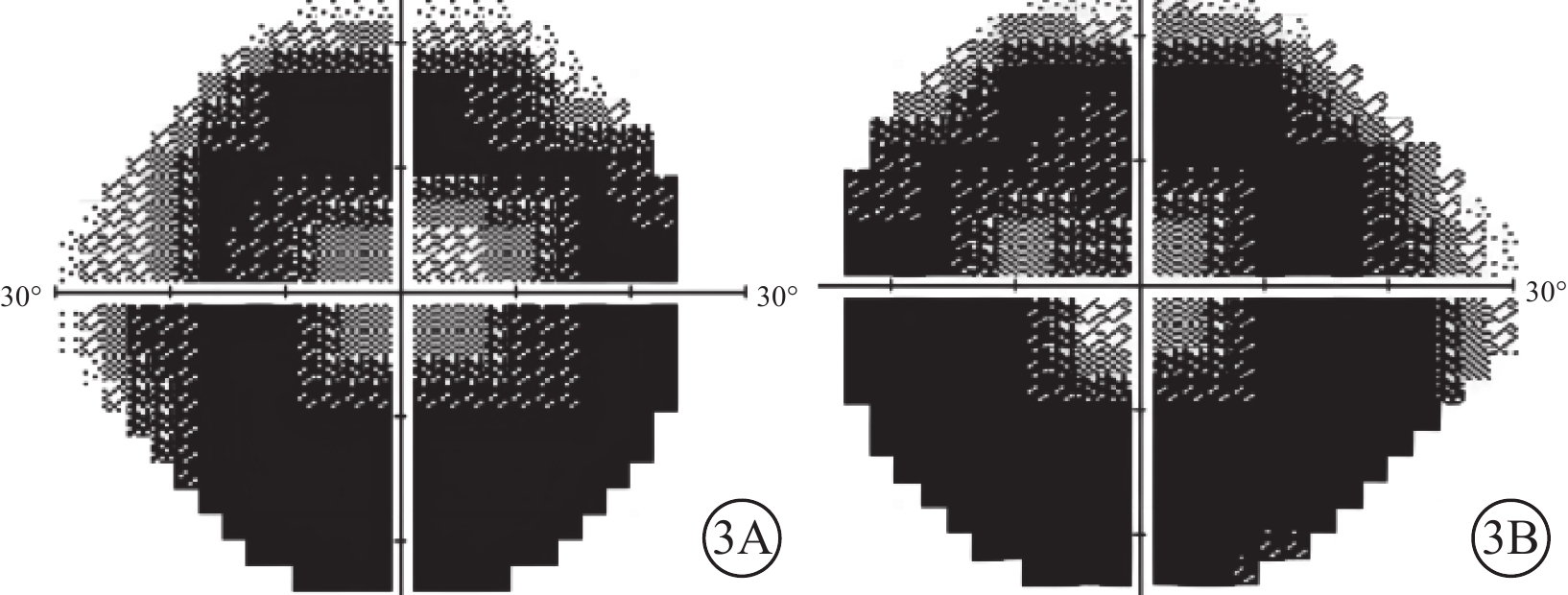 圖3
色素性視網膜炎患者30°視野檢查像 3A、3B分別示右眼、左眼,雙眼巨大環形暗點
圖3
色素性視網膜炎患者30°視野檢查像 3A、3B分別示右眼、左眼,雙眼巨大環形暗點
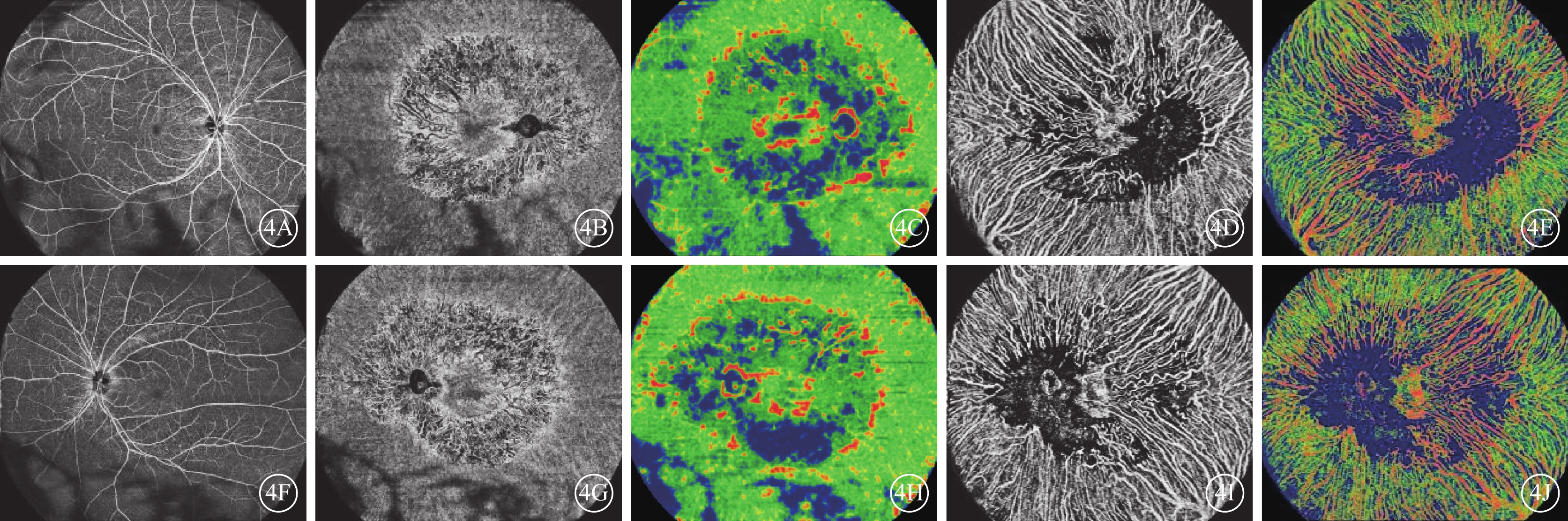 圖4
色素性視網膜炎患者光相干斷層掃描血管成像圖 4A~4E示右眼。視網膜全層血流未見明顯異常(4A);脈絡膜中小毛細血管層血管床密度(4B)、血流量明顯降低(4C);脈絡膜大血管層血管床密度(4D)、血流量中至重度降低(4E)。4F~4J示左眼,與右眼呈對稱性改變 視網膜全層:內界膜至外叢狀層;脈絡膜中小毛細血管層:Bruch膜至其下29 μm;脈絡膜大血管層:Bruch膜下29 μm至脈絡膜鞏膜交界面
圖4
色素性視網膜炎患者光相干斷層掃描血管成像圖 4A~4E示右眼。視網膜全層血流未見明顯異常(4A);脈絡膜中小毛細血管層血管床密度(4B)、血流量明顯降低(4C);脈絡膜大血管層血管床密度(4D)、血流量中至重度降低(4E)。4F~4J示左眼,與右眼呈對稱性改變 視網膜全層:內界膜至外叢狀層;脈絡膜中小毛細血管層:Bruch膜至其下29 μm;脈絡膜大血管層:Bruch膜下29 μm至脈絡膜鞏膜交界面
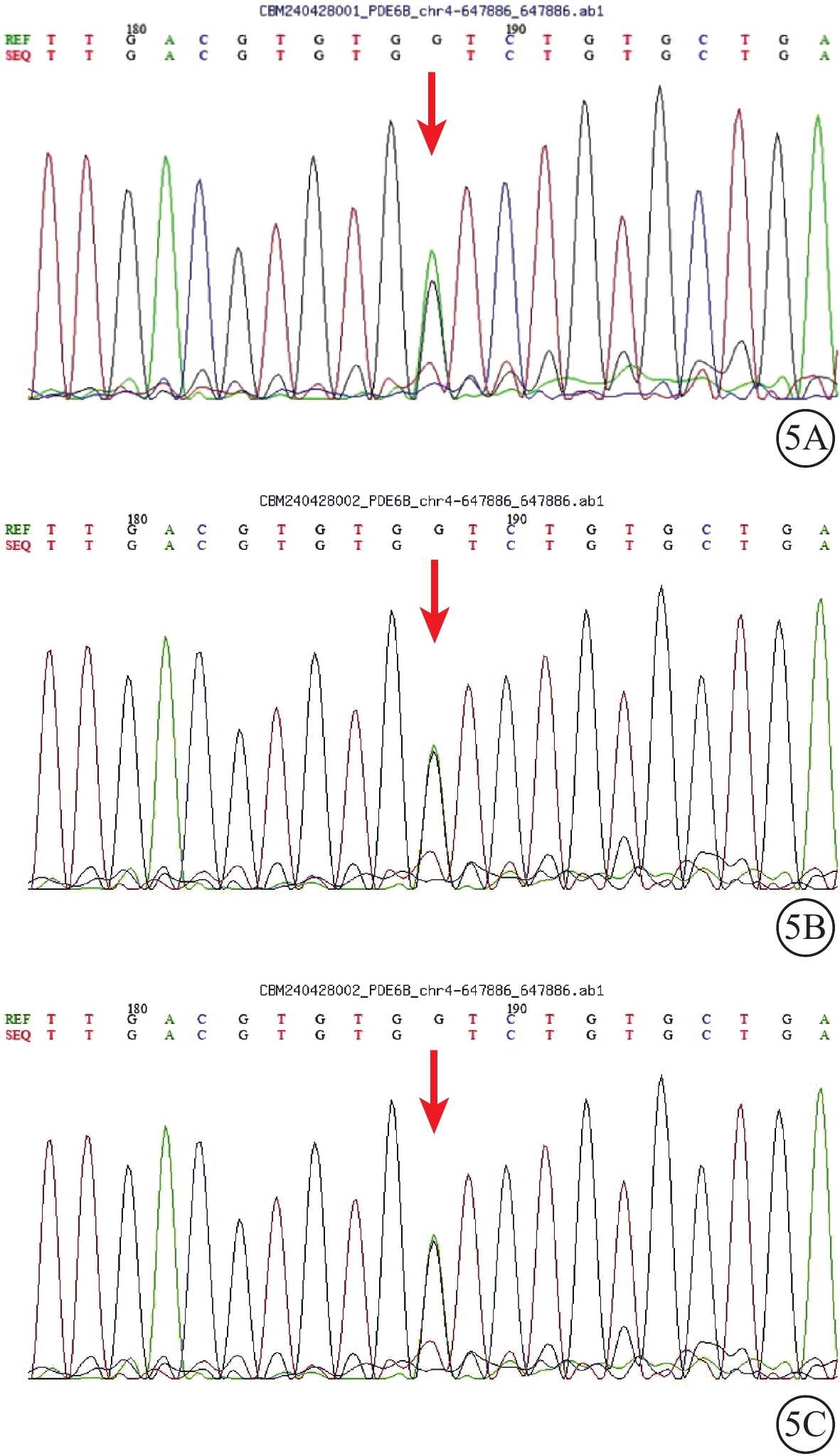
采集患者及其母親、女兒外周靜脈血2 ml,患者提取基因組DNA,行全外顯子組基因測序。對測序后的原始數據經生物信息學分析處理后,根據患者表型等信息進行變異位點篩選;對發現的致病變異位點進行Sanger驗證。參考美國醫學遺傳學和基因組學學院(ACMG)相關指南對變異位點進行致病性分析。結果顯示,患者及其母親、女兒PDE6B基因第5號外顯子均存在c.870G>A(p.W290X)雜合變異(圖5)。此變異為無義突變,PDE6B基因NM_000283轉錄本上第870個堿基由G突變為A,導致編碼色氨酸的基因序列編碼終止,可能導致肽鏈合成提前終止而影響蛋白質功能[5-7]。依據ACMG指南分析,該變異生物學致病等級被判斷為致病性變異(PVS1+PM2+PP3)。功能缺失變異導致基因功能可能喪失(PVS1);ESP數據庫(https://esp.gs.washington.edu/drupal/)、千人基因組計劃數據庫(http://www.1000genomes.org/)、外顯子組聚集聯盟數據庫(http://exac.broadinstitute.org/)中正常對照人群中未發現(或隱性遺傳病中極低頻位點)(PM2);保守性預測、進化預測、剪接位點影響等方法預測該變異會對基因或基因產物造成有害影響(PP3)。結合臨床表現、基因檢測結果,最終診斷:雙眼RP。
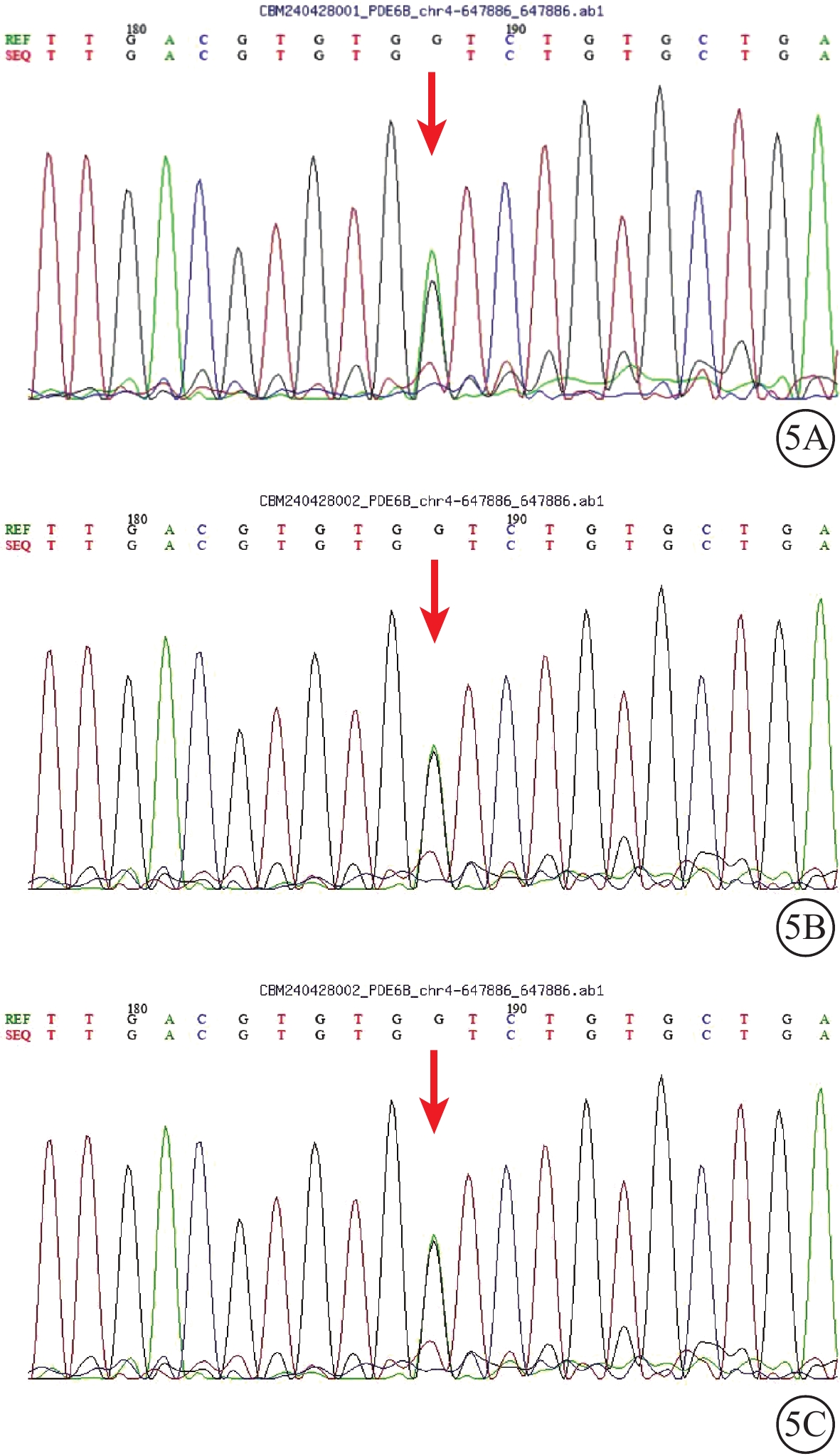 圖5
色素性視網膜炎患者及其女兒、母親基因測序圖5A~5C分別示患者及其患者女兒、母親,PDE6B基因第5號外顯子的c.870G>A雜合變異(紅箭)
圖5
色素性視網膜炎患者及其女兒、母親基因測序圖5A~5C分別示患者及其患者女兒、母親,PDE6B基因第5號外顯子的c.870G>A雜合變異(紅箭)
討論 RP是一組遺傳性、進行性光感受器細胞功能障礙、細胞丟失,以最終導致廣泛視網膜多層組織萎縮為特征,發病年齡從嬰幼兒期到中老年人[1]。RP存在多種類型,眼底表現差異較大,但共同規律是全視網膜、象限性視網膜出現大量骨細胞樣色素沉著、動靜脈血管變細、視網膜呈青灰色改變、視盤萎縮變白、視野縮窄等。即便是無色素性的RP,其視網膜色澤也較晦暗,伴血管和視盤改變;全視野ERG各波形振幅嚴重降低,甚至呈熄滅型。
McLaughlin等[2]于1993年首次發現RP患者存在PDE6B基因變異。磷酸二酯酶(PDE)是一個大家族,PDE6是其亞家族中的1種。由PDE6基因編碼的PDE蛋白水解環磷酸鳥苷(cGMP)。而cGMP是視桿細胞外節膜盤離子通道的特異性受體,同時也是脊椎動物中感光細胞將光信號轉化為電信號的重要分子。PDE6b基因缺失導致光感受器細胞內cGMP增多,引起光感受器細胞的陽離子增多導致細胞中毒而凋亡,視網膜光感受器細胞不斷破壞,從而間接影響視網膜接受光信號轉換為電信號。PDE6b基因異常是常染色體隱性遺傳RP最主要原因之一[3]。
本例患者雙眼視力正常,因近4年傍晚視物逐漸模糊,夜晚加重而就診。眼底檢查發現雙眼以視盤、黃斑為中心呈對稱性的環形視網膜、脈絡膜萎縮區,其間骨細胞樣色素沉著;周邊視網膜形態、顏色、血管床密度均正常。OCTA檢查可見萎縮區視網膜全層血管床密度和血流無明顯變化;脈絡膜中小毛細血管層血管床密度和血流均嚴重降低,脈絡膜大血管層改變相對較輕。這說明病變主要發生并集中于脈絡膜中小毛細血管層。向內波及到無血管層、RPE層,向外影響到脈絡膜大血管層。隨著RPE和脈絡膜毛細血管持續喪失,較大的脈絡膜血管可能發生變性[1]。我們推測,其發病機制是由于基因變異、脈絡膜毛細血管萎縮、血管床密度降低而導致RPE和視覺細胞層血供不足,致RPE和視細胞損害和凋亡。由于脈絡膜厚度周邊較薄,后極部較厚。故周邊部視桿細胞丟失更早、更重,患者較早即出現夜盲和視野缺失。本例患者雙眼RPE層除中心凹基本正常外,其脈絡膜萎縮區RPE層變薄、缺失,但周邊RPE層形態、厚度正常。與傳統RP表現不同,本例患者全視野ERG檢查除部分波形振幅稍降低外,其余各波形態完全正常。分析其原因是其絕大部分視網膜正常,多數視桿、視錐細胞未受累。此外,其視野呈與萎縮區一致的巨大環形暗點,周邊視野正常,也與傳統RP導致的管窺視野表現不一致。本例患者眼底表現完全不同于經典RP改變,臨床確診困難,最終經基因檢測發現PDE6B基因存在c.870G>A(p.W290X)雜合變異而確診。推測其眼底表現特殊的原因可能與PDE6B基因變異突變位點不同有關[2, 4-5]。
患者女兒、母親眼底檢查無明顯異常。基因檢測發現PDE6B基因均存在c.870G>A(p.W290X)雜合變異。推測該基因突變的變異位點導致本病發病時間約在患者40歲以后。患者女兒目前年齡29歲,其眼底檢查未發現異常可能是尚未到該病的發病年齡。臨床上應加強隨訪,同時針對此類患者的遺傳咨詢也有重要臨床意義。患者母親眼底正常,可能是臨床表型差異的原因。基因型控制生物個體的表現型,是表現型的決定性因素,但不是唯一決定性因素,與生物個體所處的特定環境條件同時發揮作用,使得表現型呈現出多樣性[6]。
本次檢測范圍為人類基因外顯子組區,不包含內含子區,不排除患者PDE6B等位基因存在內含子變異(構成復合雜合變異)從而導致疾病的發生。根據遺傳規律,常染色體隱性遺傳疾病患者的子女和父母,可能均為單個位點雜合攜帶者,這可能是母親和女兒無類似疾病表現的原因。目前已發現PDE6B基因變異與先天性靜止性夜盲2型和RP40型相關。本例患者臨床表現不符合前者,與RP40型更相似[7]。但家系不符合常染色體隱性遺傳病的遺傳規律,我們認為不排除部分變異導致的RP40型存在顯性遺傳的可能性,但該猜測證據較弱,需進一步研究。
本例患者眼底表現特殊,與傳統RP有著很大差異,應與中心性脈絡膜營養不良、視錐細胞營養不良、色素性靜脈旁視網膜脈絡膜萎縮等鑒別。中心性脈絡膜營養不良,后極部視網膜色澤較晦暗,病變主要圍繞血管弓和視神經,也可向內外蔓延,同時可累及黃斑。FAF呈與營養不良區域相一致的弱熒光區[8,本例患者眼底表現易與此混淆。視錐細胞營養不良患者疾病早期即出現視力下降,色覺障礙和眼球震顫,由CDHR1基因變異所致[9]。色素性靜脈旁視網膜脈絡膜萎縮在形態和分布上與RP有很大的差異。最后確診,除典型臨床表現外,更多應參考基因檢測結果。
志謝 感謝北京智德醫學檢驗所提供的專業基因檢測服務及其對本家族成員檢測結果的詳細解讀
患者女,54歲。因雙眼晚上視物模糊4年、加重2年(白天視物無異常),于2024年4月25日到成都愛迪眼科醫院就診。既往身體健康。家族中無類似疾病史。眼部檢查:雙眼祼眼視力1.0;右眼、左眼眼壓分別為10、13 mm Hg(1 mm Hg=0.133 kPa)。雙眼眼前節未見明顯異常。眼底檢查,雙眼視盤、視網膜色澤紅潤,黃斑區顏色、形態基本正常;后極部以視盤、黃斑為中心,邊界清楚呈對稱性環形萎縮區,色澤晦暗,其間可見骨細胞樣色素沉著,萎縮區外視網膜色澤、形態未見明顯異常(圖1A,1B)。眼底自發熒光(FAF)檢查,雙眼病灶呈邊界清晰的弱熒光區(圖1C,1D)。光相干斷層掃描(OCT)檢查,雙眼環形萎縮區橢圓體帶(EZ)連續性中斷,視網膜色素上皮(RPE)層萎縮、變薄和不規則,脈絡膜層厚度明顯變薄(圖1E,1F);周邊視網膜EZ正常。全視野視網膜電圖(ERG)檢查,各波形態正常,僅部分波形振幅不同程度降低(圖2)。30°視野檢查,雙眼呈與萎縮區相吻合的對稱性巨大環形暗點(圖3)。OCT血管成像(OCTA)檢查,雙眼萎縮區視網膜全層血流密度無明顯改變;脈絡膜中小毛細血管層可見邊界清楚的萎縮區域,血管床密度、血流量降低;脈絡膜大血管層受累相對較輕,血管床密度、血流量中至重度降低(圖4)。患者母親、兄妹、女兒均否認夜盲史。其中母親眼底檢查正常。女兒眼前節、眼底視網膜色澤、脈絡膜全層結構和血流量未見異常。臨床診斷:色素性視網膜炎(RP)。
圖1
色素性視網膜炎患者雙眼多模式影像圖 1A、1B分別示右眼、左眼彩色眼底像,雙眼后極部對稱性、邊界清楚的環形萎縮病灶,其間骨細胞樣色素沉著;黃斑區顏色相對正常。1C、1D分別示右眼、左眼眼底自發熒光像,雙眼對稱性環形弱熒光。1E、1F分別示右眼、左眼光相干斷層掃描像,中心凹外橢圓體帶、視網膜色素上皮層部分缺失、斷裂和變薄(紅箭),脈絡膜中小毛細血管層萎縮、厚度明顯變薄(白箭)
圖2
色素性視網膜炎患者全視野視網膜電圖像各波形態基本正常,暗適應10.0 a、b波和明適應3.0 a波振幅正常;其余檢查指標b波不同程度降低
圖3
色素性視網膜炎患者30°視野檢查像 3A、3B分別示右眼、左眼,雙眼巨大環形暗點
圖4
色素性視網膜炎患者光相干斷層掃描血管成像圖 4A~4E示右眼。視網膜全層血流未見明顯異常(4A);脈絡膜中小毛細血管層血管床密度(4B)、血流量明顯降低(4C);脈絡膜大血管層血管床密度(4D)、血流量中至重度降低(4E)。4F~4J示左眼,與右眼呈對稱性改變 視網膜全層:內界膜至外叢狀層;脈絡膜中小毛細血管層:Bruch膜至其下29 μm;脈絡膜大血管層:Bruch膜下29 μm至脈絡膜鞏膜交界面
采集患者及其母親、女兒外周靜脈血2 ml,患者提取基因組DNA,行全外顯子組基因測序。對測序后的原始數據經生物信息學分析處理后,根據患者表型等信息進行變異位點篩選;對發現的致病變異位點進行Sanger驗證。參考美國醫學遺傳學和基因組學學院(ACMG)相關指南對變異位點進行致病性分析。結果顯示,患者及其母親、女兒PDE6B基因第5號外顯子均存在c.870G>A(p.W290X)雜合變異(圖5)。此變異為無義突變,PDE6B基因NM_000283轉錄本上第870個堿基由G突變為A,導致編碼色氨酸的基因序列編碼終止,可能導致肽鏈合成提前終止而影響蛋白質功能[5-7]。依據ACMG指南分析,該變異生物學致病等級被判斷為致病性變異(PVS1+PM2+PP3)。功能缺失變異導致基因功能可能喪失(PVS1);ESP數據庫(https://esp.gs.washington.edu/drupal/)、千人基因組計劃數據庫(http://www.1000genomes.org/)、外顯子組聚集聯盟數據庫(http://exac.broadinstitute.org/)中正常對照人群中未發現(或隱性遺傳病中極低頻位點)(PM2);保守性預測、進化預測、剪接位點影響等方法預測該變異會對基因或基因產物造成有害影響(PP3)。結合臨床表現、基因檢測結果,最終診斷:雙眼RP。
圖5
色素性視網膜炎患者及其女兒、母親基因測序圖5A~5C分別示患者及其患者女兒、母親,PDE6B基因第5號外顯子的c.870G>A雜合變異(紅箭)
討論 RP是一組遺傳性、進行性光感受器細胞功能障礙、細胞丟失,以最終導致廣泛視網膜多層組織萎縮為特征,發病年齡從嬰幼兒期到中老年人[1]。RP存在多種類型,眼底表現差異較大,但共同規律是全視網膜、象限性視網膜出現大量骨細胞樣色素沉著、動靜脈血管變細、視網膜呈青灰色改變、視盤萎縮變白、視野縮窄等。即便是無色素性的RP,其視網膜色澤也較晦暗,伴血管和視盤改變;全視野ERG各波形振幅嚴重降低,甚至呈熄滅型。
McLaughlin等[2]于1993年首次發現RP患者存在PDE6B基因變異。磷酸二酯酶(PDE)是一個大家族,PDE6是其亞家族中的1種。由PDE6基因編碼的PDE蛋白水解環磷酸鳥苷(cGMP)。而cGMP是視桿細胞外節膜盤離子通道的特異性受體,同時也是脊椎動物中感光細胞將光信號轉化為電信號的重要分子。PDE6b基因缺失導致光感受器細胞內cGMP增多,引起光感受器細胞的陽離子增多導致細胞中毒而凋亡,視網膜光感受器細胞不斷破壞,從而間接影響視網膜接受光信號轉換為電信號。PDE6b基因異常是常染色體隱性遺傳RP最主要原因之一[3]。
本例患者雙眼視力正常,因近4年傍晚視物逐漸模糊,夜晚加重而就診。眼底檢查發現雙眼以視盤、黃斑為中心呈對稱性的環形視網膜、脈絡膜萎縮區,其間骨細胞樣色素沉著;周邊視網膜形態、顏色、血管床密度均正常。OCTA檢查可見萎縮區視網膜全層血管床密度和血流無明顯變化;脈絡膜中小毛細血管層血管床密度和血流均嚴重降低,脈絡膜大血管層改變相對較輕。這說明病變主要發生并集中于脈絡膜中小毛細血管層。向內波及到無血管層、RPE層,向外影響到脈絡膜大血管層。隨著RPE和脈絡膜毛細血管持續喪失,較大的脈絡膜血管可能發生變性[1]。我們推測,其發病機制是由于基因變異、脈絡膜毛細血管萎縮、血管床密度降低而導致RPE和視覺細胞層血供不足,致RPE和視細胞損害和凋亡。由于脈絡膜厚度周邊較薄,后極部較厚。故周邊部視桿細胞丟失更早、更重,患者較早即出現夜盲和視野缺失。本例患者雙眼RPE層除中心凹基本正常外,其脈絡膜萎縮區RPE層變薄、缺失,但周邊RPE層形態、厚度正常。與傳統RP表現不同,本例患者全視野ERG檢查除部分波形振幅稍降低外,其余各波形態完全正常。分析其原因是其絕大部分視網膜正常,多數視桿、視錐細胞未受累。此外,其視野呈與萎縮區一致的巨大環形暗點,周邊視野正常,也與傳統RP導致的管窺視野表現不一致。本例患者眼底表現完全不同于經典RP改變,臨床確診困難,最終經基因檢測發現PDE6B基因存在c.870G>A(p.W290X)雜合變異而確診。推測其眼底表現特殊的原因可能與PDE6B基因變異突變位點不同有關[2, 4-5]。
患者女兒、母親眼底檢查無明顯異常。基因檢測發現PDE6B基因均存在c.870G>A(p.W290X)雜合變異。推測該基因突變的變異位點導致本病發病時間約在患者40歲以后。患者女兒目前年齡29歲,其眼底檢查未發現異常可能是尚未到該病的發病年齡。臨床上應加強隨訪,同時針對此類患者的遺傳咨詢也有重要臨床意義。患者母親眼底正常,可能是臨床表型差異的原因。基因型控制生物個體的表現型,是表現型的決定性因素,但不是唯一決定性因素,與生物個體所處的特定環境條件同時發揮作用,使得表現型呈現出多樣性[6]。
本次檢測范圍為人類基因外顯子組區,不包含內含子區,不排除患者PDE6B等位基因存在內含子變異(構成復合雜合變異)從而導致疾病的發生。根據遺傳規律,常染色體隱性遺傳疾病患者的子女和父母,可能均為單個位點雜合攜帶者,這可能是母親和女兒無類似疾病表現的原因。目前已發現PDE6B基因變異與先天性靜止性夜盲2型和RP40型相關。本例患者臨床表現不符合前者,與RP40型更相似[7]。但家系不符合常染色體隱性遺傳病的遺傳規律,我們認為不排除部分變異導致的RP40型存在顯性遺傳的可能性,但該猜測證據較弱,需進一步研究。
本例患者眼底表現特殊,與傳統RP有著很大差異,應與中心性脈絡膜營養不良、視錐細胞營養不良、色素性靜脈旁視網膜脈絡膜萎縮等鑒別。中心性脈絡膜營養不良,后極部視網膜色澤較晦暗,病變主要圍繞血管弓和視神經,也可向內外蔓延,同時可累及黃斑。FAF呈與營養不良區域相一致的弱熒光區[8,本例患者眼底表現易與此混淆。視錐細胞營養不良患者疾病早期即出現視力下降,色覺障礙和眼球震顫,由CDHR1基因變異所致[9]。色素性靜脈旁視網膜脈絡膜萎縮在形態和分布上與RP有很大的差異。最后確診,除典型臨床表現外,更多應參考基因檢測結果。
志謝 感謝北京智德醫學檢驗所提供的專業基因檢測服務及其對本家族成員檢測結果的詳細解讀