基因之間存在多種多樣的表達調控活動,一般認為這些調控關系隱含在基因表達譜中。因此,可以根據基因表達數據對基因調控狀態進行建模,以挖掘具有生物學意義的信息及隱含在其中的基因調控關系。本文分別利用獨立成分分析(ICA)和非負矩陣分解(NMF)這兩種無監督矩陣分解技術對阿爾茨海默病(AD)基因表達數據進行顯著基因提取及基因調控網絡的構建,通過生物學分析,探討了兩種不同矩陣分解技術在挖掘潛在致病基因上的作用,通過結合兩種方法所提取的顯著基因的生物學分析,體現了炎癥反應在AD致病機制中的重要作用,為AD早期診斷、致病機制研究及基因生物標志物的探尋提供了有益的方法。
引用本文: 孔薇, 王娟, 牟曉陽. 基于矩陣分解技術的顯著基因提取及基因表達數據分析. 生物醫學工程學雜志, 2014, 31(3): 662-670. doi: 10.7507/1001-5515.20140124 復制
版權信息: ?四川大學華西醫院華西期刊社《生物醫學工程學雜志》版權所有,未經授權不得轉載、改編
引言
后基因組時代研究的重心已由對單個基因或蛋白質功能的局部性研究,轉移到以細胞內全部基因、蛋白質及代謝產物等為研究對象的“組學”時代。理論上,利用各種手段阻斷致病基因信號傳導通路中任一環節均可對疾病達到一定的治療效果。隨著DNA微陣列技術在生物學中的廣泛應用,可以得到大量的暗含著基因調控關系的基因表達譜數據,如何從高通量基因表達數據中提取有效的生物信息來探尋人類疾病產生的生物過程和患病機制是目前的主要挑戰。
阿爾茨海默病(Alzheimer’s disease,AD)是老年人癡呆疾患中最普遍的一種,由于其早期發現難、確診難和發病機制復雜的特點,至今對它的生物學機制研究仍然沒有本質上的突破。目前越來越多的學者正利用微陣列基因表達數據對AD的早期診斷、致病機制和治療進行探尋。AD的特征性病理改變主要為大腦皮層及腦區的纖維蛋白沉積,即細胞外間隙的β淀粉樣蛋白(β-amyloid,Aβ)和細胞內多聚Tau蛋白的沉積,兩者在病理形態學上分別表現為老年斑和神經纖維纏結,但其產生的因果關系尚不明確。近年來,在分子生物學領域,各國科學家在探尋Aβ淀粉樣蛋白生成和蓄積的原因和機制方面,取得了卓有成效的研究成果。Ray等[1]發現血漿中的18種信號蛋白可以檢測和分類AD以及輕微的認知損傷;Ray等[2]識別出了與AD相關的6個共表達基因模塊;Wang等[3]發現了與APP共表達基因:Gsk3b,Falz,Mef2a,Tlk2,Rtn和Prkca等;Zhang等[4]提取了36個與AD密切相關的基因。
利用生物信息學方法進行特征基因選擇是信號傳導通路分析及基因調控網絡重構的基礎,也是后續進行生物學實驗的依據。目前,在特征基因選擇及特征提取方面多采用無監督聚類方法,提出了很多識別共表達基因以及在不同實驗條件下表達相似的模式,如K均值、自組織映射、層次聚類等。這些方法在分析微陣列基因表達數據時有兩個缺陷:① 基于全局相似性對基因進行分組聚類;② 單個基因只能聚到單獨某一類中。這與基因通常會參與多個信號傳導通路的生物學特性十分不符,而且由于基因數量大以及基因相互之間的從屬關系,生物學家難以根據這些聚類算法的結果作出合理的生物學解釋。為了避免傳統聚類算法的缺陷,近年來許多學者提出了雙向聚類方法,它可以對基因和樣本同時進行聚類,并將基因聚到不同類中,更為符合基因之間調控關系的生物學特性。
近年來,無監督分析方法的矩陣分解技術,如主成分分析(principle component analysis,PCA)、獨立成分分析(independent component analysis,ICA)和非負矩陣分解(nonnegative matrix factorization,NMF)等已經成功地應用到生物醫學數據分析中,并且從微陣列數據中發現了許多具有生物意義的表達模式。2001年,Hori等[5]首次提出利用ICA方法對孢子形成階段的酵母菌基因表達數據進行分類研究。Liebermeister[6]也利用ICA方法提取基因表達數據的表達模式,而且每一個表達模式都是潛變量的線性組合。另外,ICA還廣泛地應用于各類癌癥基因表達數據的特征提取中,如卵巢癌[7]、乳腺癌[8]、子宮癌[9]、結腸癌[10]和前列腺癌[11]等。近年來,許多學者提出了改進方法。Han等[12]提出了非負主成分分析方法方法與支持向量機相結合進行微陣列數據的特征基因選擇,成功地應用于白血病及神經管細胞瘤特異性基因標志物的探尋中。Zhang等[13]提出了利用奇異值分解方法對微陣列數據進行特征提取,以辨識共表達模塊。Han等[14]提出了多重獨立成分分析方法進行基因表達數據的局部特征提取,大大提高了癌癥樣本分類效果和生物標志物探尋的有效性。
與ICA相比,NMF把輸入矩陣分解為兩個非負矩陣。Lee和Seung首先將NMF用于人臉圖像分解[15],通過線性相加來獲得具有實際物理意義的局部特征,增強了數據的可解釋性。目前,NMF也有效地應用于基因表達數據特征提取和降維中,并且針對傳統NMF算法分解結果的不唯一性,近幾年通過提高分解矩陣的稀疏性,學者們提出了許多改進方法,如局部NMF方法、非負稀疏編碼方法、稀疏NMF方法、稀疏約束的NMF方法和非平滑NMF[16-20]等。
如上所述,無監督矩陣分解技術已應用于微陣列基因表達數據中并能夠提取具有一定生物學意義的顯著基因或生物過程,目前的研究工作大多數注重于各類算法自身的改進,以便提取更精確的致病基因,尚沒有對不同算法間的橫向比較分析。不同的矩陣分解技術應用于同一數據集時所提取的特征基因有何不同、所挖掘的信號傳導通路是否相似、所構建的基因調控網絡是否一致,這些都需要結合分子生物學進行系統的仿真與分析。本研究首先改進了ICA和NMF算法,在此基礎上比較和分析這兩種不同的矩陣分解技術在AD基因表達數據中顯著基因提取及基因調控網絡重構中的作用,結合生物學分析討論兩者的異同。然后,將ICA和NMF分別提取的顯著基因及潛在的生物過程有機結合,構建了AD致病的基因調控網絡,給出了與AD密切相關的顯著基因及信號傳導通路的生物學分析,有效地協助了生物學家進一步分析AD致病的信號傳導通路及探尋AD早期診斷的基因生物標志物。
1 矩陣分解技術的基因表達數據模型
1.1 微陣列基因表達數據的ICA模型
近年來學者們已使用ICA從微陣列數據中提取出許多特征基因及有用的生物信息。通常假設基因表達數據是某些具有特定生物學意義的獨立成分的線性組合。設基因表達數據表示為m×n維的矩陣X,其中m為樣本個數或者表示不同的實驗條件,n為基因個數,一般來說基因數目n遠遠大于樣本數目m,即n>>m。基因表達譜X的列表示基因在樣本/條件中的表達值,行表示某一樣本n個基因的表達值。矩陣X中的任意元素xij表示為第j個基因在第i個樣本/條件中的表達值。假設數據集通過預處理和歸一化,具有零均值和標準方差,基因表達譜數據的ICA模型可以表示為
| $X=AS ,$ |
ICA算法將基因表達譜數據X分解為矩陣A和S。其中,A=[a1,a2,…,an]為m×m維,A的列表示微陣列基因數據的m個基向量,S為m×n維基因標記矩陣,S的每行之間是相互統計獨立的,稱作基因標記向量或基因表達模式。每個基因表達譜(X的行)被認為是統計獨立的各基因表達模式的線性組合,A中第i行的元素則提供了第i個觀測樣本所需的m個基因表達模式的線性組合權重。為了得到矩陣S和A,ICA算法的解混合模型可以表示為
| $Y=WX=W\cdot AS=DP{{A}^{-1}}\cdot AS=\hat{S}$ |
ICA算法的求解過程即求解混合矩陣W的過程,理想狀況下求解出的W逼近于A-1,由于矩陣S和A均未知,只能得到WA= DP,DP表示單位陣的標量變換,即WA為單位陣的線性變換。根據考察Y的行向量之間獨立性方法的不同,目前已形成了多種多樣的ICA算法,其中以FastICA算法的速度最佳[21]。本課題采用FastICA算法進行AD基因表達數據的矩陣分解分析,同時針對基因數據量較大的特點本文改進了FastICA方法。
傳統FastICA算法中給出了三種非線性函數:G1(y)=log cosha1y,G2(y)=-exp,G3(y)y4,這些非線性函數的作用在于其導數是對分解所得的獨立成分,即S的行向量分布的概率密度函數的估計,估計得越準確ICA分解效果越好。傳統FastICA算法中采用的非線性函數均具有較好的穩健性,但它們都是指數類函數,在微陣列基因表達數據中應用時,運算量大,運算時間也非常長,會影響結果的準確性和特異性基因的有效提取。為此本文提出了采用Tukey雙權函數作為FastICA算法的非線性函數,該函數只涉及到乘法和加法,降低了微陣列數據的運算量和運算時間,同時也提高了結果的準確性和特異性基因的有效提取,實驗也證明其穩健性更佳。Tukey雙權函數如下:
| $g\left( y \right)=\left\{ \begin{align} & y(1-{{\left( \frac{{{y}^{2}}}{{{a}^{2}}} \right)}^{2}},\left| y \right|\le a \\ & 0,\left| y \right|>a \\ \end{align} \right.~,$ |
其中 a為門限值。
| $g\prime \left( y \right)=\left\{ \begin{align} & \left( 1-\frac{{{y}^{2}}}{{{a}^{2}}} \right)\left( 1-5\frac{{{y}^{2}}}{{{a}^{2}}} \right),\left| y \right|\le a \\ & 0,\left| y \right|>a \\ \end{align} \right.,$ |
式中g′(y)為Tukey雙權函數的一階導數。將Tukey雙權函數及其一階導數代入FastICA算法的迭代過程中,可得到基于Tukey雙權函數的改進FastICA算法。
本課題采用數據集來自美國國家生物技術信息中心(National Center for Biotechnology Information,NCBI) 基因表達數據庫中的GSE5281系列,選取其中與AD的產生和發展密切相關的人腦海馬區數據進行仿真實驗。該數據塊共23個樣本,54 675個基因表達值。包括13個正常對照樣本和10個AD患病樣本。經過t檢測及SAM(significance analysis of microarray)方法的預處理,我們將該數據集基因篩選至7 500個,剔除了冗余噪聲及對分類沒有貢獻的基因。
ICA矩陣分解后,基因表達數據可以認為是相互統計獨立的基因表達模式(S的行)的線性組合。矩陣乘積關系中可以看出,A的某列固定和S的某行相乘,因此利用A矩陣的列所產生的類別信息可以確定起主要作用的基因表達模式,從而得到S行向量中表達顯著的基因即為與疾病密切相關的顯著基因。A矩陣第i行的值表示m個基因表達模式(S的行)對第i個樣本的表達權重,每一行A可以表征所對應的每一個原樣本,因此對原樣本的分類等同于對A的行的分類。如上所述,矩陣A的第j列包含第j行S對所有觀測者(原樣本)的權值,因此,對于不同的類別這一列的值的大小應該有所差異。反言之,對于針對不同類別權值差異較大的矩陣A的列,可以獲得相應的對分類貢獻大的基因表達模式。同樣在這些基因表達模式中,表達水平明顯過高或過低的基因對辨識疾病具有重大作用,這些基因所形成的信號傳導網絡能更好地解釋疾病的生物調控過程。
1.2 基因表達數據的NMF模型
與ICA分解方法不同,NMF算法不要求分解向量間相互統計獨立,它要求分解矩陣均為非負值。NMF算法可用于數據降維及可視化,同時由于其分解值的非負特性更有利于數據的可解釋性。NMF矩陣分解可表示為
| $V≈WH ,$ |
式中V為n×m的非負矩陣,在基因表達數據中,n為基因個數,m為樣本數或者不同的實驗條件。矩陣W為n×k維非負基向量,H(k×m)為非負系數矩陣,NMF把數據分解為k個聚類,k代表著W×H特征子空間的維數,也是降維的維數或分類的類別數。基因表達數據V則為非負基矩陣W和系數矩陣H的線性組合。
由于NMF方法具有分解不唯一的缺陷,近年來有許多改進算法,克服了NMF算法在不同應用領域的缺點。這些改進方法大致分為兩類:第一類改進主要使算法能收斂到全局最優點;第二類改進是提高算法的稀疏度。其中非平滑NMF(nonsmooth NMF,nsNMF)算法的主要目的是為了得到全局的稀疏性,即W和H矩陣都具稀疏性[17]。由于混合模型的乘法本質,如果要求W和H同時具有一定稀疏度,那么必然會惡化模型對數據的擬合效果。因此,Montano等將一個“平滑”矩陣S∈Rk×k引入到混合模型中,表示為
| $V≈WSH ,$ |
式中的S∈Rk×k是一個正平滑矩陣,其定義為
| $S=(1-\theta )I+\frac{\theta }{q}{{11}^{T}}$ |
其中I為單位矩陣,1表示元素為1的列向量,11T是一個k×k矩陣,所有元素為1,參數θ滿足0≤θ≤1,控制模型的稀疏度。平滑矩陣S的調節作用可解釋如下:假設X為一個非負向量,乘以平滑矩陣S得到Y=SX,如果θ=0,則Y=X,矩陣X為非平滑的;而隨著θ→1,Y趨向于常數向量,矩陣Y中的每一個元素趨近于X元素的平均值,這是最平滑的向量,因為向量Y中所有的數值都等于相同的非零值,而不是有一些大的數值和接近于0的離散值。進一步觀察nsNMF模型,可以等效為:
| $V=(WS)H=W(SH)$ |
這樣的結合方式在算法迭代過程中十分有利,當迭代H時可用WS代替W,同理,迭代W時用SH替換H,在算法收斂時,就可以同時保證矩陣W和H均具有較高的稀疏性。我們采用式(9)所示的散度(熵)為目標函數:
| $D\left( V\|WSH \right)=\sum\limits_{ij}{{{V}_{ij}}}log\frac{{{V}_{ij}}}{{{\left( WSH \right)}_{ij}}}-{{V}_{ij}}+{{\left( WSH \right)}_{ij}})~,$ |
分別對W、H和S求偏導,求得的W、H和S的迭代式如下:
| ${{W}_{la}}={{W}_{la}}\frac{\sum\limits_{u}{{{\left( SH \right)}_{au}}}{{V}_{lu}}/{{\left( WH \right)}_{lu}}}{\sum\limits_{u}{{{H}_{au}}}}$ |
| ${{H}_{au}}={{H}_{aU}}\frac{\sum\limits_{l}{{{\left( WS \right)}_{la}}{{V}_{lu}}}/{{\left( WH \right)}_{lu}}}{\sum\limits_{k}{{{W}_{ka}}}}$ |
| ${{S}_{uv}}\leftarrow {{S}_{uv}}\frac{\sum\limits_{i=1}{\sum\limits_{j=1}{{{V}_{ij}}{{W}_{iu}}{{H}_{vj}}}}/\text{ }\Sigma \Sigma WSH}{\sum\limits_{i=1}{\sum\limits_{j=1}{{{W}_{lu}}{{H}_{vj}}}}}$ |
nsNMF算法的優勢在于同時增強了基向量矩陣W和系數矩陣H的稀疏性。分解結果中,利用矩陣H中對應樣本的系數值高低即可將m個樣本進行分類。NMF分解后,第i個原樣本可表示為基向量(k個W的列)與對應系數矩陣(H的第i列)的線性組合。顯然,基向量及系數矩陣中表達值大的基因及系數對原數據值起著重要作用。圖 1給出了NMF分解的例子,輸入數據仍為上節ICA方法中使用的AD數據集。在NMF分解中,V=XT,V為7 500×23維基因表達數據,包括13個正常對照樣本和10個AD樣本,每個樣本有7 500個基因表達值。
 圖1
AD微陣列基因表達數據的nsNMF分解模型
圖1
AD微陣列基因表達數據的nsNMF分解模型
樣本矩陣V(7 500×23)分解為非負矩陣W(7 500×2)及系數矩陣H(2×7 500)
Figure1. nsNMF decomposition model of AD gene expression dataV (7 500×23) into metagene matrix W (7 500×2) and coefficient matrix H (2×7 500)
2 實驗結果
為了比較FastICA和nsNMF算法在AD微陣列基因表達數據分析中的作用,我們分別使用這兩種方法的改進算法對上述AD患者大腦海馬區細胞組織的微陣列基因表達數據集進行了矩陣分解,提取了顯著基因。
2.1 FastICA分解結果
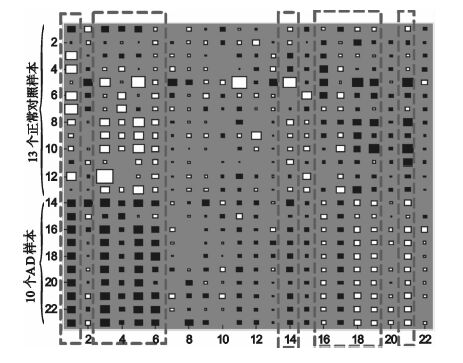
FastICA分解后得到A矩陣(23×23)及S矩陣(23×7 500)。如圖 2所示為矩陣A的Hinton圖,能夠比較直觀地反映A矩陣中每一個值的大小及正負。由于上述矩陣分解原理可知,A的行對應于樣本矩陣X的行,因此X矩陣的類別信息與A矩陣完全對應。從圖 2中可以看出,A矩陣中第1、3、4、5、6、14、16、17、18、19和21列的分類信息十分明顯(虛線框內),它們的前13項和后10項的數值大小及符號有顯著不同,可以明顯區分正常樣本和AD樣本。因此,A矩陣這些行所對應的S的行即是對樣本分類起著重要作用的行向量,同樣,在這些行向量中,表達值(絕對值)大的基因是對分類具有重要貢獻的基因。
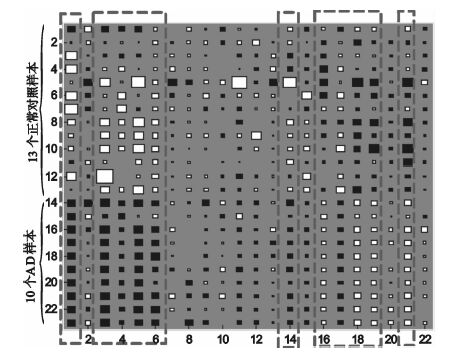 圖2
ICA分解后得到A矩陣的Hinton 圖
圖2
ICA分解后得到A矩陣的Hinton 圖
圖中每個正方形的大小與
the size of each square corresponds to the amount
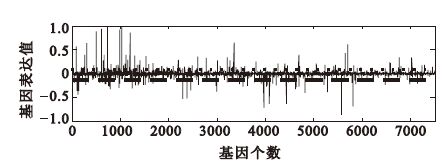
通過設置閾值,可選取表達值的絕對值超過閾值的基因作為診斷AD的顯著基因,如圖 3所示為A矩陣第6列所對應的S中的第6行基因標記向量值,這里基因表達值的正、負表示基因的高表達和低表達,閾值取為0.12。在本實驗中我們共提取了264個顯著基因。
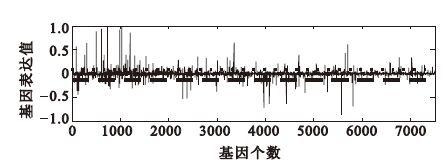 圖3
A矩陣第6列所對應的S矩陣第6行基因標記向量
Figure3.
The 6th row of the gene expression mode matrix S corresponding to 6th column of A
圖3
A矩陣第6列所對應的S矩陣第6行基因標記向量
Figure3.
The 6th row of the gene expression mode matrix S corresponding to 6th column of A
2.2 nsNMF分解結果
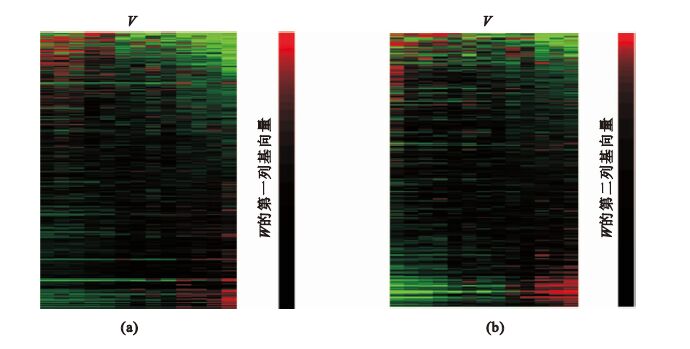
nsNMF分解過程中,考慮到該數據集為兩類問題,我們直接將k值取為2,迭代2 000次算法收斂。通過nsNMF獲得基向量矩陣W具有較好的稀疏性,即只有小部分項具有非零值,使基因表達局部化。之后,分別根據W的列從大到小排序,將原數據矩陣所對應W不同的列進行基因重新排序,就可以得到基因及樣本的局部表達特性,如圖 4所示。系數矩陣H的列與樣本相對應,當某一類的樣本與某些局部生物過程密切相關時,系數值將較高,反之較低,因此可以根據矩陣H的值來進行樣本分類。圖 4為原基因數據矩陣V分別根據W的第一列和第二列基向量排序后重新排列的結果。圖中紅色表示基因表達上調,綠色表示基因表達下調,可以看出在重新排序的基因表達矩陣V中,兩類樣本在頂部和底部均有差異明顯的基因。結合H矩陣的樣本類別信息,這些表達差異的基因與類別具有明顯關聯,即這些基因與AD密切相關。本實驗通過nsNMF提取了與AD密切相關的1 400個顯著基因。
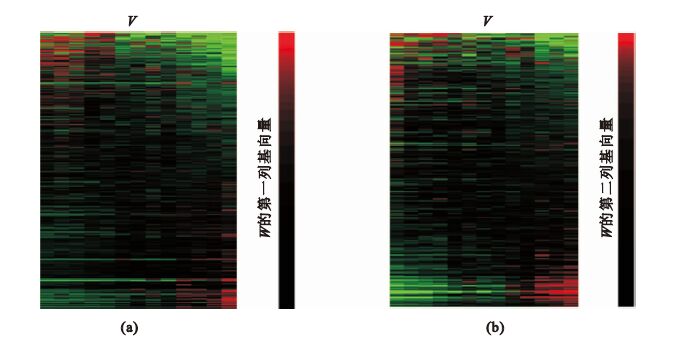 圖4
通過nsNMF基向量對原始數據進行重新排序
圖4
通過nsNMF基向量對原始數據進行重新排序
(a)根據W基向量第一列對原始數據進行排序;(b)根據W基向量第二列對原始數據進行排序
Figure4. Raw input data were reconstructed by nsNMF decomposition(a) according to the first sorted metagene (the first column of W); (b) according to the second sorted metagene (the second column of W)
3 生物學分析
我們通過FastICA和nsNMF兩種無監督算法分別獲得了與AD致病密切相關的顯著基因。利用FastICA方法,根據對所選取基因標志向量的不同閾值的選擇,從所選取的11個基因表達模式向量中共獲取264個顯著基因,這些顯著基因在與AD相關的信號傳導通路中起著重要作用。利用nsNMF方法,根據算法結果的局部特性和雙向聚類的效果,共提取了1 400個顯著基因。可以看出這兩種算法所提取的顯著基因數目差距較大,主要由于在ICA算法中可以調節閾值的大小來縮小和擴大選取顯著基因的數目,由于高表達和低表達基因在ICA主成分中較為明顯,且只需提取較少量的顯著基因就能使樣本精確分類。而在NMF算法中,算法更傾向于基因和樣本兩方面的雙向聚類并具有局部特性,各局部聚類的顯著基因累加以后數目較多。
如表 1所示,我們給出了部分FastICA方法所提取的與AD致病密切相關的上調和下調顯著基因。
表 1中可以看出,FastICA分解結果中AD樣本中表達上調的基因包括免疫反應蛋白、金屬蛋白、膜蛋白、脂蛋白、神經肽蛋白、細胞骨架蛋白,綁定蛋白和核糖體蛋白等。在免疫反應中,AMIGO2、BTG1、CD24、CD44、CDC42EP4、IFITM1、IFITM2、IRF7、IFI44L、IL4R、IRAK1 和 NFKBIA在AD樣本中都是表達上調的。與強烈的炎癥反應有關的Aβ的細胞外沉積是AD病理特征,神經炎癥也成為AD發病重要原因。表 1還顯示許多與金屬蛋白相關的基因有所上調,它們的代謝水平與AD緊密關聯。如:在Aβ增加后,鈣離子穩態的變化可能會影響一些能夠改善神經失調的生理反應,鈣離子大量涌入,導致了鈣離子/依賴性鈣調蛋白激酶Ⅱ的激活和突觸的積聚。另外,轉錄成蛋白質 CABP1、CACNG3、CAMK2B、CAMK1G、CAPZB的鈣離子,在AD中具有低表達特性。一些參與調解早期神經系統發育的初級特異性神經元,它們的轉錄調節是以鋅的變化為基礎的。膽固醇代謝異常已被證實與AD和其他神經系統疾病有關。氧化型膽固醇的相對溶解度促進了大腦各區室和血腦屏障出口的脂質通量,在表 1中可以看出APOC2/APOC4、APOE和ABCA1處于高表達水平。此外,ICA方法在AD樣本中也提取了許多神經肽蛋白、核蛋白、脂蛋白、捆綁蛋白和膜蛋白等的一些重要基因,在AD中高表達,而致癌基因及磷酸蛋白均是低表達,如PIP3-E、PLD3、PTPRT、ABCA2、ATP6V0C、ATP13A2及BCAS1等。
NMF分解結果中,所提取的顯著基因在細胞凋亡、生物合成、代謝過程、神經系統等相關生物過程起著重要的作用。其中有的與Aβ的聚集有關,有的與神經遞質的傳輸有關或與神經元的形成發展有關,還有的與金屬的代謝相關(如鋅離子和鈣離子)。這些表達上調和下調的基因伴隨著細胞的炎癥反應,導致神經元損害,引起記憶減退和認知障礙,產生癡呆癥狀。NMF所提取的顯著基因中還發現大量與炎癥相關的基因在AD樣本中表現為上調,以及許多抑制細胞生長和促進細胞凋亡的基因表達均為上調,如Programmed cell death 6、deleted in liver cancer 1、cyclin N-terminal domain containing 2、 killer cell immunoglobulin-like receptor、 p21、p53等。同時,那些促進細胞周期、細胞分裂和細胞修復的基因均表現為下調。還提取出了與線粒體功能障礙、轉錄翻譯、氧化磷酸化過程相關的基因簇。此外,NMF算法提取出了ATF4、AXIN1、CALCOCO1、CD81、CHP、CREBBP、CSNK1A1、CTBP1、CTBP2、EEF1D、FGF1、FGF2、FGFR3、FKBP1A、G3BP2、GNG12、HRAS、JUND、KIT、KRAS、LITAF、MAP2K1、MAP2K4、MAP3K5、MAP4K4、MAPK9、MAPKAPK2、MAPT、MAX、NFATC4等與AD密切相關的信號傳導通路的基因,以及MAPK信號通路、NF-κB信號傳導通路、Wnt信號傳導通路等的相關基因。另外,還提取出了直接參與AD信號傳導通路的重要基因,包括APP、APOE、THRA、TF、BIN1、CLU、SORL1、DAPK1、NFKBIA、SNCA、BACE1、NAE1等。
在本研究中,考慮到ICA及NMF結果重復性不高,同時又均與AD密切相關,具有互補性,我們將這兩種方法所提取的顯著基因融合在一起,利用Cytoscape軟件構建了AD的基因表達調控網絡,如圖 5所示。
 圖5
結合FastICA及nsNMF提取的顯著基因構建AD的基因調控網絡
Figure5.
Gene regulatory network reconstructed by the significant genes extracted by both FastICA and nsNMF
圖5
結合FastICA及nsNMF提取的顯著基因構建AD的基因調控網絡
Figure5.
Gene regulatory network reconstructed by the significant genes extracted by both FastICA and nsNMF
由于Cytoscape軟件數據庫中不一定涵蓋所有導入的顯著基因,因此在構建基因調控網絡的過程中會對輸入的顯著基因進行進一步的篩選和剔除。圖 5中每一個節點表示相對應的基因;不同顏色表示該基因所屬的生物功能;節點的形狀表示其上下調屬性,上調基因為方形,下調基因為圓形;每條邊為數據庫中存儲基因的相互作用。圖 5中基因顏色的不同表示分別屬于不同的生物過程:大紅色表示線粒體功能障礙,深紫色表示細胞凋亡,淡黃色表示鈣金屬元素作用機制,深藍色表示神經組織,粉色代表氧化磷酸化過程,綠色代表信號傳導通路,淺紫色代表轉錄翻譯淺藍色代表其他生物過程。
在此基礎上,我們利用基因本體(gene ontology,GO)數據庫,將所有顯著基因名稱分別導入其中,對每個基因的生物學過程、分子功能及細胞組分的相關信息進行采集并分類。經觀察篩選,兩種算法所提取的AD顯著基因信號傳導通路中與APP有直接相關通路關系的基因有21個,分別是:LDLRAP1、MAPK8IP1、TP53BP2、NUMBL、TGFB2、SNCB、IDE、CLU、CHRNA7、SHC3、MME、F12、TM2D1、KLC1、COL1A2、NF1、DAB1、APPBP2、NUMB、HSPG2、CALR;與NF-κB有直接相關通路關系的基因有18個,分別是:IL8、UBE2D3、HMGA2、TRAF6、RUVBL2、SUCLG1、MAP3K7IP1、BCL3、FBXW11、NCOR2、TPR、TP53BP1、IL2RA、UCP2、TIMM50、MRCL3、TNIP2及BTRC。
值得一提的是,不論是ICA方法還是NMF方法,均提取出了大量與炎癥反應密切相關的過表達基因,以及涉及炎癥反應的信號傳導通路,進一步證實了AD的產生和發展與炎癥反應密切相關。
從以上分子生物學分析可以看出,這兩種算法提取了與AD致病密切相關的不同類型、不同信號傳導通路中的顯著基因。對比ICA所提取的264個顯著基因 和NMF提取的1 400個顯著基因,發現兩組幾乎沒有重復。從算法理論及迭代過程上看,ICA方法假設基因表達譜由相互統計獨立的生物過程線性組合而成,但是到目前為止,數據的仿真實驗還不能確定和驗證這一假設的完全成立。同時,相互統計獨立這一約束條件對于生物過程來說過于苛刻。NMF方法則是將基因表達譜分解成非負向量的線性累加,從NMF在其它自然數據如語音、圖像等信號的應用上看,它具有較好的自然可解釋性。但NMF算法本身也具有結果不唯一、稀疏性不強等缺陷,使得其結果與真實生物過程的對應關系還需深入研究。
4 分析與結論
本文運用無監督矩陣分解技術ICA和NMF進行微陣列基因表達譜數據的分析。通過ICA提取的基因表達模式矩陣(S矩陣)及NMF提取的基向量矩陣(W矩陣)都能夠將基因歸類于不同的生物過程,實現了基因和樣本同時進行雙向聚類的功能。通過比較ICA和NMF兩種算法提取的顯著基因可知,它們相互重復的基因很少,這就意味著通過ICA提取的顯著基因即表達模式并不一定在NMF算法中表現為顯著基因,出現這種情況的原因可能是算法本身及收斂條件的限制。ICA假設表達模式統計獨立,而NMF需要非負性和稀疏約束以及形成局部基因表達譜。因此,我們可以分析和結合不同算法來尋找更多的顯著基因進行疾病的分析。通過實驗和生物學分析我們證明了ICA方法和NMF方法的有效性,通過ICA表達模式和NMF的局部聚類過程提取的表達上調和下調基因都與AD密切相關,并與臨床癥狀相吻合。同時,大量與炎癥反應相關的基因及信號傳導通路的進一步發現,可以幫助生物學家分析AD的致病信號傳導通路及其調控關系,從而為早期診斷、治療、預防AD以及新藥物的研發提供重要的參考價值。
引言
后基因組時代研究的重心已由對單個基因或蛋白質功能的局部性研究,轉移到以細胞內全部基因、蛋白質及代謝產物等為研究對象的“組學”時代。理論上,利用各種手段阻斷致病基因信號傳導通路中任一環節均可對疾病達到一定的治療效果。隨著DNA微陣列技術在生物學中的廣泛應用,可以得到大量的暗含著基因調控關系的基因表達譜數據,如何從高通量基因表達數據中提取有效的生物信息來探尋人類疾病產生的生物過程和患病機制是目前的主要挑戰。
阿爾茨海默病(Alzheimer’s disease,AD)是老年人癡呆疾患中最普遍的一種,由于其早期發現難、確診難和發病機制復雜的特點,至今對它的生物學機制研究仍然沒有本質上的突破。目前越來越多的學者正利用微陣列基因表達數據對AD的早期診斷、致病機制和治療進行探尋。AD的特征性病理改變主要為大腦皮層及腦區的纖維蛋白沉積,即細胞外間隙的β淀粉樣蛋白(β-amyloid,Aβ)和細胞內多聚Tau蛋白的沉積,兩者在病理形態學上分別表現為老年斑和神經纖維纏結,但其產生的因果關系尚不明確。近年來,在分子生物學領域,各國科學家在探尋Aβ淀粉樣蛋白生成和蓄積的原因和機制方面,取得了卓有成效的研究成果。Ray等[1]發現血漿中的18種信號蛋白可以檢測和分類AD以及輕微的認知損傷;Ray等[2]識別出了與AD相關的6個共表達基因模塊;Wang等[3]發現了與APP共表達基因:Gsk3b,Falz,Mef2a,Tlk2,Rtn和Prkca等;Zhang等[4]提取了36個與AD密切相關的基因。
利用生物信息學方法進行特征基因選擇是信號傳導通路分析及基因調控網絡重構的基礎,也是后續進行生物學實驗的依據。目前,在特征基因選擇及特征提取方面多采用無監督聚類方法,提出了很多識別共表達基因以及在不同實驗條件下表達相似的模式,如K均值、自組織映射、層次聚類等。這些方法在分析微陣列基因表達數據時有兩個缺陷:① 基于全局相似性對基因進行分組聚類;② 單個基因只能聚到單獨某一類中。這與基因通常會參與多個信號傳導通路的生物學特性十分不符,而且由于基因數量大以及基因相互之間的從屬關系,生物學家難以根據這些聚類算法的結果作出合理的生物學解釋。為了避免傳統聚類算法的缺陷,近年來許多學者提出了雙向聚類方法,它可以對基因和樣本同時進行聚類,并將基因聚到不同類中,更為符合基因之間調控關系的生物學特性。
近年來,無監督分析方法的矩陣分解技術,如主成分分析(principle component analysis,PCA)、獨立成分分析(independent component analysis,ICA)和非負矩陣分解(nonnegative matrix factorization,NMF)等已經成功地應用到生物醫學數據分析中,并且從微陣列數據中發現了許多具有生物意義的表達模式。2001年,Hori等[5]首次提出利用ICA方法對孢子形成階段的酵母菌基因表達數據進行分類研究。Liebermeister[6]也利用ICA方法提取基因表達數據的表達模式,而且每一個表達模式都是潛變量的線性組合。另外,ICA還廣泛地應用于各類癌癥基因表達數據的特征提取中,如卵巢癌[7]、乳腺癌[8]、子宮癌[9]、結腸癌[10]和前列腺癌[11]等。近年來,許多學者提出了改進方法。Han等[12]提出了非負主成分分析方法方法與支持向量機相結合進行微陣列數據的特征基因選擇,成功地應用于白血病及神經管細胞瘤特異性基因標志物的探尋中。Zhang等[13]提出了利用奇異值分解方法對微陣列數據進行特征提取,以辨識共表達模塊。Han等[14]提出了多重獨立成分分析方法進行基因表達數據的局部特征提取,大大提高了癌癥樣本分類效果和生物標志物探尋的有效性。
與ICA相比,NMF把輸入矩陣分解為兩個非負矩陣。Lee和Seung首先將NMF用于人臉圖像分解[15],通過線性相加來獲得具有實際物理意義的局部特征,增強了數據的可解釋性。目前,NMF也有效地應用于基因表達數據特征提取和降維中,并且針對傳統NMF算法分解結果的不唯一性,近幾年通過提高分解矩陣的稀疏性,學者們提出了許多改進方法,如局部NMF方法、非負稀疏編碼方法、稀疏NMF方法、稀疏約束的NMF方法和非平滑NMF[16-20]等。
如上所述,無監督矩陣分解技術已應用于微陣列基因表達數據中并能夠提取具有一定生物學意義的顯著基因或生物過程,目前的研究工作大多數注重于各類算法自身的改進,以便提取更精確的致病基因,尚沒有對不同算法間的橫向比較分析。不同的矩陣分解技術應用于同一數據集時所提取的特征基因有何不同、所挖掘的信號傳導通路是否相似、所構建的基因調控網絡是否一致,這些都需要結合分子生物學進行系統的仿真與分析。本研究首先改進了ICA和NMF算法,在此基礎上比較和分析這兩種不同的矩陣分解技術在AD基因表達數據中顯著基因提取及基因調控網絡重構中的作用,結合生物學分析討論兩者的異同。然后,將ICA和NMF分別提取的顯著基因及潛在的生物過程有機結合,構建了AD致病的基因調控網絡,給出了與AD密切相關的顯著基因及信號傳導通路的生物學分析,有效地協助了生物學家進一步分析AD致病的信號傳導通路及探尋AD早期診斷的基因生物標志物。
1 矩陣分解技術的基因表達數據模型
1.1 微陣列基因表達數據的ICA模型
近年來學者們已使用ICA從微陣列數據中提取出許多特征基因及有用的生物信息。通常假設基因表達數據是某些具有特定生物學意義的獨立成分的線性組合。設基因表達數據表示為m×n維的矩陣X,其中m為樣本個數或者表示不同的實驗條件,n為基因個數,一般來說基因數目n遠遠大于樣本數目m,即n>>m。基因表達譜X的列表示基因在樣本/條件中的表達值,行表示某一樣本n個基因的表達值。矩陣X中的任意元素xij表示為第j個基因在第i個樣本/條件中的表達值。假設數據集通過預處理和歸一化,具有零均值和標準方差,基因表達譜數據的ICA模型可以表示為
| $X=AS ,$ |
ICA算法將基因表達譜數據X分解為矩陣A和S。其中,A=[a1,a2,…,an]為m×m維,A的列表示微陣列基因數據的m個基向量,S為m×n維基因標記矩陣,S的每行之間是相互統計獨立的,稱作基因標記向量或基因表達模式。每個基因表達譜(X的行)被認為是統計獨立的各基因表達模式的線性組合,A中第i行的元素則提供了第i個觀測樣本所需的m個基因表達模式的線性組合權重。為了得到矩陣S和A,ICA算法的解混合模型可以表示為
| $Y=WX=W\cdot AS=DP{{A}^{-1}}\cdot AS=\hat{S}$ |
ICA算法的求解過程即求解混合矩陣W的過程,理想狀況下求解出的W逼近于A-1,由于矩陣S和A均未知,只能得到WA= DP,DP表示單位陣的標量變換,即WA為單位陣的線性變換。根據考察Y的行向量之間獨立性方法的不同,目前已形成了多種多樣的ICA算法,其中以FastICA算法的速度最佳[21]。本課題采用FastICA算法進行AD基因表達數據的矩陣分解分析,同時針對基因數據量較大的特點本文改進了FastICA方法。
傳統FastICA算法中給出了三種非線性函數:G1(y)=log cosha1y,G2(y)=-exp,G3(y)y4,這些非線性函數的作用在于其導數是對分解所得的獨立成分,即S的行向量分布的概率密度函數的估計,估計得越準確ICA分解效果越好。傳統FastICA算法中采用的非線性函數均具有較好的穩健性,但它們都是指數類函數,在微陣列基因表達數據中應用時,運算量大,運算時間也非常長,會影響結果的準確性和特異性基因的有效提取。為此本文提出了采用Tukey雙權函數作為FastICA算法的非線性函數,該函數只涉及到乘法和加法,降低了微陣列數據的運算量和運算時間,同時也提高了結果的準確性和特異性基因的有效提取,實驗也證明其穩健性更佳。Tukey雙權函數如下:
| $g\left( y \right)=\left\{ \begin{align} & y(1-{{\left( \frac{{{y}^{2}}}{{{a}^{2}}} \right)}^{2}},\left| y \right|\le a \\ & 0,\left| y \right|>a \\ \end{align} \right.~,$ |
其中 a為門限值。
| $g\prime \left( y \right)=\left\{ \begin{align} & \left( 1-\frac{{{y}^{2}}}{{{a}^{2}}} \right)\left( 1-5\frac{{{y}^{2}}}{{{a}^{2}}} \right),\left| y \right|\le a \\ & 0,\left| y \right|>a \\ \end{align} \right.,$ |
式中g′(y)為Tukey雙權函數的一階導數。將Tukey雙權函數及其一階導數代入FastICA算法的迭代過程中,可得到基于Tukey雙權函數的改進FastICA算法。
本課題采用數據集來自美國國家生物技術信息中心(National Center for Biotechnology Information,NCBI) 基因表達數據庫中的GSE5281系列,選取其中與AD的產生和發展密切相關的人腦海馬區數據進行仿真實驗。該數據塊共23個樣本,54 675個基因表達值。包括13個正常對照樣本和10個AD患病樣本。經過t檢測及SAM(significance analysis of microarray)方法的預處理,我們將該數據集基因篩選至7 500個,剔除了冗余噪聲及對分類沒有貢獻的基因。
ICA矩陣分解后,基因表達數據可以認為是相互統計獨立的基因表達模式(S的行)的線性組合。矩陣乘積關系中可以看出,A的某列固定和S的某行相乘,因此利用A矩陣的列所產生的類別信息可以確定起主要作用的基因表達模式,從而得到S行向量中表達顯著的基因即為與疾病密切相關的顯著基因。A矩陣第i行的值表示m個基因表達模式(S的行)對第i個樣本的表達權重,每一行A可以表征所對應的每一個原樣本,因此對原樣本的分類等同于對A的行的分類。如上所述,矩陣A的第j列包含第j行S對所有觀測者(原樣本)的權值,因此,對于不同的類別這一列的值的大小應該有所差異。反言之,對于針對不同類別權值差異較大的矩陣A的列,可以獲得相應的對分類貢獻大的基因表達模式。同樣在這些基因表達模式中,表達水平明顯過高或過低的基因對辨識疾病具有重大作用,這些基因所形成的信號傳導網絡能更好地解釋疾病的生物調控過程。
1.2 基因表達數據的NMF模型
與ICA分解方法不同,NMF算法不要求分解向量間相互統計獨立,它要求分解矩陣均為非負值。NMF算法可用于數據降維及可視化,同時由于其分解值的非負特性更有利于數據的可解釋性。NMF矩陣分解可表示為
| $V≈WH ,$ |
式中V為n×m的非負矩陣,在基因表達數據中,n為基因個數,m為樣本數或者不同的實驗條件。矩陣W為n×k維非負基向量,H(k×m)為非負系數矩陣,NMF把數據分解為k個聚類,k代表著W×H特征子空間的維數,也是降維的維數或分類的類別數。基因表達數據V則為非負基矩陣W和系數矩陣H的線性組合。
由于NMF方法具有分解不唯一的缺陷,近年來有許多改進算法,克服了NMF算法在不同應用領域的缺點。這些改進方法大致分為兩類:第一類改進主要使算法能收斂到全局最優點;第二類改進是提高算法的稀疏度。其中非平滑NMF(nonsmooth NMF,nsNMF)算法的主要目的是為了得到全局的稀疏性,即W和H矩陣都具稀疏性[17]。由于混合模型的乘法本質,如果要求W和H同時具有一定稀疏度,那么必然會惡化模型對數據的擬合效果。因此,Montano等將一個“平滑”矩陣S∈Rk×k引入到混合模型中,表示為
| $V≈WSH ,$ |
式中的S∈Rk×k是一個正平滑矩陣,其定義為
| $S=(1-\theta )I+\frac{\theta }{q}{{11}^{T}}$ |
其中I為單位矩陣,1表示元素為1的列向量,11T是一個k×k矩陣,所有元素為1,參數θ滿足0≤θ≤1,控制模型的稀疏度。平滑矩陣S的調節作用可解釋如下:假設X為一個非負向量,乘以平滑矩陣S得到Y=SX,如果θ=0,則Y=X,矩陣X為非平滑的;而隨著θ→1,Y趨向于常數向量,矩陣Y中的每一個元素趨近于X元素的平均值,這是最平滑的向量,因為向量Y中所有的數值都等于相同的非零值,而不是有一些大的數值和接近于0的離散值。進一步觀察nsNMF模型,可以等效為:
| $V=(WS)H=W(SH)$ |
這樣的結合方式在算法迭代過程中十分有利,當迭代H時可用WS代替W,同理,迭代W時用SH替換H,在算法收斂時,就可以同時保證矩陣W和H均具有較高的稀疏性。我們采用式(9)所示的散度(熵)為目標函數:
| $D\left( V\|WSH \right)=\sum\limits_{ij}{{{V}_{ij}}}log\frac{{{V}_{ij}}}{{{\left( WSH \right)}_{ij}}}-{{V}_{ij}}+{{\left( WSH \right)}_{ij}})~,$ |
分別對W、H和S求偏導,求得的W、H和S的迭代式如下:
| ${{W}_{la}}={{W}_{la}}\frac{\sum\limits_{u}{{{\left( SH \right)}_{au}}}{{V}_{lu}}/{{\left( WH \right)}_{lu}}}{\sum\limits_{u}{{{H}_{au}}}}$ |
| ${{H}_{au}}={{H}_{aU}}\frac{\sum\limits_{l}{{{\left( WS \right)}_{la}}{{V}_{lu}}}/{{\left( WH \right)}_{lu}}}{\sum\limits_{k}{{{W}_{ka}}}}$ |
| ${{S}_{uv}}\leftarrow {{S}_{uv}}\frac{\sum\limits_{i=1}{\sum\limits_{j=1}{{{V}_{ij}}{{W}_{iu}}{{H}_{vj}}}}/\text{ }\Sigma \Sigma WSH}{\sum\limits_{i=1}{\sum\limits_{j=1}{{{W}_{lu}}{{H}_{vj}}}}}$ |
nsNMF算法的優勢在于同時增強了基向量矩陣W和系數矩陣H的稀疏性。分解結果中,利用矩陣H中對應樣本的系數值高低即可將m個樣本進行分類。NMF分解后,第i個原樣本可表示為基向量(k個W的列)與對應系數矩陣(H的第i列)的線性組合。顯然,基向量及系數矩陣中表達值大的基因及系數對原數據值起著重要作用。圖 1給出了NMF分解的例子,輸入數據仍為上節ICA方法中使用的AD數據集。在NMF分解中,V=XT,V為7 500×23維基因表達數據,包括13個正常對照樣本和10個AD樣本,每個樣本有7 500個基因表達值。
圖1
AD微陣列基因表達數據的nsNMF分解模型
樣本矩陣V(7 500×23)分解為非負矩陣W(7 500×2)及系數矩陣H(2×7 500)
Figure1. nsNMF decomposition model of AD gene expression dataV (7 500×23) into metagene matrix W (7 500×2) and coefficient matrix H (2×7 500)
2 實驗結果
為了比較FastICA和nsNMF算法在AD微陣列基因表達數據分析中的作用,我們分別使用這兩種方法的改進算法對上述AD患者大腦海馬區細胞組織的微陣列基因表達數據集進行了矩陣分解,提取了顯著基因。
2.1 FastICA分解結果
FastICA分解后得到A矩陣(23×23)及S矩陣(23×7 500)。如圖 2所示為矩陣A的Hinton圖,能夠比較直觀地反映A矩陣中每一個值的大小及正負。由于上述矩陣分解原理可知,A的行對應于樣本矩陣X的行,因此X矩陣的類別信息與A矩陣完全對應。從圖 2中可以看出,A矩陣中第1、3、4、5、6、14、16、17、18、19和21列的分類信息十分明顯(虛線框內),它們的前13項和后10項的數值大小及符號有顯著不同,可以明顯區分正常樣本和AD樣本。因此,A矩陣這些行所對應的S的行即是對樣本分類起著重要作用的行向量,同樣,在這些行向量中,表達值(絕對值)大的基因是對分類具有重要貢獻的基因。
圖2
ICA分解后得到A矩陣的Hinton 圖
圖中每個正方形的大小與
the size of each square corresponds to the amount
通過設置閾值,可選取表達值的絕對值超過閾值的基因作為診斷AD的顯著基因,如圖 3所示為A矩陣第6列所對應的S中的第6行基因標記向量值,這里基因表達值的正、負表示基因的高表達和低表達,閾值取為0.12。在本實驗中我們共提取了264個顯著基因。
圖3
A矩陣第6列所對應的S矩陣第6行基因標記向量
Figure3.
The 6th row of the gene expression mode matrix S corresponding to 6th column of A
2.2 nsNMF分解結果
nsNMF分解過程中,考慮到該數據集為兩類問題,我們直接將k值取為2,迭代2 000次算法收斂。通過nsNMF獲得基向量矩陣W具有較好的稀疏性,即只有小部分項具有非零值,使基因表達局部化。之后,分別根據W的列從大到小排序,將原數據矩陣所對應W不同的列進行基因重新排序,就可以得到基因及樣本的局部表達特性,如圖 4所示。系數矩陣H的列與樣本相對應,當某一類的樣本與某些局部生物過程密切相關時,系數值將較高,反之較低,因此可以根據矩陣H的值來進行樣本分類。圖 4為原基因數據矩陣V分別根據W的第一列和第二列基向量排序后重新排列的結果。圖中紅色表示基因表達上調,綠色表示基因表達下調,可以看出在重新排序的基因表達矩陣V中,兩類樣本在頂部和底部均有差異明顯的基因。結合H矩陣的樣本類別信息,這些表達差異的基因與類別具有明顯關聯,即這些基因與AD密切相關。本實驗通過nsNMF提取了與AD密切相關的1 400個顯著基因。
圖4
通過nsNMF基向量對原始數據進行重新排序
(a)根據W基向量第一列對原始數據進行排序;(b)根據W基向量第二列對原始數據進行排序
Figure4. Raw input data were reconstructed by nsNMF decomposition(a) according to the first sorted metagene (the first column of W); (b) according to the second sorted metagene (the second column of W)
3 生物學分析
我們通過FastICA和nsNMF兩種無監督算法分別獲得了與AD致病密切相關的顯著基因。利用FastICA方法,根據對所選取基因標志向量的不同閾值的選擇,從所選取的11個基因表達模式向量中共獲取264個顯著基因,這些顯著基因在與AD相關的信號傳導通路中起著重要作用。利用nsNMF方法,根據算法結果的局部特性和雙向聚類的效果,共提取了1 400個顯著基因。可以看出這兩種算法所提取的顯著基因數目差距較大,主要由于在ICA算法中可以調節閾值的大小來縮小和擴大選取顯著基因的數目,由于高表達和低表達基因在ICA主成分中較為明顯,且只需提取較少量的顯著基因就能使樣本精確分類。而在NMF算法中,算法更傾向于基因和樣本兩方面的雙向聚類并具有局部特性,各局部聚類的顯著基因累加以后數目較多。
如表 1所示,我們給出了部分FastICA方法所提取的與AD致病密切相關的上調和下調顯著基因。
表 1中可以看出,FastICA分解結果中AD樣本中表達上調的基因包括免疫反應蛋白、金屬蛋白、膜蛋白、脂蛋白、神經肽蛋白、細胞骨架蛋白,綁定蛋白和核糖體蛋白等。在免疫反應中,AMIGO2、BTG1、CD24、CD44、CDC42EP4、IFITM1、IFITM2、IRF7、IFI44L、IL4R、IRAK1 和 NFKBIA在AD樣本中都是表達上調的。與強烈的炎癥反應有關的Aβ的細胞外沉積是AD病理特征,神經炎癥也成為AD發病重要原因。表 1還顯示許多與金屬蛋白相關的基因有所上調,它們的代謝水平與AD緊密關聯。如:在Aβ增加后,鈣離子穩態的變化可能會影響一些能夠改善神經失調的生理反應,鈣離子大量涌入,導致了鈣離子/依賴性鈣調蛋白激酶Ⅱ的激活和突觸的積聚。另外,轉錄成蛋白質 CABP1、CACNG3、CAMK2B、CAMK1G、CAPZB的鈣離子,在AD中具有低表達特性。一些參與調解早期神經系統發育的初級特異性神經元,它們的轉錄調節是以鋅的變化為基礎的。膽固醇代謝異常已被證實與AD和其他神經系統疾病有關。氧化型膽固醇的相對溶解度促進了大腦各區室和血腦屏障出口的脂質通量,在表 1中可以看出APOC2/APOC4、APOE和ABCA1處于高表達水平。此外,ICA方法在AD樣本中也提取了許多神經肽蛋白、核蛋白、脂蛋白、捆綁蛋白和膜蛋白等的一些重要基因,在AD中高表達,而致癌基因及磷酸蛋白均是低表達,如PIP3-E、PLD3、PTPRT、ABCA2、ATP6V0C、ATP13A2及BCAS1等。
NMF分解結果中,所提取的顯著基因在細胞凋亡、生物合成、代謝過程、神經系統等相關生物過程起著重要的作用。其中有的與Aβ的聚集有關,有的與神經遞質的傳輸有關或與神經元的形成發展有關,還有的與金屬的代謝相關(如鋅離子和鈣離子)。這些表達上調和下調的基因伴隨著細胞的炎癥反應,導致神經元損害,引起記憶減退和認知障礙,產生癡呆癥狀。NMF所提取的顯著基因中還發現大量與炎癥相關的基因在AD樣本中表現為上調,以及許多抑制細胞生長和促進細胞凋亡的基因表達均為上調,如Programmed cell death 6、deleted in liver cancer 1、cyclin N-terminal domain containing 2、 killer cell immunoglobulin-like receptor、 p21、p53等。同時,那些促進細胞周期、細胞分裂和細胞修復的基因均表現為下調。還提取出了與線粒體功能障礙、轉錄翻譯、氧化磷酸化過程相關的基因簇。此外,NMF算法提取出了ATF4、AXIN1、CALCOCO1、CD81、CHP、CREBBP、CSNK1A1、CTBP1、CTBP2、EEF1D、FGF1、FGF2、FGFR3、FKBP1A、G3BP2、GNG12、HRAS、JUND、KIT、KRAS、LITAF、MAP2K1、MAP2K4、MAP3K5、MAP4K4、MAPK9、MAPKAPK2、MAPT、MAX、NFATC4等與AD密切相關的信號傳導通路的基因,以及MAPK信號通路、NF-κB信號傳導通路、Wnt信號傳導通路等的相關基因。另外,還提取出了直接參與AD信號傳導通路的重要基因,包括APP、APOE、THRA、TF、BIN1、CLU、SORL1、DAPK1、NFKBIA、SNCA、BACE1、NAE1等。
在本研究中,考慮到ICA及NMF結果重復性不高,同時又均與AD密切相關,具有互補性,我們將這兩種方法所提取的顯著基因融合在一起,利用Cytoscape軟件構建了AD的基因表達調控網絡,如圖 5所示。
圖5
結合FastICA及nsNMF提取的顯著基因構建AD的基因調控網絡
Figure5.
Gene regulatory network reconstructed by the significant genes extracted by both FastICA and nsNMF
由于Cytoscape軟件數據庫中不一定涵蓋所有導入的顯著基因,因此在構建基因調控網絡的過程中會對輸入的顯著基因進行進一步的篩選和剔除。圖 5中每一個節點表示相對應的基因;不同顏色表示該基因所屬的生物功能;節點的形狀表示其上下調屬性,上調基因為方形,下調基因為圓形;每條邊為數據庫中存儲基因的相互作用。圖 5中基因顏色的不同表示分別屬于不同的生物過程:大紅色表示線粒體功能障礙,深紫色表示細胞凋亡,淡黃色表示鈣金屬元素作用機制,深藍色表示神經組織,粉色代表氧化磷酸化過程,綠色代表信號傳導通路,淺紫色代表轉錄翻譯淺藍色代表其他生物過程。
在此基礎上,我們利用基因本體(gene ontology,GO)數據庫,將所有顯著基因名稱分別導入其中,對每個基因的生物學過程、分子功能及細胞組分的相關信息進行采集并分類。經觀察篩選,兩種算法所提取的AD顯著基因信號傳導通路中與APP有直接相關通路關系的基因有21個,分別是:LDLRAP1、MAPK8IP1、TP53BP2、NUMBL、TGFB2、SNCB、IDE、CLU、CHRNA7、SHC3、MME、F12、TM2D1、KLC1、COL1A2、NF1、DAB1、APPBP2、NUMB、HSPG2、CALR;與NF-κB有直接相關通路關系的基因有18個,分別是:IL8、UBE2D3、HMGA2、TRAF6、RUVBL2、SUCLG1、MAP3K7IP1、BCL3、FBXW11、NCOR2、TPR、TP53BP1、IL2RA、UCP2、TIMM50、MRCL3、TNIP2及BTRC。
值得一提的是,不論是ICA方法還是NMF方法,均提取出了大量與炎癥反應密切相關的過表達基因,以及涉及炎癥反應的信號傳導通路,進一步證實了AD的產生和發展與炎癥反應密切相關。
從以上分子生物學分析可以看出,這兩種算法提取了與AD致病密切相關的不同類型、不同信號傳導通路中的顯著基因。對比ICA所提取的264個顯著基因 和NMF提取的1 400個顯著基因,發現兩組幾乎沒有重復。從算法理論及迭代過程上看,ICA方法假設基因表達譜由相互統計獨立的生物過程線性組合而成,但是到目前為止,數據的仿真實驗還不能確定和驗證這一假設的完全成立。同時,相互統計獨立這一約束條件對于生物過程來說過于苛刻。NMF方法則是將基因表達譜分解成非負向量的線性累加,從NMF在其它自然數據如語音、圖像等信號的應用上看,它具有較好的自然可解釋性。但NMF算法本身也具有結果不唯一、稀疏性不強等缺陷,使得其結果與真實生物過程的對應關系還需深入研究。
4 分析與結論
本文運用無監督矩陣分解技術ICA和NMF進行微陣列基因表達譜數據的分析。通過ICA提取的基因表達模式矩陣(S矩陣)及NMF提取的基向量矩陣(W矩陣)都能夠將基因歸類于不同的生物過程,實現了基因和樣本同時進行雙向聚類的功能。通過比較ICA和NMF兩種算法提取的顯著基因可知,它們相互重復的基因很少,這就意味著通過ICA提取的顯著基因即表達模式并不一定在NMF算法中表現為顯著基因,出現這種情況的原因可能是算法本身及收斂條件的限制。ICA假設表達模式統計獨立,而NMF需要非負性和稀疏約束以及形成局部基因表達譜。因此,我們可以分析和結合不同算法來尋找更多的顯著基因進行疾病的分析。通過實驗和生物學分析我們證明了ICA方法和NMF方法的有效性,通過ICA表達模式和NMF的局部聚類過程提取的表達上調和下調基因都與AD密切相關,并與臨床癥狀相吻合。同時,大量與炎癥反應相關的基因及信號傳導通路的進一步發現,可以幫助生物學家分析AD的致病信號傳導通路及其調控關系,從而為早期診斷、治療、預防AD以及新藥物的研發提供重要的參考價值。