遺傳性視網膜變性(IRD)是一組具有高度遺傳異質性和臨床異質性的眼底病,目前已發現超過300種基因突變與IRD有關。胞內第二信使環磷酸鳥苷(cGMP)的失調在多種IRD的發生和發展中發揮重要作用。cGMP在光感受器細胞中參與光轉導過程,當cGMP水平異常增高,則會過度激活蛋白激酶G和環核苷酸門控通道,分別引起蛋白質磷酸化和Ca2+超載,這兩種依賴cGMP的途徑可能單獨或共同驅動光感受器細胞退行性病變和死亡;因此,減少cGMP合成或阻止cGMP下游信號傳導可作為疾病的治療策略。研究cGMP失調在光感受器細胞退化過程中的分子機制可以更加全面地闡述IRD發病原因,同時為尋找新治療靶點和設計治療方案提供思路。
引用本文: 劉子實, 李彤, 孫曉東. 環磷酸鳥苷在遺傳性視網膜變性中的研究進展. 中華眼底病雜志, 2024, 40(11): 898-904. doi: 10.3760/cma.j.cn511434-20240701-00246 復制
版權信息: ?四川大學華西醫院華西期刊社《中華眼底病雜志》版權所有,未經授權不得轉載、改編
遺傳性視網膜變性(IRD)是一組罕見的視網膜疾病并導致視力逐漸喪失,其具有高度的遺傳異質性和臨床異質性且發病機制尚未完全闡明。環磷酸鳥苷(cGMP)的失調可能是多種IRD的共同通路[1-3]。高水平的cGMP過度激活蛋白激酶G(PKG)和環核苷酸門控通道(CNGC),分別引起蛋白質磷酸化和Ca2+超載,這兩種依賴cGMP的途徑可能單獨或共同驅動光感受器細胞退行性病變和死亡。現就cGMP在IRD中的研究進展作一綜述,以期為進一步理解和研究光感受器變性機制、找到有效的干預靶點提供理論基礎。
1 cGMP概述
cGMP作為第二信使廣泛存在于不同類型細胞,同時靶向多種下游分子并引發不同細胞效應[4]。光感受器細胞是高度分化的神經元,它們能夠靈敏捕獲光子并將光信號轉化為電信號,而cGMP在光感受器細胞中主要參與光轉導這一過程。
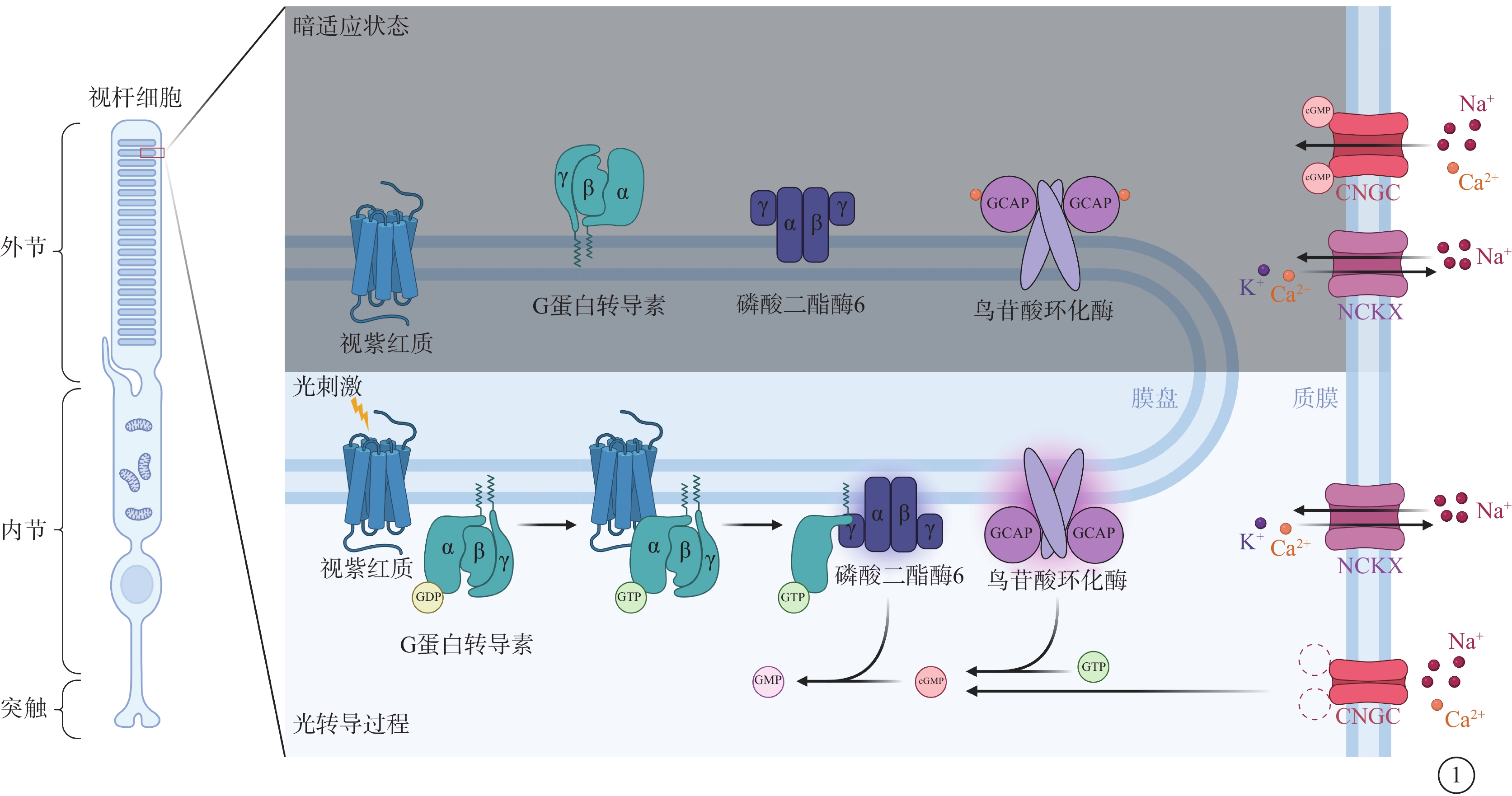
cGMP由視網膜特異性鳥苷酸環化酶(retGC)催化三磷酸鳥苷(GTP)生成,而retGC的活性受到Ca2+依賴的鳥苷酸環化酶激活蛋白(GCAP)的調節(圖1)。當Ca2+濃度低時,GCAP促進retGC產生cGMP;相反,在高Ca2+濃度下GCAP抑制retGC。光感受器細胞外節中Ca2+濃度取決于CNGC的開放程度,而CNGC的開放與高水平的cGMP相關,這種負反饋環路將cGMP的生理水平限制在一定范圍[5-7]。在光轉導過程中,光子使得視蛋白中的11-順式視黃醛異構化,并引發G蛋白轉導素和磷酸二酯酶6(PDE6)的連續激活。PDE6位于光感受器細胞外節的膜盤中,能夠水解cGMP并導致CNGC關閉和光感受器細胞超極化,進而突觸停止釋放谷氨酸。CNGC的關閉同樣降低了外節Ca2+水平,低水平的Ca2+能夠促進視紫紅質磷酸化并通過上述負反饋環路維持cGMP的水平,從而產生光適應[8]。原始光信號在光轉導級聯的每一步中被不斷放大,從而保證了光感受器細胞、尤其是視桿細胞具有顯著的單光子敏感性[9]。因此,cGMP和Ca2+的變化是光轉導級聯所涉及的眾多相關蛋白分子共同作用的結果,而影響cGMP相關調控分子的突變通常會導致cGMP和(或)Ca2+的失調,引發一系列下游效應,最終引起光感受器細胞變性。不同致病基因所引起的IRD及發病比例不同(表1)。
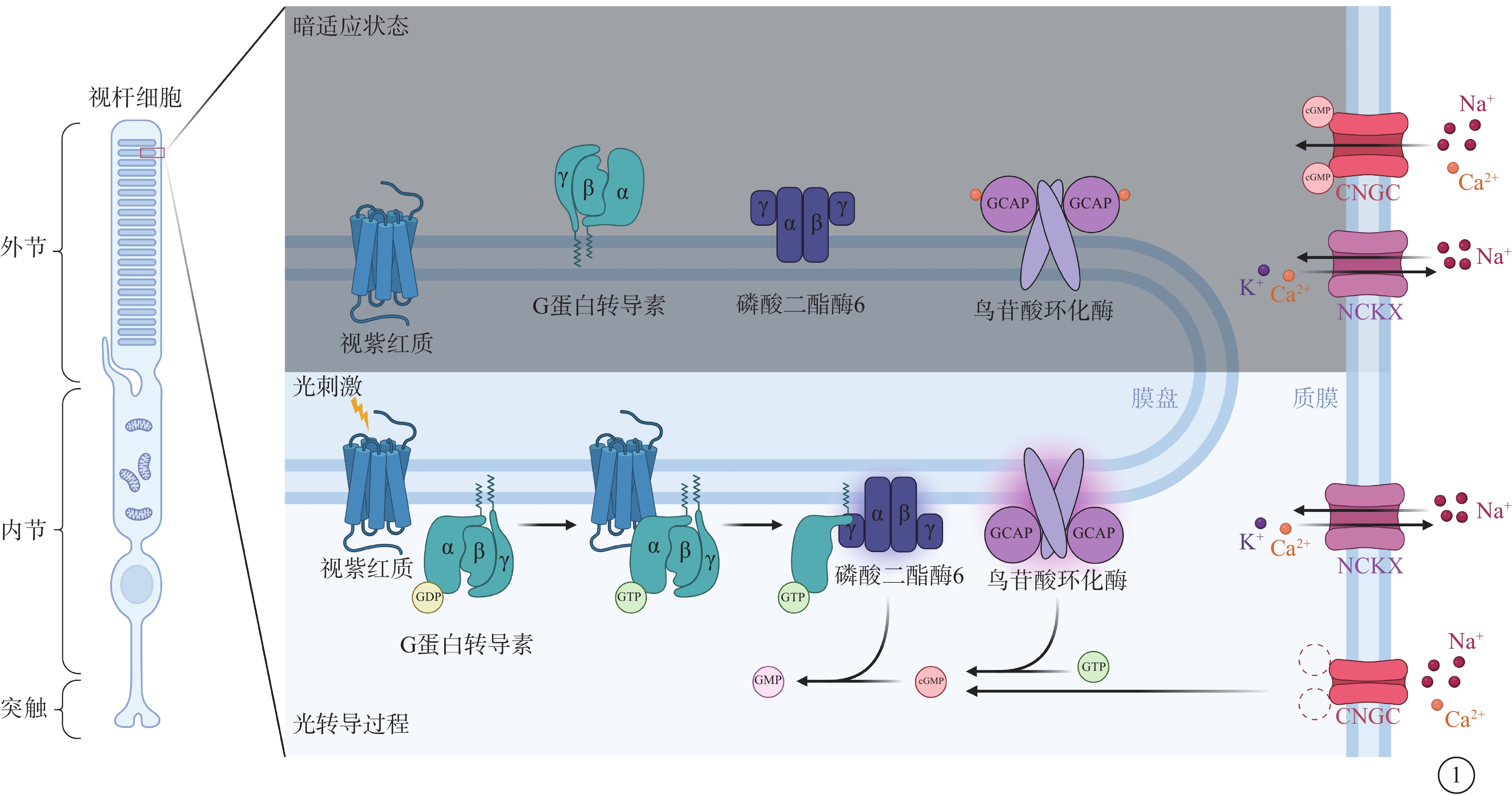 圖1
視桿細胞中暗適應狀態和光轉導過程示意圖 在暗適應狀態下,細胞內高水平cGMP結合CNGC并使其保持開放狀態,Na+和Ca2+流入,高水平的Ca2+與GCAP結合抑制其對retGC的激活并限制cGMP的生成,同時Ca2+通過NCKX流出以保持胞內Ca2+水平恒定。光轉導過程中,光刺激使得視紫紅質異構,從而激活G蛋白轉導素的α亞基脫離并與磷酸二酯酶6中的γ亞基結合,這使得磷酸二酯酶6被激活并催化cGMP水解,cGMP水平的降低使CNGC關閉,Ca2+流入減少,GCAP在無Ca2+結合的狀態下激活retGC 生成cGMP以保持胞內cGMP水平恒定 CNGC:環核苷酸門控通道;NCKX:K+依賴的Na+/Ca2+交換器;GCAP:鳥苷酸環化酶激活蛋白;retGC:視網膜特異性鳥苷酸環化酶;GTP:三磷酸鳥苷;GDP:二磷酸鳥苷;GMP:單磷酸鳥苷;cGMP:環磷酸鳥苷
圖1
視桿細胞中暗適應狀態和光轉導過程示意圖 在暗適應狀態下,細胞內高水平cGMP結合CNGC并使其保持開放狀態,Na+和Ca2+流入,高水平的Ca2+與GCAP結合抑制其對retGC的激活并限制cGMP的生成,同時Ca2+通過NCKX流出以保持胞內Ca2+水平恒定。光轉導過程中,光刺激使得視紫紅質異構,從而激活G蛋白轉導素的α亞基脫離并與磷酸二酯酶6中的γ亞基結合,這使得磷酸二酯酶6被激活并催化cGMP水解,cGMP水平的降低使CNGC關閉,Ca2+流入減少,GCAP在無Ca2+結合的狀態下激活retGC 生成cGMP以保持胞內cGMP水平恒定 CNGC:環核苷酸門控通道;NCKX:K+依賴的Na+/Ca2+交換器;GCAP:鳥苷酸環化酶激活蛋白;retGC:視網膜特異性鳥苷酸環化酶;GTP:三磷酸鳥苷;GDP:二磷酸鳥苷;GMP:單磷酸鳥苷;cGMP:環磷酸鳥苷
2 cGMP與相關調控分子
光感受器細胞中cGMP的水平受到嚴格的分子調控,包括生成和水解兩個部分。其中,cGMP的生成依賴于GCAP/retGC復合物,水解依賴于PDE6。同時通道開放程度與cGMP結合有關的CNGC也能通過引起外節Ca2+水平變化來間接影響GCAP/retGC復合物活性,從而影響cGMP水平。
2.1 GCAP/retGC復合物
GCAP/retGC復合物直接調控cGMP的生成。其中retGC有兩種亞型:retGC1(GUCY2D基因編碼)和retGC2(GUCY2F基因編碼)。retGC1在視桿細胞和視錐細胞中均有表達并負責大約70%以上cGMP的催化生成,而retGC2在視桿細胞中表達并負責剩余cGMP的生成[26]。其均由7個結構域組成:前導序列、胞外結構域、跨膜結構域、近膜結構域、激酶同源結構域、二聚結構域和催化結構域;retGC通過在二聚結構域形成同型二聚體并在催化結構域與兩個GTP底物結合發揮作用[27]。GCAP同樣有兩種亞型:GCAP1(GUCA1A基因編碼)和GCAP2(GUCA1B基因編碼)。GCAP1存在于視桿細胞和視錐細胞中并主要調控retGC1,而GCAP2主要存在于視桿細胞中調控retGC1和retGC2。GCAP的同型二聚體與retGC的同型二聚體結合形成GCAP/retGC復合物,該復合體的催化活性依賴于GCAP金屬結合域上的配體類型。當金屬結合域被Mg2+占據時,則激活retGC并刺激cGMP合成,反之被Ca2+占據時則抑制retGC并減少cGMP合成[28]。除了GCAP外,retGC的活性還受到視網膜變性蛋白3(RD3)的調節,在retGC內節合成之后到運輸至外節之前,RD3抑制GCAP對retGC的激活[29]。
目前已經在GUCY2D基因中鑒定出超過100種不同突變,包括功能喪失性突變引起的常染色體隱性遺傳的Leber先天性黑矇(arLCA)和功能獲得性突變引起的常染色體顯性遺傳的錐桿細胞營養不良(adCORD)[30]。在arLCA中,光感受器細胞因retGC1催化生成cGMP水平大幅度降低而不表現出光反應;而在adCORD中,突變導致retGC1功能增強,提高了對GCAP的敏感性并引起cGMP過量產生[31]。位于retGC1二聚化結構域上的R838S突變導致GCAP/retGC復合體的過度激活并引發adCORD,而通過在小鼠上敲除GCAP基因的表達則改善這一表型[32]。編碼GCAP的GUCA1A和GUCA1B基因也受到大量不同突變影響,功能喪失性突變(如Y99C和G86R突變)通常減少了Ca2+與GCAP的結合,導致GCAP對retGC的抑制作用缺乏和cGMP的過量產生[33-34]。此外RD3基因的功能喪失性突變也產生了類似的影響,在內節中retGC失去RD3基因對GCAP的抑制并導致cGMP過量產生[35]。因此retGC基因的功能獲得性突變與GCAP和RD3基因的功能喪失性產生了相同的效應,即共同引發了光感受器細胞中cGMP的過量產生和下游的CNGC通道開放、Ca2+內流增加。
2.2 PDE6
PDE6直接調控cGMP的水解。視桿細胞中PDE6由催化性的α亞基(PDE6A基因編碼)和β亞基(PDE6B基因編碼),以及兩個抑制性γ亞基(PDE6G編碼)組成;視錐細胞中 PDE6由兩個催化性的α亞基(PDE6C基因編碼)和兩個抑制性γ亞基(PDE6H基因編碼)組成。熱休克蛋白90和作為伴侶蛋白的芳烴受體結合蛋白樣1(AIPL1)負責將這些亞基組合成PDE6全酶[36-38]。在暗適應的光感受器細胞中,γ亞基持續抑制PDE6的催化活性;在光刺激下,G蛋白轉導素的α亞基(視桿細胞中由GNAT1基因編碼,視錐細胞中由GNAT2基因編碼)脫離并結合抑制PDE6中的γ亞基,從而使得PDE6被激活并催化cGMP水解[39]。
在PDE6相關基因中,PDE6A、PDE6B、PDE6G基因突變會導致高水平的cGMP,當致使視桿細胞死亡以及隨后的視錐細胞繼發性死亡時,通常會引起色素性視網膜炎(RP)。以PDE6B的7號外顯子無義突變為代表的rd1小鼠的視桿細胞中cGMP含量極高,在出生后第7天即出現視桿細胞外節紊亂,第21天視桿細胞完全消失;而與之對應的PDE6B的13號外顯子錯義突變為代表的rd10小鼠在表型上則要溫和許多,出生后第18天視桿細胞才開始退化,至第30天大部分死亡,這與rd10相較于rd1小鼠中較低的cGMP升高水平一致[40-42]。同樣的,在與RP患者PDE6A同源點突(V685M和R562W)的小鼠模型中也發現了cGMP的累積,并且在表型上顯示出與rd1和rd10小鼠相當的光感受器細胞損失速率[43]。而已知一些PDE6B突變僅導致視桿細胞光轉導功能喪失而非細胞死亡,從而引起常染色體顯性遺傳的先天性靜止性夜盲癥(adCSNB),但目前在PDE6B突變的adCSNB小鼠模型上尚未有cGMP水平的報道[44-45]。PDE6C和PDE6H突變與錐體營養不良和全色盲(ACHM)相關,以PDE6C移碼突變為代表的cpfl1小鼠顯示有大量的cGMP累積,視錐細胞在出生后21 d開始退化并在第5個月大部分死亡[1, 17, 46]。同樣的,AIPL1突變阻礙了PDE6全酶的功能組裝并導致cGMP積累和arLCA;而G蛋白轉導素中的GNAT1和GNAT2突變影響PDE6的激活并分別導致RP和ACHM[19, 47-48]。總而言之,PDE6相關基因中,與PDE6組成(PDE6A、PDE6B、PDE6C、PDE6G、PDE6H)、組裝(AIPL1)、激活(GNAT1、GNAT2)有關的突變幾乎均為功能喪失性,并直接或間接導致PDE6酶水解cGMP能力下降,引起cGMP累積和隨后的細胞退化。
2.3 CNGC
cGMP水平可以被CNGC通過Ca2+間接調控。光感受器細胞中CNGC是由三個負責通道特性的α亞基和一個負責外節定位的β亞基組成的異源四聚體,視桿細胞中為CNGA1亞基(CNGA1編碼)和CNGB1亞基(CNGB1編碼),視錐細胞中為CNGA3亞基(CNGA3編碼)和CNGB3亞基(CNGB3編碼)[49-50]。作為嚴格的環核苷酸門控的離子通道,光感受器細胞中CNGC需要與cGMP結合維持開放,Na+和Ca2+通過CNGC流入使細胞質膜去極化并促進突觸釋放谷氨酸,這一過程發生在暗適應的光感受器細胞中,因此Na+和Ca2+流入引發的電流變化也被稱為暗電流;同時胞內較高水平的Ca2+通過GCAP/retGC復合物抑制cGMP合成,這間接維持了離子流入和流出的平衡。當光感受器細胞受到光刺激時這一平衡被打破,cGMP水解導致CNGC關閉,Ca2+水平降低,隨后激活GCAP/retGC復合物。因此,CNGC在cGMP和Ca2+的反饋環路中起到了關鍵作用[50]。
與PDE6類似,CNGC相關基因中的突變大多數是功能喪失性突變。CNGA1和CNGB1突變與RP相關,而CNGA3和CNGB3基因突變與ACHM、CORD相關[51-52]。目前缺少對CNGA1小鼠模型中cGMP水平的報道,但在出生后第3周的CNGB1-/-的小鼠中發現了大量cGMP累積并與其外核層的退化過程一致[53]。由于視錐細胞僅占野生型小鼠視網膜總光感受器細胞的2%~3%,因此對CNGA3和CNGB3的研究集中在Nrl-/-背景的小鼠上,這類小鼠的視桿細胞被形態結構和生化分子近似于視錐細胞的光感受器細胞所代替。在CNGA3全敲除的Nrl-/-小鼠中,視錐細胞外節Ca2+濃度降低并在出生后第8天出現cGMP累積[54]。與PDE6相關基因突變使得cGMP水解減少來增加cGMP水平相反,CNGC相關基因中的突變通過引發Ca2+水平降低以及隨后的GCAP/retGC復合物持續激活使得cGMP過量產生。
2.4 其他潛在調控分子
與cGMP生成或水解不相關的兩種基因突變的動物模型中也觀察到了高水平的cGMP。視紫紅質(RHO基因編碼)中的P23H和S334ter基因突變在小鼠的光感受器細胞中顯示出大量cGMP累積[1]。這可能與突變的RHO無法通過激活G蛋白轉導素來激活下游PDE6有關。外周蛋白2(PRPH2基因編碼)的突變同樣觀察到了cGMP的累積,PRPH2基因突變與外節缺失有關,這可能進一步導致了cGMP調節相關分子的表達失調或異常激活[1, 55]。
3 cGMP與光感受器細胞變性
3.1 PKG信號過度激活
PKG作為cGMP唯一靶向的酶,在體內多種組織如心血管、大腦、視網膜中廣泛參與各種生理過程。PKG的表達和活性上調已經在多種cGMP相關調控分子缺陷的小鼠上得到證明[54, 56-57]。對野生型小鼠給予能夠選擇性激活PKG的cGMP類似物,同樣觀察到了嚴重的光感受器細胞變性[55]。對cGMP失調小鼠進行PKG抑制,雖然不能阻止光感受器細胞繼續變性,但能夠延緩退化時間[2, 55]。PKG過度激活引發的下游效應包括內質網穩態受損和多聚ADP核糖聚合酶(PARP)激活。為了避免高水平Ca2+對光感受器細胞變性帶來的額外影響,這類研究通常集中在CNGC缺陷的小鼠上。CNGC缺陷小鼠內質網穩態受損主要體現在相關的標志蛋白如葡萄糖調節蛋白(GRP78/BiP)、真核翻譯起始因子2α的增加,這些應激因子啟動了未折疊蛋白反應,最終導致細胞死亡[58-59]。同時內質網穩態破壞使得位于內質網上的半胱氨酸蛋白酶7和半胱氨酸蛋白酶12的相繼激活,這進一步促進了細胞死亡[60]。PARP是一種DNA修復酶,其激活會導致細胞程序性死亡,通過使用組蛋白去乙酰化酶(HDAC)抑制劑證明了PARP激活與HDAC的相關[61]。目前認為PKG是HDAC的上游催化劑,但PKG和HDAC激活的中間過程尚未闡明。
3.2 Ca2+超載
除了CNGC相關基因的突變外,cGMP的失調性升高將導致CNGC的持續開放和隨后的胞內Ca2+超載。Ca2+超載引起的光感受器細胞變性被認為與鈣蛋白酶的過度激活有關,從而降解細胞骨架、膜蛋白以及各種激酶以及轉錄因子[62]。同時鈣蛋白酶的激活還誘導線粒體釋放凋亡誘導因子,引發DNA片段化和染色質濃縮,進一步促進了上述過程[63]。通過敲除CNGA1、CNGB1延緩了相應cGMP失調突變小鼠的光感受器細胞死亡;但敲除CNGC相關基因后Ca2+水平降低,cGMP仍然處于病理性的高水平[53, 64]。盡管光感受器細胞的死亡進程并不會被阻止,但其存活時間和外節形態都有極大的改善,同時cGMP失調突變小鼠中鈣蛋白酶活性的變化與細胞死亡程度同步,這提示Ca2+超載可能相較PKG信號過度激活在光感受器細胞死亡中的作用更大[65]。
4 靶向cGMP的治療開發
4.1 減少cGMP合成
考慮到cGMP作為第二信使廣泛參與體內不同組織細胞的生理活動,所有減少cGMP合成的治療必須要具有光感受器細胞特異性,同時一些導致cGMP失調的基因僅在視錐細胞或視桿細胞中表達,這在治療中也要區分開。一種方法是抑制retGC,Garger等[66]發現Rp-GTPαS對retGC具有很高的特異性,但是Rp-GTPαS攜帶3個負電荷,具有高度的膜滲透性,因此其應用還需要高特異性的藥物遞送系統實現。另一種方法是減少cGMP生成所需要的底物GTP,次黃嘌呤單核苷酸脫氫酶(IMPDH)是GTP生成的催化酶,而麥考酚酸酯(MMF)能夠選擇性抑制IMPDH從而減少GTP的生成[67]。MMF目前在rd1和rd10小鼠中被證明能夠降低光感受器細胞cGMP水平并顯著延緩光感受器細胞變性,目前還需要大量的臨床前數據來推進其在臨床上的應用開發[68]。
4.2 阻斷cGMP信號傳導
cGMP信號的下游效應器PKG和CNGC,分別導致PKG信號過度激活和Ca2+超載,也是治療開發需要關注的靶點。與Rp-GTPαS類似,多項研究報道了專門設計的抑制性cGMP類似物來與PKG和(或)CNGC結合,并在rd1、rd2和rd10小鼠中表現出顯著的神經保護特性[2, 69-71]。其他幾種抑制劑包括寡肽DT-2、KT5823、N46等缺少在動物模型中的驗證[72-73]。由于目前PKG信號過度激活和Ca2+超載所引發光感受器細胞變性的具體機制未被完全闡明,同時缺少對關鍵分子的探索,這使得相關治療的開發仍然有限。
5 小結與展望
IRD中光感受器細胞變性的機制復雜且具有交叉性,而cGMP相關基因在IRD中的發病比例非常可觀,這提示cGMP失調是光感受器細胞變性不可忽視的一個原因。基于對cGMP失調和其引發的下游PKG信號過度激活和Ca2+超載的研究,減少cGMP合成和阻止下游信號傳導是兩項重要的治療開發策略。然而目前對cGMP引發光感受器細胞死亡的研究尚不完善,缺乏動物實驗以及臨床試驗數據。進一步明確IRD中cGMP所引起的光感受器細胞死亡機制中各條通路的相互作用關系、下游的關鍵分子以及上游始動機制等均有助于了解IRD的病理生理過程及找到有效的藥物治療策略以挽救或延緩光感受器細胞變性。
遺傳性視網膜變性(IRD)是一組罕見的視網膜疾病并導致視力逐漸喪失,其具有高度的遺傳異質性和臨床異質性且發病機制尚未完全闡明。環磷酸鳥苷(cGMP)的失調可能是多種IRD的共同通路[1-3]。高水平的cGMP過度激活蛋白激酶G(PKG)和環核苷酸門控通道(CNGC),分別引起蛋白質磷酸化和Ca2+超載,這兩種依賴cGMP的途徑可能單獨或共同驅動光感受器細胞退行性病變和死亡。現就cGMP在IRD中的研究進展作一綜述,以期為進一步理解和研究光感受器變性機制、找到有效的干預靶點提供理論基礎。
1 cGMP概述
cGMP作為第二信使廣泛存在于不同類型細胞,同時靶向多種下游分子并引發不同細胞效應[4]。光感受器細胞是高度分化的神經元,它們能夠靈敏捕獲光子并將光信號轉化為電信號,而cGMP在光感受器細胞中主要參與光轉導這一過程。
cGMP由視網膜特異性鳥苷酸環化酶(retGC)催化三磷酸鳥苷(GTP)生成,而retGC的活性受到Ca2+依賴的鳥苷酸環化酶激活蛋白(GCAP)的調節(圖1)。當Ca2+濃度低時,GCAP促進retGC產生cGMP;相反,在高Ca2+濃度下GCAP抑制retGC。光感受器細胞外節中Ca2+濃度取決于CNGC的開放程度,而CNGC的開放與高水平的cGMP相關,這種負反饋環路將cGMP的生理水平限制在一定范圍[5-7]。在光轉導過程中,光子使得視蛋白中的11-順式視黃醛異構化,并引發G蛋白轉導素和磷酸二酯酶6(PDE6)的連續激活。PDE6位于光感受器細胞外節的膜盤中,能夠水解cGMP并導致CNGC關閉和光感受器細胞超極化,進而突觸停止釋放谷氨酸。CNGC的關閉同樣降低了外節Ca2+水平,低水平的Ca2+能夠促進視紫紅質磷酸化并通過上述負反饋環路維持cGMP的水平,從而產生光適應[8]。原始光信號在光轉導級聯的每一步中被不斷放大,從而保證了光感受器細胞、尤其是視桿細胞具有顯著的單光子敏感性[9]。因此,cGMP和Ca2+的變化是光轉導級聯所涉及的眾多相關蛋白分子共同作用的結果,而影響cGMP相關調控分子的突變通常會導致cGMP和(或)Ca2+的失調,引發一系列下游效應,最終引起光感受器細胞變性。不同致病基因所引起的IRD及發病比例不同(表1)。
圖1
視桿細胞中暗適應狀態和光轉導過程示意圖 在暗適應狀態下,細胞內高水平cGMP結合CNGC并使其保持開放狀態,Na+和Ca2+流入,高水平的Ca2+與GCAP結合抑制其對retGC的激活并限制cGMP的生成,同時Ca2+通過NCKX流出以保持胞內Ca2+水平恒定。光轉導過程中,光刺激使得視紫紅質異構,從而激活G蛋白轉導素的α亞基脫離并與磷酸二酯酶6中的γ亞基結合,這使得磷酸二酯酶6被激活并催化cGMP水解,cGMP水平的降低使CNGC關閉,Ca2+流入減少,GCAP在無Ca2+結合的狀態下激活retGC 生成cGMP以保持胞內cGMP水平恒定 CNGC:環核苷酸門控通道;NCKX:K+依賴的Na+/Ca2+交換器;GCAP:鳥苷酸環化酶激活蛋白;retGC:視網膜特異性鳥苷酸環化酶;GTP:三磷酸鳥苷;GDP:二磷酸鳥苷;GMP:單磷酸鳥苷;cGMP:環磷酸鳥苷
2 cGMP與相關調控分子
光感受器細胞中cGMP的水平受到嚴格的分子調控,包括生成和水解兩個部分。其中,cGMP的生成依賴于GCAP/retGC復合物,水解依賴于PDE6。同時通道開放程度與cGMP結合有關的CNGC也能通過引起外節Ca2+水平變化來間接影響GCAP/retGC復合物活性,從而影響cGMP水平。
2.1 GCAP/retGC復合物
GCAP/retGC復合物直接調控cGMP的生成。其中retGC有兩種亞型:retGC1(GUCY2D基因編碼)和retGC2(GUCY2F基因編碼)。retGC1在視桿細胞和視錐細胞中均有表達并負責大約70%以上cGMP的催化生成,而retGC2在視桿細胞中表達并負責剩余cGMP的生成[26]。其均由7個結構域組成:前導序列、胞外結構域、跨膜結構域、近膜結構域、激酶同源結構域、二聚結構域和催化結構域;retGC通過在二聚結構域形成同型二聚體并在催化結構域與兩個GTP底物結合發揮作用[27]。GCAP同樣有兩種亞型:GCAP1(GUCA1A基因編碼)和GCAP2(GUCA1B基因編碼)。GCAP1存在于視桿細胞和視錐細胞中并主要調控retGC1,而GCAP2主要存在于視桿細胞中調控retGC1和retGC2。GCAP的同型二聚體與retGC的同型二聚體結合形成GCAP/retGC復合物,該復合體的催化活性依賴于GCAP金屬結合域上的配體類型。當金屬結合域被Mg2+占據時,則激活retGC并刺激cGMP合成,反之被Ca2+占據時則抑制retGC并減少cGMP合成[28]。除了GCAP外,retGC的活性還受到視網膜變性蛋白3(RD3)的調節,在retGC內節合成之后到運輸至外節之前,RD3抑制GCAP對retGC的激活[29]。
目前已經在GUCY2D基因中鑒定出超過100種不同突變,包括功能喪失性突變引起的常染色體隱性遺傳的Leber先天性黑矇(arLCA)和功能獲得性突變引起的常染色體顯性遺傳的錐桿細胞營養不良(adCORD)[30]。在arLCA中,光感受器細胞因retGC1催化生成cGMP水平大幅度降低而不表現出光反應;而在adCORD中,突變導致retGC1功能增強,提高了對GCAP的敏感性并引起cGMP過量產生[31]。位于retGC1二聚化結構域上的R838S突變導致GCAP/retGC復合體的過度激活并引發adCORD,而通過在小鼠上敲除GCAP基因的表達則改善這一表型[32]。編碼GCAP的GUCA1A和GUCA1B基因也受到大量不同突變影響,功能喪失性突變(如Y99C和G86R突變)通常減少了Ca2+與GCAP的結合,導致GCAP對retGC的抑制作用缺乏和cGMP的過量產生[33-34]。此外RD3基因的功能喪失性突變也產生了類似的影響,在內節中retGC失去RD3基因對GCAP的抑制并導致cGMP過量產生[35]。因此retGC基因的功能獲得性突變與GCAP和RD3基因的功能喪失性產生了相同的效應,即共同引發了光感受器細胞中cGMP的過量產生和下游的CNGC通道開放、Ca2+內流增加。
2.2 PDE6
PDE6直接調控cGMP的水解。視桿細胞中PDE6由催化性的α亞基(PDE6A基因編碼)和β亞基(PDE6B基因編碼),以及兩個抑制性γ亞基(PDE6G編碼)組成;視錐細胞中 PDE6由兩個催化性的α亞基(PDE6C基因編碼)和兩個抑制性γ亞基(PDE6H基因編碼)組成。熱休克蛋白90和作為伴侶蛋白的芳烴受體結合蛋白樣1(AIPL1)負責將這些亞基組合成PDE6全酶[36-38]。在暗適應的光感受器細胞中,γ亞基持續抑制PDE6的催化活性;在光刺激下,G蛋白轉導素的α亞基(視桿細胞中由GNAT1基因編碼,視錐細胞中由GNAT2基因編碼)脫離并結合抑制PDE6中的γ亞基,從而使得PDE6被激活并催化cGMP水解[39]。
在PDE6相關基因中,PDE6A、PDE6B、PDE6G基因突變會導致高水平的cGMP,當致使視桿細胞死亡以及隨后的視錐細胞繼發性死亡時,通常會引起色素性視網膜炎(RP)。以PDE6B的7號外顯子無義突變為代表的rd1小鼠的視桿細胞中cGMP含量極高,在出生后第7天即出現視桿細胞外節紊亂,第21天視桿細胞完全消失;而與之對應的PDE6B的13號外顯子錯義突變為代表的rd10小鼠在表型上則要溫和許多,出生后第18天視桿細胞才開始退化,至第30天大部分死亡,這與rd10相較于rd1小鼠中較低的cGMP升高水平一致[40-42]。同樣的,在與RP患者PDE6A同源點突(V685M和R562W)的小鼠模型中也發現了cGMP的累積,并且在表型上顯示出與rd1和rd10小鼠相當的光感受器細胞損失速率[43]。而已知一些PDE6B突變僅導致視桿細胞光轉導功能喪失而非細胞死亡,從而引起常染色體顯性遺傳的先天性靜止性夜盲癥(adCSNB),但目前在PDE6B突變的adCSNB小鼠模型上尚未有cGMP水平的報道[44-45]。PDE6C和PDE6H突變與錐體營養不良和全色盲(ACHM)相關,以PDE6C移碼突變為代表的cpfl1小鼠顯示有大量的cGMP累積,視錐細胞在出生后21 d開始退化并在第5個月大部分死亡[1, 17, 46]。同樣的,AIPL1突變阻礙了PDE6全酶的功能組裝并導致cGMP積累和arLCA;而G蛋白轉導素中的GNAT1和GNAT2突變影響PDE6的激活并分別導致RP和ACHM[19, 47-48]。總而言之,PDE6相關基因中,與PDE6組成(PDE6A、PDE6B、PDE6C、PDE6G、PDE6H)、組裝(AIPL1)、激活(GNAT1、GNAT2)有關的突變幾乎均為功能喪失性,并直接或間接導致PDE6酶水解cGMP能力下降,引起cGMP累積和隨后的細胞退化。
2.3 CNGC
cGMP水平可以被CNGC通過Ca2+間接調控。光感受器細胞中CNGC是由三個負責通道特性的α亞基和一個負責外節定位的β亞基組成的異源四聚體,視桿細胞中為CNGA1亞基(CNGA1編碼)和CNGB1亞基(CNGB1編碼),視錐細胞中為CNGA3亞基(CNGA3編碼)和CNGB3亞基(CNGB3編碼)[49-50]。作為嚴格的環核苷酸門控的離子通道,光感受器細胞中CNGC需要與cGMP結合維持開放,Na+和Ca2+通過CNGC流入使細胞質膜去極化并促進突觸釋放谷氨酸,這一過程發生在暗適應的光感受器細胞中,因此Na+和Ca2+流入引發的電流變化也被稱為暗電流;同時胞內較高水平的Ca2+通過GCAP/retGC復合物抑制cGMP合成,這間接維持了離子流入和流出的平衡。當光感受器細胞受到光刺激時這一平衡被打破,cGMP水解導致CNGC關閉,Ca2+水平降低,隨后激活GCAP/retGC復合物。因此,CNGC在cGMP和Ca2+的反饋環路中起到了關鍵作用[50]。
與PDE6類似,CNGC相關基因中的突變大多數是功能喪失性突變。CNGA1和CNGB1突變與RP相關,而CNGA3和CNGB3基因突變與ACHM、CORD相關[51-52]。目前缺少對CNGA1小鼠模型中cGMP水平的報道,但在出生后第3周的CNGB1-/-的小鼠中發現了大量cGMP累積并與其外核層的退化過程一致[53]。由于視錐細胞僅占野生型小鼠視網膜總光感受器細胞的2%~3%,因此對CNGA3和CNGB3的研究集中在Nrl-/-背景的小鼠上,這類小鼠的視桿細胞被形態結構和生化分子近似于視錐細胞的光感受器細胞所代替。在CNGA3全敲除的Nrl-/-小鼠中,視錐細胞外節Ca2+濃度降低并在出生后第8天出現cGMP累積[54]。與PDE6相關基因突變使得cGMP水解減少來增加cGMP水平相反,CNGC相關基因中的突變通過引發Ca2+水平降低以及隨后的GCAP/retGC復合物持續激活使得cGMP過量產生。
2.4 其他潛在調控分子
與cGMP生成或水解不相關的兩種基因突變的動物模型中也觀察到了高水平的cGMP。視紫紅質(RHO基因編碼)中的P23H和S334ter基因突變在小鼠的光感受器細胞中顯示出大量cGMP累積[1]。這可能與突變的RHO無法通過激活G蛋白轉導素來激活下游PDE6有關。外周蛋白2(PRPH2基因編碼)的突變同樣觀察到了cGMP的累積,PRPH2基因突變與外節缺失有關,這可能進一步導致了cGMP調節相關分子的表達失調或異常激活[1, 55]。
3 cGMP與光感受器細胞變性
3.1 PKG信號過度激活
PKG作為cGMP唯一靶向的酶,在體內多種組織如心血管、大腦、視網膜中廣泛參與各種生理過程。PKG的表達和活性上調已經在多種cGMP相關調控分子缺陷的小鼠上得到證明[54, 56-57]。對野生型小鼠給予能夠選擇性激活PKG的cGMP類似物,同樣觀察到了嚴重的光感受器細胞變性[55]。對cGMP失調小鼠進行PKG抑制,雖然不能阻止光感受器細胞繼續變性,但能夠延緩退化時間[2, 55]。PKG過度激活引發的下游效應包括內質網穩態受損和多聚ADP核糖聚合酶(PARP)激活。為了避免高水平Ca2+對光感受器細胞變性帶來的額外影響,這類研究通常集中在CNGC缺陷的小鼠上。CNGC缺陷小鼠內質網穩態受損主要體現在相關的標志蛋白如葡萄糖調節蛋白(GRP78/BiP)、真核翻譯起始因子2α的增加,這些應激因子啟動了未折疊蛋白反應,最終導致細胞死亡[58-59]。同時內質網穩態破壞使得位于內質網上的半胱氨酸蛋白酶7和半胱氨酸蛋白酶12的相繼激活,這進一步促進了細胞死亡[60]。PARP是一種DNA修復酶,其激活會導致細胞程序性死亡,通過使用組蛋白去乙酰化酶(HDAC)抑制劑證明了PARP激活與HDAC的相關[61]。目前認為PKG是HDAC的上游催化劑,但PKG和HDAC激活的中間過程尚未闡明。
3.2 Ca2+超載
除了CNGC相關基因的突變外,cGMP的失調性升高將導致CNGC的持續開放和隨后的胞內Ca2+超載。Ca2+超載引起的光感受器細胞變性被認為與鈣蛋白酶的過度激活有關,從而降解細胞骨架、膜蛋白以及各種激酶以及轉錄因子[62]。同時鈣蛋白酶的激活還誘導線粒體釋放凋亡誘導因子,引發DNA片段化和染色質濃縮,進一步促進了上述過程[63]。通過敲除CNGA1、CNGB1延緩了相應cGMP失調突變小鼠的光感受器細胞死亡;但敲除CNGC相關基因后Ca2+水平降低,cGMP仍然處于病理性的高水平[53, 64]。盡管光感受器細胞的死亡進程并不會被阻止,但其存活時間和外節形態都有極大的改善,同時cGMP失調突變小鼠中鈣蛋白酶活性的變化與細胞死亡程度同步,這提示Ca2+超載可能相較PKG信號過度激活在光感受器細胞死亡中的作用更大[65]。
4 靶向cGMP的治療開發
4.1 減少cGMP合成
考慮到cGMP作為第二信使廣泛參與體內不同組織細胞的生理活動,所有減少cGMP合成的治療必須要具有光感受器細胞特異性,同時一些導致cGMP失調的基因僅在視錐細胞或視桿細胞中表達,這在治療中也要區分開。一種方法是抑制retGC,Garger等[66]發現Rp-GTPαS對retGC具有很高的特異性,但是Rp-GTPαS攜帶3個負電荷,具有高度的膜滲透性,因此其應用還需要高特異性的藥物遞送系統實現。另一種方法是減少cGMP生成所需要的底物GTP,次黃嘌呤單核苷酸脫氫酶(IMPDH)是GTP生成的催化酶,而麥考酚酸酯(MMF)能夠選擇性抑制IMPDH從而減少GTP的生成[67]。MMF目前在rd1和rd10小鼠中被證明能夠降低光感受器細胞cGMP水平并顯著延緩光感受器細胞變性,目前還需要大量的臨床前數據來推進其在臨床上的應用開發[68]。
4.2 阻斷cGMP信號傳導
cGMP信號的下游效應器PKG和CNGC,分別導致PKG信號過度激活和Ca2+超載,也是治療開發需要關注的靶點。與Rp-GTPαS類似,多項研究報道了專門設計的抑制性cGMP類似物來與PKG和(或)CNGC結合,并在rd1、rd2和rd10小鼠中表現出顯著的神經保護特性[2, 69-71]。其他幾種抑制劑包括寡肽DT-2、KT5823、N46等缺少在動物模型中的驗證[72-73]。由于目前PKG信號過度激活和Ca2+超載所引發光感受器細胞變性的具體機制未被完全闡明,同時缺少對關鍵分子的探索,這使得相關治療的開發仍然有限。
5 小結與展望
IRD中光感受器細胞變性的機制復雜且具有交叉性,而cGMP相關基因在IRD中的發病比例非常可觀,這提示cGMP失調是光感受器細胞變性不可忽視的一個原因。基于對cGMP失調和其引發的下游PKG信號過度激活和Ca2+超載的研究,減少cGMP合成和阻止下游信號傳導是兩項重要的治療開發策略。然而目前對cGMP引發光感受器細胞死亡的研究尚不完善,缺乏動物實驗以及臨床試驗數據。進一步明確IRD中cGMP所引起的光感受器細胞死亡機制中各條通路的相互作用關系、下游的關鍵分子以及上游始動機制等均有助于了解IRD的病理生理過程及找到有效的藥物治療策略以挽救或延緩光感受器細胞變性。