版權信息: ?四川大學華西醫院華西期刊社《中華眼底病雜志》版權所有,未經授權不得轉載、改編
患者女,38歲。因雙眼不明原因中心視力緩慢進展性下降8年加重1年,于2022年8月24日到河北省邯鄲市眼科醫院(邯鄲市第三醫院)就診。既往身體健康,否認其他全身系統性疾病史。眼科檢查:右眼、左眼最佳矯正視力(BCVA)分別為0.15、0.12。右眼、左眼眼壓分別為14、15 mm Hg(1 mm Hg=0.133 kPa)。雙眼眼前節檢查未見明顯異常。眼底檢查,近紅外成像(NIR)檢查,雙眼黃斑中心區反射信號弱(圖1A、1B)。藍光自發熒光(BAF)檢查,黃斑區可見高自發熒光(圖1C、1D)。光相干斷層掃描(OCT)檢查,雙眼黃斑區橢圓體帶(EZ)、嵌合體帶(IZ)反射信號減弱、紊亂(圖1E、1F)。OCT血管成像(OCTA)定量分析發現,淺層毛細血管叢(SCP)和深層毛細血管叢(DCP)的血流密度均降低(圖1G、1H)。Humphrey視野檢查,雙眼視野中心暗點(圖1I、1J)。多焦點視網膜電圖(ERG)檢查,雙眼黃斑區中心凹尖峰振幅密度下降(圖1K、1L)。閃光視覺誘發電位檢查,雙眼各波潛伏期稍有延長(圖1M、1N)。征求患者及父親和姑姑同意后行全外顯子基因檢測。結果顯示,患者攜帶RP1L1基因錯義突變及NM_178857:exon4:c.2880G>T:(p.Trp960Cys)突變點位。其父親和姑姑均無基因變異。診斷:隱匿性黃斑營養不良(OMD)(圖2)。對患者進行隨訪觀察,2年后患者于我院復查,OCT檢查,雙眼中央視網膜厚度變薄(圖3A、3B)。
 圖1
RP1L1基因變異相關隱匿性黃斑營養不良患者初診雙眼眼部檢查像 1A、1B分別示右眼、左眼近紅外成像,黃斑中心區反射信號稍有減弱。1C、1D分別示藍光自發熒光像,黃斑區可見斑點狀強自發熒光。1E、1F分別示右眼、左眼光相干斷層掃描像,左圖為掃描方向和部位,右圖為檢查結果,黃斑區橢圓體帶、嵌合體帶反射信號減弱、紊亂。1G、1H分別示右眼、左眼光相干斷層掃描血管成像,左圖為淺層毛細血管叢像,右圖為深層毛細血管叢像,黃斑區淺層毛細血管叢、深層毛細血管叢血流密度均有降低。1I、1J分別示Humphrey視野檢查結果,視野表現為中心暗點。1K、1L分別示右眼、左眼多焦點視網膜電圖,黃斑區中心凹尖峰振幅密度下降。1M、1N分別示右眼、左眼閃光視覺誘發電位,各波潛伏期稍有延長
圖1
RP1L1基因變異相關隱匿性黃斑營養不良患者初診雙眼眼部檢查像 1A、1B分別示右眼、左眼近紅外成像,黃斑中心區反射信號稍有減弱。1C、1D分別示藍光自發熒光像,黃斑區可見斑點狀強自發熒光。1E、1F分別示右眼、左眼光相干斷層掃描像,左圖為掃描方向和部位,右圖為檢查結果,黃斑區橢圓體帶、嵌合體帶反射信號減弱、紊亂。1G、1H分別示右眼、左眼光相干斷層掃描血管成像,左圖為淺層毛細血管叢像,右圖為深層毛細血管叢像,黃斑區淺層毛細血管叢、深層毛細血管叢血流密度均有降低。1I、1J分別示Humphrey視野檢查結果,視野表現為中心暗點。1K、1L分別示右眼、左眼多焦點視網膜電圖,黃斑區中心凹尖峰振幅密度下降。1M、1N分別示右眼、左眼閃光視覺誘發電位,各波潛伏期稍有延長
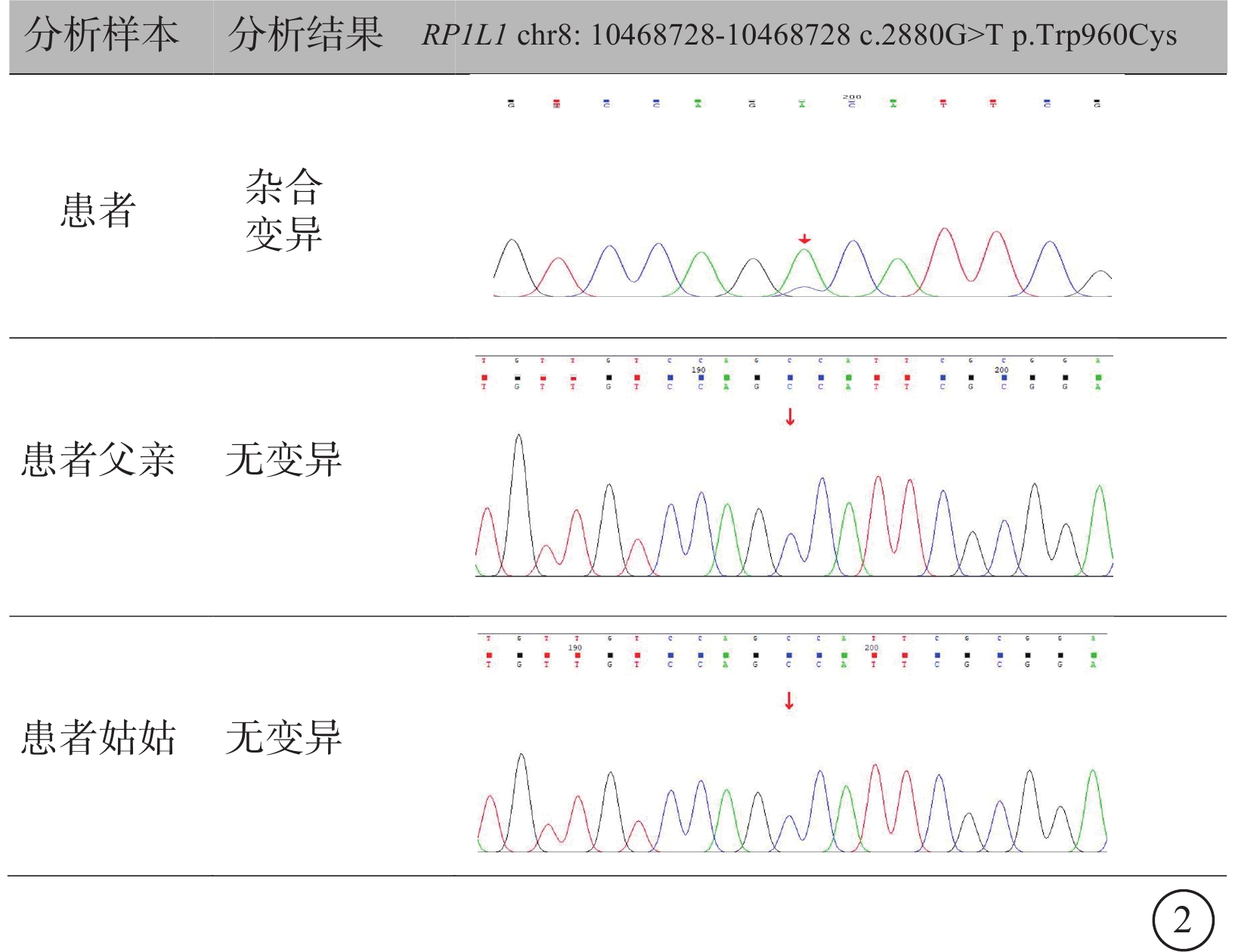
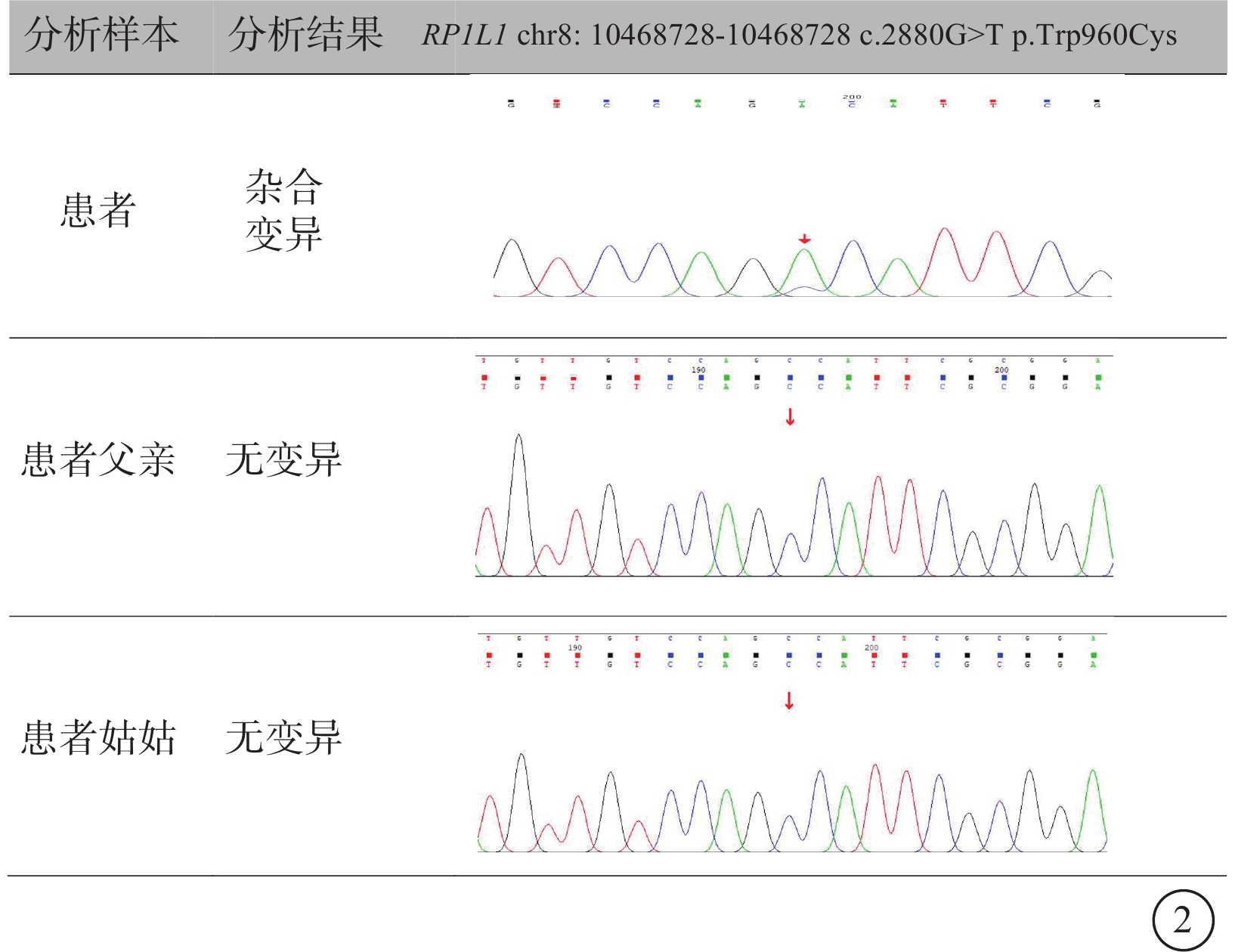 圖2
RP1L1基因變異相關隱匿性黃斑營養不良一家系基因測序圖 患者攜帶RP1L1基因錯義突變及NM_178857: c.2880G>TP.Trp960Cys突變點位;患者父親和姑姑均無基因變異
圖2
RP1L1基因變異相關隱匿性黃斑營養不良一家系基因測序圖 患者攜帶RP1L1基因錯義突變及NM_178857: c.2880G>TP.Trp960Cys突變點位;患者父親和姑姑均無基因變異
 圖3
RP1L1基因變異相關隱匿性黃斑營養不良患者復查雙眼光相干斷層掃描像 3A、3B分別示右眼、左眼, 左圖為掃描方向和部位,右圖為檢查結果,雙眼中央視網膜厚度變薄
圖3
RP1L1基因變異相關隱匿性黃斑營養不良患者復查雙眼光相干斷層掃描像 3A、3B分別示右眼、左眼, 左圖為掃描方向和部位,右圖為檢查結果,雙眼中央視網膜厚度變薄
討論 OMD是一種遺傳性黃斑營養不良癥,罕見的常染色體顯性遺傳病。1989年,Miyake等[1]首次描述該病。1996年,Miyake等[2]將其命名OMD。RP1L1基因變異與OMD和視網膜色素變性有關[3]。RP1L1基因突變是OMD中最常見的病理突變,最常見的變異位點是c.133C>T[4-5]。
OMD臨床特征表現視力進行性下降[6]。本例患者主要臨床表現為雙眼中心視力進行性下降就診。NIR檢查可見雙眼黃斑中心區局限性弱反射信號,由于感光細胞破壞[7]。OCT檢查,雙眼黃斑區EZ、IZ反射信號明顯減弱、紊亂、變薄,錐細胞密度降低,RP1L1基因突變引起視錐細胞營養不良致使視錐細胞功能障礙,視野表現為中心暗點[8-9]。OCTA檢查,患者雙眼視網膜內毛細血管叢受損,脈絡膜毛細血管血流相對少的情況造成SCP和DCP的血流密度均降低[10]。
OMD需與Stargardt病、球后視神經炎及卵黃樣黃斑營養不良(Best)等疾病相鑒別。Stargardt病黃斑區呈牛眼狀典型特征及視網膜下脂褐素樣沉積物[11];而OMD眼底檢查多正常,即使晚期光感受器也不完全萎縮,視力多維持在0.1~0.2或更高[12-13]。球后視神經炎患者多單眼急性視力下降,伴眼球轉動痛,晚期視神經萎縮,糖皮質激素治療有效;OMD則表現為緩慢視力下降,無眼球轉動痛,對糖皮質激素治療無效,多焦ERG檢查可見微小改變[14-15]。OCT、眼底自發熒光檢查可見Best患者黃斑區卵黃樣堆積物;OMD患者無上述典型改變。
本例患者RP1L1基因上檢測到一個雜合錯義變異NM_178857:exon4:c.2880G>T: (p.Trp960Cys),位于染色體8p23.1。該變異位點未被ClinVar數據庫及人類基因突變數據庫數據庫收錄。根據基因組聚合數據庫數據庫的分析,該基因變異在人群中極為罕見。該變異導致第960位氨基酸由甘氨酸變異為半胱氨酸。色氨酸具有特殊的體積和結構特性,在關鍵位點的替換如氨基酸半胱氨酸可能會干擾蛋白質折疊或功能域的相互作用。家系基因測序,患者父親、姑姑均不攜帶該變異,母親因去世未檢測,提示致病基因可能來自于母親,或為基因突變。
本病例為臨床上罕見疾病,目前尚無根治性治療方法,其診斷和治療仍存在諸多挑戰。在診斷方面,OMD的早期癥狀較為隱匿,患者多表現為緩慢視力下降,且眼底檢查常無明顯異常,易導致漏診或誤診。雖然多模式影像檢查如OCT、ERG,在早期OMD中可顯示微小異常,是目前重要的診斷工具,但初期影像學表現可能不夠典型,需通過定期隨訪觀察病變區域的進展速度,提高診斷敏感性。基因檢測已成為OMD診斷的重要輔助手段,通過檢測RP1L1等相關基因,可以明確診斷并進行家系篩查。然而,因基因檢測費用高、技術普及率有限,其臨床應用仍存在阻礙。OMD易與其他視網膜病變如Stargardt病、Best病混淆,因此,結合影像學、遺傳學及臨床表現進行綜合鑒別尤為重要。在治療方面,目前尚無針對OMD的特異性療法。患者的管理主要依賴于對癥處理和病情監測。RP1L1基因不僅對光感受器的功能維持至關重要,其在腫瘤發生發展中的潛在作用也引起關注[16]。未來有必要針對該基因在OMD發病機制中的具體作用展開深入研究,以探索可能的靶向治療策略。此外,應加強對疾病病理機制的研究,重點關注光感受器的慢性損傷機制以及視網膜色素上皮功能改變的進展路徑,尋找延緩疾病進展的方法。
患者女,38歲。因雙眼不明原因中心視力緩慢進展性下降8年加重1年,于2022年8月24日到河北省邯鄲市眼科醫院(邯鄲市第三醫院)就診。既往身體健康,否認其他全身系統性疾病史。眼科檢查:右眼、左眼最佳矯正視力(BCVA)分別為0.15、0.12。右眼、左眼眼壓分別為14、15 mm Hg(1 mm Hg=0.133 kPa)。雙眼眼前節檢查未見明顯異常。眼底檢查,近紅外成像(NIR)檢查,雙眼黃斑中心區反射信號弱(圖1A、1B)。藍光自發熒光(BAF)檢查,黃斑區可見高自發熒光(圖1C、1D)。光相干斷層掃描(OCT)檢查,雙眼黃斑區橢圓體帶(EZ)、嵌合體帶(IZ)反射信號減弱、紊亂(圖1E、1F)。OCT血管成像(OCTA)定量分析發現,淺層毛細血管叢(SCP)和深層毛細血管叢(DCP)的血流密度均降低(圖1G、1H)。Humphrey視野檢查,雙眼視野中心暗點(圖1I、1J)。多焦點視網膜電圖(ERG)檢查,雙眼黃斑區中心凹尖峰振幅密度下降(圖1K、1L)。閃光視覺誘發電位檢查,雙眼各波潛伏期稍有延長(圖1M、1N)。征求患者及父親和姑姑同意后行全外顯子基因檢測。結果顯示,患者攜帶RP1L1基因錯義突變及NM_178857:exon4:c.2880G>T:(p.Trp960Cys)突變點位。其父親和姑姑均無基因變異。診斷:隱匿性黃斑營養不良(OMD)(圖2)。對患者進行隨訪觀察,2年后患者于我院復查,OCT檢查,雙眼中央視網膜厚度變薄(圖3A、3B)。
圖1
RP1L1基因變異相關隱匿性黃斑營養不良患者初診雙眼眼部檢查像 1A、1B分別示右眼、左眼近紅外成像,黃斑中心區反射信號稍有減弱。1C、1D分別示藍光自發熒光像,黃斑區可見斑點狀強自發熒光。1E、1F分別示右眼、左眼光相干斷層掃描像,左圖為掃描方向和部位,右圖為檢查結果,黃斑區橢圓體帶、嵌合體帶反射信號減弱、紊亂。1G、1H分別示右眼、左眼光相干斷層掃描血管成像,左圖為淺層毛細血管叢像,右圖為深層毛細血管叢像,黃斑區淺層毛細血管叢、深層毛細血管叢血流密度均有降低。1I、1J分別示Humphrey視野檢查結果,視野表現為中心暗點。1K、1L分別示右眼、左眼多焦點視網膜電圖,黃斑區中心凹尖峰振幅密度下降。1M、1N分別示右眼、左眼閃光視覺誘發電位,各波潛伏期稍有延長
圖2
RP1L1基因變異相關隱匿性黃斑營養不良一家系基因測序圖 患者攜帶RP1L1基因錯義突變及NM_178857: c.2880G>TP.Trp960Cys突變點位;患者父親和姑姑均無基因變異
圖3
RP1L1基因變異相關隱匿性黃斑營養不良患者復查雙眼光相干斷層掃描像 3A、3B分別示右眼、左眼, 左圖為掃描方向和部位,右圖為檢查結果,雙眼中央視網膜厚度變薄
討論 OMD是一種遺傳性黃斑營養不良癥,罕見的常染色體顯性遺傳病。1989年,Miyake等[1]首次描述該病。1996年,Miyake等[2]將其命名OMD。RP1L1基因變異與OMD和視網膜色素變性有關[3]。RP1L1基因突變是OMD中最常見的病理突變,最常見的變異位點是c.133C>T[4-5]。
OMD臨床特征表現視力進行性下降[6]。本例患者主要臨床表現為雙眼中心視力進行性下降就診。NIR檢查可見雙眼黃斑中心區局限性弱反射信號,由于感光細胞破壞[7]。OCT檢查,雙眼黃斑區EZ、IZ反射信號明顯減弱、紊亂、變薄,錐細胞密度降低,RP1L1基因突變引起視錐細胞營養不良致使視錐細胞功能障礙,視野表現為中心暗點[8-9]。OCTA檢查,患者雙眼視網膜內毛細血管叢受損,脈絡膜毛細血管血流相對少的情況造成SCP和DCP的血流密度均降低[10]。
OMD需與Stargardt病、球后視神經炎及卵黃樣黃斑營養不良(Best)等疾病相鑒別。Stargardt病黃斑區呈牛眼狀典型特征及視網膜下脂褐素樣沉積物[11];而OMD眼底檢查多正常,即使晚期光感受器也不完全萎縮,視力多維持在0.1~0.2或更高[12-13]。球后視神經炎患者多單眼急性視力下降,伴眼球轉動痛,晚期視神經萎縮,糖皮質激素治療有效;OMD則表現為緩慢視力下降,無眼球轉動痛,對糖皮質激素治療無效,多焦ERG檢查可見微小改變[14-15]。OCT、眼底自發熒光檢查可見Best患者黃斑區卵黃樣堆積物;OMD患者無上述典型改變。
本例患者RP1L1基因上檢測到一個雜合錯義變異NM_178857:exon4:c.2880G>T: (p.Trp960Cys),位于染色體8p23.1。該變異位點未被ClinVar數據庫及人類基因突變數據庫數據庫收錄。根據基因組聚合數據庫數據庫的分析,該基因變異在人群中極為罕見。該變異導致第960位氨基酸由甘氨酸變異為半胱氨酸。色氨酸具有特殊的體積和結構特性,在關鍵位點的替換如氨基酸半胱氨酸可能會干擾蛋白質折疊或功能域的相互作用。家系基因測序,患者父親、姑姑均不攜帶該變異,母親因去世未檢測,提示致病基因可能來自于母親,或為基因突變。
本病例為臨床上罕見疾病,目前尚無根治性治療方法,其診斷和治療仍存在諸多挑戰。在診斷方面,OMD的早期癥狀較為隱匿,患者多表現為緩慢視力下降,且眼底檢查常無明顯異常,易導致漏診或誤診。雖然多模式影像檢查如OCT、ERG,在早期OMD中可顯示微小異常,是目前重要的診斷工具,但初期影像學表現可能不夠典型,需通過定期隨訪觀察病變區域的進展速度,提高診斷敏感性。基因檢測已成為OMD診斷的重要輔助手段,通過檢測RP1L1等相關基因,可以明確診斷并進行家系篩查。然而,因基因檢測費用高、技術普及率有限,其臨床應用仍存在阻礙。OMD易與其他視網膜病變如Stargardt病、Best病混淆,因此,結合影像學、遺傳學及臨床表現進行綜合鑒別尤為重要。在治療方面,目前尚無針對OMD的特異性療法。患者的管理主要依賴于對癥處理和病情監測。RP1L1基因不僅對光感受器的功能維持至關重要,其在腫瘤發生發展中的潛在作用也引起關注[16]。未來有必要針對該基因在OMD發病機制中的具體作用展開深入研究,以探索可能的靶向治療策略。此外,應加強對疾病病理機制的研究,重點關注光感受器的慢性損傷機制以及視網膜色素上皮功能改變的進展路徑,尋找延緩疾病進展的方法。