為提高蛋白質點檢測的效率,利用圖像處理單元(GPU)在并行計算和內存管理方面的優勢,提出一種基于CUDA的蛋白質點檢測快速實現方法。首先,對蛋白質點檢測算法中最耗時的圖像預處理、蛋白質點粗檢測和重疊蛋白質點分割三部分進行并行化設計;然后,根據CUDA單指令多線程的執行方式對數據空間進行二維分塊,利用共享寄存器和二維紋理內存的內存管理措施實現了蛋白質點快速檢測。通過本文方法與中央處理器(CPU)串行方法進行真實凝膠圖像的檢測對比實驗,結果表明,本文方法的執行效率明顯高于CPU串行方法,并且隨著圖像大小的增加,效率也隨之提高,對于2 048×2 048大小的圖像數據,CPU串行實現時間為52 641 ms,GPU則為4 384 ms,效率提高了11倍。
引用本文: 熊邦書, 葉毅嘉, 歐巧鳳, 張郝東. 基于CUDA的蛋白質點檢測快速實現方法. 生物醫學工程學雜志, 2016, 33(1): 83-88. doi: 10.7507/1001-5515.20160016 復制
版權信息: ?四川大學華西醫院華西期刊社《生物醫學工程學雜志》版權所有,未經授權不得轉載、改編
引言
差異蛋白質點提取是二維凝膠技術[1]的核心內容,是目前蛋白質組學的重要研究方法之一,能夠為疾病診斷、藥物研究和環境污染分析提供依據。蛋白質點檢測是差異蛋白質點提取的關鍵步驟,現有檢測方法主要分為兩類:一類是基于數學模型的檢測方法[2-3],如混合模型、擴散模型和高斯模型,這類方法對一些形狀比較規則的蛋白質點檢測效果好,但無法檢測出重疊蛋白質點。另一類是基于分水嶺變換的檢測方法及其改進方法[4-8],Pleissner等[4]首次將分水嶺方法用于凝膠圖像中,取得較好的效果,但存在過分割現象及弱蛋白質點、重疊蛋白質點檢測效果不佳的問題;其后,Srinark等[5]提出了基于區域合并的分水嶺方法,減少了過分割的現象;Sun等[6]提出了內外標記控制分水嶺算法,改善了分水嶺的過分割問題,但存在重疊蛋白質點不能分離的問題;Kim等[7]提出了累積梯度的分水嶺方法,改善了重疊蛋白質點難分離的問題;本課題組此前曾針對弱點及重疊點難以分離的問題進行研究,并取得較好的效果[8],而本文則針對此類算法效率不高的問題,開展了并行化設計研究。
分水嶺及其相關改進檢測方法是目前蛋白質點檢測中的主流方法,但存在計算耗時多、效率不高的問題。近些年,計算正從中央處理器(central processing unit,CPU)的“中央處理”模式向CPU與圖像處理單元(graphic processing unit,GPU)“協同處理”的方向發展,NVIDIA公司通過引入新的GPU架構和CUDA(Compute Unified Device Architecture,統一計算架構)編程模型[9],將GPU應用在通用計算中,為圖像處理算法的高效實現提供了新途徑。據此,本文提出一種基于CUDA的蛋白質點檢測快速實現方法,利用CUDA架構并行計算和內存管理方面的優勢,提高算法的效率。
1 方法
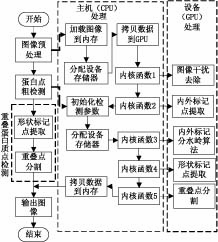
主要對蛋白質點檢測中最耗時的圖像預處理、蛋白質點粗檢測和重疊蛋白質點分割三部分進行并行化設計。蛋白質點檢測算法的快速實現流程如圖 1所示。
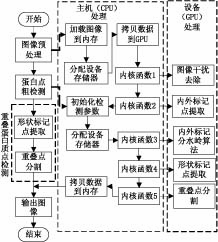 圖1
基于CUDA的蛋白質點檢測快速實現方法流程圖
Figure1.
Flow chart of quick-achieved protein spots detection based on CUDA
圖1
基于CUDA的蛋白質點檢測快速實現方法流程圖
Figure1.
Flow chart of quick-achieved protein spots detection based on CUDA
1.1 二維網格和線程塊設計
CUDA并行程序中,網格和線程塊的設計是其中關鍵,而網格和線程塊都是dim3類型的參數,也就是具有三維數據的類型。其中,Dim3 Grid(x,y,1)表示每行中有x個線程塊,每列中有y個線程塊,而Grid的維度只能是一維或者二維的,所以其第三維默認為1;而Dim block(x,y,z)表示有一個三維尺寸的線程塊,每行有x個線程,每列有y個線程,在深度方向有z個線程。但本文所使用的凝膠圖像都是二維圖像,所以最多只需設置其中的前2個參數即可。而不同型號的GPU中能開辟的線程塊與線程數量都是有限制的,本文采用的是NIVIDA公司型號為GT605的GPU,其整個網格中最多有65 535個線程塊,而每個線程塊中最多能啟用512個線程。
線程的劃分與數據大小有關,為了充分利用GPU中的晶體單元,經過大量實驗證明,當線程塊中的線程數量為16的整數倍時,能夠得到較高的計算效率,因此本文二維線程塊大小定為16×16,即每個線程塊中有256個線程。x方向網格維度為(圖像寬+線程塊在x方向上的維度+1)/線程塊在x方向上的維度,y方向網格維度為(圖像高+線程塊在y方向上的維度+1)/線程塊在y方向上的維度。在上式中加1是為了防止取整計算中,分配的線程數不足,造成圖像邊緣的像素點得不到處理。
在經過網格和線程塊分配后,每個線程塊和線程都對應了一個索引標識號,通過該標識號,可以計算出像元在圖像中的坐標,其中x坐標為線程塊維度.x×線程塊索引.x+線程索引值.x,y坐標為線程塊維度.y×線程塊索引.y+線程索引值.y。由此通過計算實現了圖像中像元與處理線程之間的一一對應。
1.2 圖像預處理
二維凝膠在制作及成像過程中存在一定人為因素和干擾,造成圖像噪聲和背景干擾。常采用高斯濾波和中值濾波去除噪聲干擾,采用top-hat變換[10]消除背景干擾。采用CUDA技術進行高斯濾波、中值濾波和top-hat變換的并行化設計,主要實現過程如下:
步驟1:中值濾波。首先根據圖像大小分配線程,并將圖像數據綁定紋理內存。綁定紋理內存是為了減少線程對內存的請求,以便更有效地利用內存帶寬。其次用線程塊和線程索引確定當前處理像元數據在圖像中的坐標和在內存中的偏移量,從二維紋理內存中讀出以當前像元為中心3×3鄰域內像元的像素值,取它們的中值替換當前像元,并將結果存入全局內存。
步驟2:高斯濾波。將經過中值濾波的圖像數據綁定紋理內存,采用分離濾波器[11]的方式進行高斯濾波。原理是將二維濾波過程分解成兩次一維濾波的方式進行處理,即分成行、列兩次濾波。首先進行行濾波,由于各個像元高斯濾波過程都是獨立的,所以可以采用一個線程計算相對應一個像元,將每個像素與其周圍的8個像素點進行加權計算出濾波結果。列濾波過程與行濾波過程相似,兩者的區別是列濾波過程中每個線程號的增加跨過了濾波區域。由于每個線程束中的每個線程訪問相同的數據內存空間,而連續線程訪問連續共享內存地址,所以在數據處理過程中不會存在紋理內存和共享內存的訪問沖突。
步驟3:top-hat變換。將經過圖像形態學開、閉的圖像α(x,y)、β(x,y)分別綁定紋理內存。之后將進行過濾波處理的圖像數據F(x,y)與對應位置的α(x,y)、β(x,y)按式(1)進行運算。在線程支出過程中,每個線程束中的線程只需一次顯存讀寫操作。
| $\left\{ \begin{align} & W\left( x,y \right)=F\left( x,y \right)-\alpha \left( x,y \right) \\ & H\left( x,y \right)=\beta \left( x,y \right)-F\left( x,y \right) \\ \end{align} \right.,$ |
1.3 蛋白質點預檢測
圖像預處理完成后,采用基于內外標記分水嶺算法[6]對蛋白質點進行預檢測,其并行化設計的主要實現步驟如下:
步驟1:內標記點提取。 根據凝膠圖像,選定圖像最小點來初始化檢測參數,得到最小點半徑minR。以半徑minR×minR為移動窗口遍歷所有線程中的像元數據,尋找圖像中的峰值點,用原子操作數atomicAdd進行計數,并將峰值點數據存入全局內存中。計算兩兩峰值間的距離,若兩者距離小于minR,則將灰度值大的點去除,同時原子操作數atomicAdd減1,則此灰度值小的峰值點即為內標記點。
步驟2:外標記點提取。首先將內標記圖像中屬于同一個內標記的用同一個數值進行編號(即標記),且每個內標記的數值都不相同;然后將不同內標記區域圖像數據放在不同block(線程塊)中的線程;其次各個block中的每個thread(線程)處理經過標記了的數值,從每個標記了數值的點開始向其周圍上下左右的點開始擴展,如果上下左右的點都是未標記過的點則將這些點標記為與該點相同的數值,并繼續對這些新的標記數值點進行擴展;當擴展時,如果發現待標記的點已被其他數值標記過,且標記值與該標記不同,也不是特殊標記,則停止擴展,并把該標記點改成特殊標記,作為分水嶺脊線。最后將未處理的數據按上述方法繼續擴展;重復上述步驟直到圖像上的所有點都已被標記過。將得到的分水嶺脊線像素值記為0,即為凝膠圖像的外標記。
步驟3:配置執行參數。定義一個bool類型的指針,大小與圖像大小相同,將全部元素置為0。每個block分配16個thread,則分配的block個數為:atomicAdd/16+1。由于每個蛋白質點區域的漲水過程互不相關,則每個block可單獨處理一個內標記點所在的蛋白質點區域。
步驟4:內外標記分水嶺算法。首先定義一個與圖像大小相同的bool類型的指針,分配空間后置0,從全局內存中讀出標記點,計算標記點在圖像中的坐標(startX,startY),以標記點坐標為起始坐標在各個蛋白質點區域進行擴展。若擴展點是未標記點則將其標記為與內標記相同的數值存入二維內存中,若擴展點是標記點且其數值與該區域的內標點數值不同,則該點為蛋白質點的輪廓邊緣并將其對應的bool變量置1,待所有線程處理完畢之后,將二維內存中的數據進行并行方式排序,取出其中像素值最小的像元數據按上面方法操作,二維內存中數據為空。最后將bool型指針數據與圖像數據結合,得到檢測結果圖。利用上述粗檢測方法對凝膠圖像進行蛋白質點檢測,其效果如圖 2所示。
 圖2
蛋白質點粗檢測
Figure2.
Protein spots coarse detection
圖2
蛋白質點粗檢測
Figure2.
Protein spots coarse detection
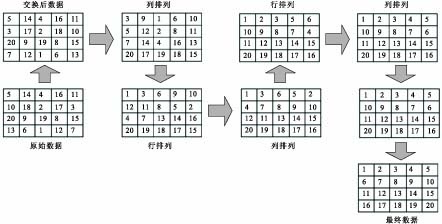
像元數據并行排序:主要是對二維數組進行排序,并行排序方法是建立在雙調排序的基礎上,通過對二維數組的行列交替進行排序。首先對線程進行分配,每個線程塊使用512個線程,需要一個共享寄存器同步所有的線程數據,而共享寄存器只能同步屬于同一個線程塊中線程的數據,每次最多只能處理1 024×1 024的二維數組,如數據較多則可分多次進行。首先將待排序的數據進行格式轉換,以便于排序;其次,啟用線程,每個線程處理一列數據,對列數據進行并行的升序排序,得出排序結果存入共享寄存器中并調用_synchreads()函數同步各個線程;待所有線程執行至_synchreads()函數時,開始對行數據進行并行排序,其中偶數行進行升序排序,奇數行進行降序排序,得出排序結果存入共享寄存器中并調用_synchreads()函數線程同步;重復上述步驟循環,共循環log(m),其中m為二維數組的行數;最后,處理得到的數據是一個蛇形一樣的二維數組(像元并行排序示意圖如圖 3所示)。
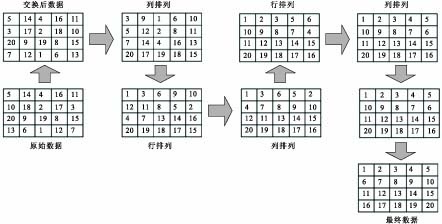 圖3
像元排序示意圖
Figure3.
Process of pixel sort
圖3
像元排序示意圖
Figure3.
Process of pixel sort
1.4 重疊蛋白質點檢測
蛋白質點粗檢測后,大部分蛋白質點已被檢測出來,但存在重疊蛋白質點檢測效果不佳的問題,如圖 2粗檢測結果中所示的1和2處。針對此問題,采用文獻[8]方法進行重疊蛋白質點分割,主要包括形狀標記點提取和重疊蛋白質點分割兩個過程。并行化設計的主要步驟如下:
步驟1:區域生長法確定凝膠圖像二值圖像重建[12-13]。首先確定種子點,利用預檢測蛋白質點區域的幾何中心做為種子點,以蛋白質點外圍輪廓作為增長的終止規則。每個像素對應一個線程,定義兩個bool類型的變量A、B(原始值都為0),將種子點(即區域極小值)所對應位置的A中值置為1。調用內核函數,對A中為1所對應的像素點取其3×3鄰域進行判斷,并將對應的A值置為0,若其為預檢測蛋白質點邊緣輪廓則將其對應的B中值置為1,反之將A置為1。之后判斷B中數據有無變化,若B中數據無變化,則代表區域生長完成,若有變化,則重復上述步驟。
步驟2:形狀標記點提取。本文采用自適應閾值的H-極小值變換方法(如式2所示),采用一維線程分配方法,每個線程塊分配256個,則其對應的網格線程塊的分配為(蛋白質點連通域個數+線程塊在x方向上的維度+1)/線程塊在x方向上的維度。采用一個線程塊處理一個蛋白質點區域,因為每個蛋白質點區域的像素點數據需相互通信。每個線程塊處理一個單獨的蛋白質點連通區域,以第i個連通域(線程塊)為例,首先從閾值h=1進行極小值變換,計算對應的極小值個數Ni(h),若其小于或等于1,則該連通域中沒有重疊蛋白質點;反之,含有重疊點。重復增長h,增長步長為1,計算Ni(h),直至其小于1。根據各個線程求得的閾值h計算該線程處理蛋白質點區域的形狀標記點。
| $H\left( c,h \right)={{R}^{\delta }}_{f}\left( c+h \right)-c,$ |
式中,c為經過粗檢測后的凝膠圖像C(x,y),h為極小值深度閾值,Rfδ(c+h)為對圖像c進行極小值強加得到的圖像數據,H(c,h)為圖像c的區域極小值。
步驟3:距離變換。線程分配仍用上一步的方法,每個線程處理一個蛋白質點連通區域,形狀標記距離變換是計算形狀標記點到該連通區域中任意像素點的最短路徑。采用8-鄰域的距離變換方法[14],首先對蛋白質點連通區域的位置進行統計,CUDA架構可以利用線程的唯一ID來標識統計,將統計后的數據利用均值法求出該區域的中心。對蛋白質點區域的所有像素點進行位置統計,利用均值法求出其幾何中心;判斷蛋白質點區域中任意像素點相對幾何中心的位置,如A點在其左上,則計算距離的方向順序是:左、左上、上,將這3個方向上的最小值Lmin保存;計算A點的其他5個方向到邊界的距離L,以Lmin為最高的循環次數,在該循環次數內,如果在某方向上計算的距離L小于Lmin,則用L的值替換Lmin的值,否則,停止該方向上的距離計算。重復上述步驟,完成所有像素計算。
步驟4:對形狀距離圖像進行分水嶺變換[15]。由于每個蛋白質點區域的分水嶺算法互不相干,則可以采用每個線程塊負責一個蛋白質點區域,并將各個蛋白質點區域進行數值標記,用線程標識號進行數值標記。在此以第i個蛋白質點連通區域為例,其他像素類似。先以該區域的中心點進行3×3鄰域像素的擴展,將這些數據存入全局寄存器中,再采用設備端的Shear方法對二維數組中的數據進行排序(這是由于分水嶺算法在進行數據“泛洪”時只能先處理像素值小的點),并取出最小的像素值,繼續對這些像素進行3×3數據擴展,對擴展的數據進行判斷,若發現有些點的標記值與該點區域的標記數值不同,則該點為區域的邊緣輪廓點;重復上述步驟,直至遍歷該區域內的所有蛋白質點。將數據存入全局寄存器中,并將其拷貝至主機內存,顯示圖像。
利用上述重疊點分割方法對圖 2粗檢測所示凝膠圖像進行重疊蛋白質點分割。以圖 2結果圖中的重疊點1為檢測對象,其效果如圖 4所示。
 圖4
重疊蛋白質點分割
Figure4.
Overlapping protein spots segmentation
圖4
重疊蛋白質點分割
Figure4.
Overlapping protein spots segmentation
2 實驗結果及分析
為驗證本文方法的有效性,在硬件配置為NVIDIA GeForce 605圖形處理器、主機內存為4.0G、Pentium(R) Dual-Core CPU E5800 @ 3.20 GHz 雙核下,以及軟件環境為WIN7操作系統、CUDA6.0 32 bit版本、Visual Studio 2010 32 bit和GEFORCE 344.11顯卡驅動版本下,分別實現蛋白質點檢測算法在CPU上運行的串行方法和在CUDA上運行的并行方法。
為驗證圖像大小對算法效率提高的影響,選取6組不同分辨率的凝膠圖像進行檢測對比實驗,算法耗時取多次運行時間的平均值(取3次)。檢測算法在CPU和GPU上實現的性能比較如表 1所示,其中加速比為CPU耗時與GPU耗時之比。
可以看出,蛋白質點檢測算法在GPU上運行并行方法的效率明顯高于在CPU上運行的串行方法,并且隨著圖像像素的增大,檢測效率提高越明顯。這是因為采用CPU串行實現時,需用循環對每個像素進行遍歷,像素越多,耗時越多,而采用GPU并行實現時,為每一像素開辟一個線程數并行實現每個像素的遍歷,這樣明顯提高了算法的效率。
3 結論
本文提出了基于CUDA的蛋白質點檢測快速實現方法。該方法根據凝膠圖像的特點,利用CUDA單指令多線程指令確定了適合凝膠圖像的線程分配方式,加快了凝膠圖像預處理算法、蛋白質點預檢測及重疊蛋白質點檢測算法的實現,提高了檢測算法的運行效率,同時使用CUDA共享內存和紋理內存的內存管理方式,減少了重復訪問,進一步提高檢測效率。通過不同像素大小的真實凝膠圖像檢測實驗,驗證了本文快速實現方法的有效性。
引言
差異蛋白質點提取是二維凝膠技術[1]的核心內容,是目前蛋白質組學的重要研究方法之一,能夠為疾病診斷、藥物研究和環境污染分析提供依據。蛋白質點檢測是差異蛋白質點提取的關鍵步驟,現有檢測方法主要分為兩類:一類是基于數學模型的檢測方法[2-3],如混合模型、擴散模型和高斯模型,這類方法對一些形狀比較規則的蛋白質點檢測效果好,但無法檢測出重疊蛋白質點。另一類是基于分水嶺變換的檢測方法及其改進方法[4-8],Pleissner等[4]首次將分水嶺方法用于凝膠圖像中,取得較好的效果,但存在過分割現象及弱蛋白質點、重疊蛋白質點檢測效果不佳的問題;其后,Srinark等[5]提出了基于區域合并的分水嶺方法,減少了過分割的現象;Sun等[6]提出了內外標記控制分水嶺算法,改善了分水嶺的過分割問題,但存在重疊蛋白質點不能分離的問題;Kim等[7]提出了累積梯度的分水嶺方法,改善了重疊蛋白質點難分離的問題;本課題組此前曾針對弱點及重疊點難以分離的問題進行研究,并取得較好的效果[8],而本文則針對此類算法效率不高的問題,開展了并行化設計研究。
分水嶺及其相關改進檢測方法是目前蛋白質點檢測中的主流方法,但存在計算耗時多、效率不高的問題。近些年,計算正從中央處理器(central processing unit,CPU)的“中央處理”模式向CPU與圖像處理單元(graphic processing unit,GPU)“協同處理”的方向發展,NVIDIA公司通過引入新的GPU架構和CUDA(Compute Unified Device Architecture,統一計算架構)編程模型[9],將GPU應用在通用計算中,為圖像處理算法的高效實現提供了新途徑。據此,本文提出一種基于CUDA的蛋白質點檢測快速實現方法,利用CUDA架構并行計算和內存管理方面的優勢,提高算法的效率。
1 方法
主要對蛋白質點檢測中最耗時的圖像預處理、蛋白質點粗檢測和重疊蛋白質點分割三部分進行并行化設計。蛋白質點檢測算法的快速實現流程如圖 1所示。
圖1
基于CUDA的蛋白質點檢測快速實現方法流程圖
Figure1.
Flow chart of quick-achieved protein spots detection based on CUDA
1.1 二維網格和線程塊設計
CUDA并行程序中,網格和線程塊的設計是其中關鍵,而網格和線程塊都是dim3類型的參數,也就是具有三維數據的類型。其中,Dim3 Grid(x,y,1)表示每行中有x個線程塊,每列中有y個線程塊,而Grid的維度只能是一維或者二維的,所以其第三維默認為1;而Dim block(x,y,z)表示有一個三維尺寸的線程塊,每行有x個線程,每列有y個線程,在深度方向有z個線程。但本文所使用的凝膠圖像都是二維圖像,所以最多只需設置其中的前2個參數即可。而不同型號的GPU中能開辟的線程塊與線程數量都是有限制的,本文采用的是NIVIDA公司型號為GT605的GPU,其整個網格中最多有65 535個線程塊,而每個線程塊中最多能啟用512個線程。
線程的劃分與數據大小有關,為了充分利用GPU中的晶體單元,經過大量實驗證明,當線程塊中的線程數量為16的整數倍時,能夠得到較高的計算效率,因此本文二維線程塊大小定為16×16,即每個線程塊中有256個線程。x方向網格維度為(圖像寬+線程塊在x方向上的維度+1)/線程塊在x方向上的維度,y方向網格維度為(圖像高+線程塊在y方向上的維度+1)/線程塊在y方向上的維度。在上式中加1是為了防止取整計算中,分配的線程數不足,造成圖像邊緣的像素點得不到處理。
在經過網格和線程塊分配后,每個線程塊和線程都對應了一個索引標識號,通過該標識號,可以計算出像元在圖像中的坐標,其中x坐標為線程塊維度.x×線程塊索引.x+線程索引值.x,y坐標為線程塊維度.y×線程塊索引.y+線程索引值.y。由此通過計算實現了圖像中像元與處理線程之間的一一對應。
1.2 圖像預處理
二維凝膠在制作及成像過程中存在一定人為因素和干擾,造成圖像噪聲和背景干擾。常采用高斯濾波和中值濾波去除噪聲干擾,采用top-hat變換[10]消除背景干擾。采用CUDA技術進行高斯濾波、中值濾波和top-hat變換的并行化設計,主要實現過程如下:
步驟1:中值濾波。首先根據圖像大小分配線程,并將圖像數據綁定紋理內存。綁定紋理內存是為了減少線程對內存的請求,以便更有效地利用內存帶寬。其次用線程塊和線程索引確定當前處理像元數據在圖像中的坐標和在內存中的偏移量,從二維紋理內存中讀出以當前像元為中心3×3鄰域內像元的像素值,取它們的中值替換當前像元,并將結果存入全局內存。
步驟2:高斯濾波。將經過中值濾波的圖像數據綁定紋理內存,采用分離濾波器[11]的方式進行高斯濾波。原理是將二維濾波過程分解成兩次一維濾波的方式進行處理,即分成行、列兩次濾波。首先進行行濾波,由于各個像元高斯濾波過程都是獨立的,所以可以采用一個線程計算相對應一個像元,將每個像素與其周圍的8個像素點進行加權計算出濾波結果。列濾波過程與行濾波過程相似,兩者的區別是列濾波過程中每個線程號的增加跨過了濾波區域。由于每個線程束中的每個線程訪問相同的數據內存空間,而連續線程訪問連續共享內存地址,所以在數據處理過程中不會存在紋理內存和共享內存的訪問沖突。
步驟3:top-hat變換。將經過圖像形態學開、閉的圖像α(x,y)、β(x,y)分別綁定紋理內存。之后將進行過濾波處理的圖像數據F(x,y)與對應位置的α(x,y)、β(x,y)按式(1)進行運算。在線程支出過程中,每個線程束中的線程只需一次顯存讀寫操作。
| $\left\{ \begin{align} & W\left( x,y \right)=F\left( x,y \right)-\alpha \left( x,y \right) \\ & H\left( x,y \right)=\beta \left( x,y \right)-F\left( x,y \right) \\ \end{align} \right.,$ |
1.3 蛋白質點預檢測
圖像預處理完成后,采用基于內外標記分水嶺算法[6]對蛋白質點進行預檢測,其并行化設計的主要實現步驟如下:
步驟1:內標記點提取。 根據凝膠圖像,選定圖像最小點來初始化檢測參數,得到最小點半徑minR。以半徑minR×minR為移動窗口遍歷所有線程中的像元數據,尋找圖像中的峰值點,用原子操作數atomicAdd進行計數,并將峰值點數據存入全局內存中。計算兩兩峰值間的距離,若兩者距離小于minR,則將灰度值大的點去除,同時原子操作數atomicAdd減1,則此灰度值小的峰值點即為內標記點。
步驟2:外標記點提取。首先將內標記圖像中屬于同一個內標記的用同一個數值進行編號(即標記),且每個內標記的數值都不相同;然后將不同內標記區域圖像數據放在不同block(線程塊)中的線程;其次各個block中的每個thread(線程)處理經過標記了的數值,從每個標記了數值的點開始向其周圍上下左右的點開始擴展,如果上下左右的點都是未標記過的點則將這些點標記為與該點相同的數值,并繼續對這些新的標記數值點進行擴展;當擴展時,如果發現待標記的點已被其他數值標記過,且標記值與該標記不同,也不是特殊標記,則停止擴展,并把該標記點改成特殊標記,作為分水嶺脊線。最后將未處理的數據按上述方法繼續擴展;重復上述步驟直到圖像上的所有點都已被標記過。將得到的分水嶺脊線像素值記為0,即為凝膠圖像的外標記。
步驟3:配置執行參數。定義一個bool類型的指針,大小與圖像大小相同,將全部元素置為0。每個block分配16個thread,則分配的block個數為:atomicAdd/16+1。由于每個蛋白質點區域的漲水過程互不相關,則每個block可單獨處理一個內標記點所在的蛋白質點區域。
步驟4:內外標記分水嶺算法。首先定義一個與圖像大小相同的bool類型的指針,分配空間后置0,從全局內存中讀出標記點,計算標記點在圖像中的坐標(startX,startY),以標記點坐標為起始坐標在各個蛋白質點區域進行擴展。若擴展點是未標記點則將其標記為與內標記相同的數值存入二維內存中,若擴展點是標記點且其數值與該區域的內標點數值不同,則該點為蛋白質點的輪廓邊緣并將其對應的bool變量置1,待所有線程處理完畢之后,將二維內存中的數據進行并行方式排序,取出其中像素值最小的像元數據按上面方法操作,二維內存中數據為空。最后將bool型指針數據與圖像數據結合,得到檢測結果圖。利用上述粗檢測方法對凝膠圖像進行蛋白質點檢測,其效果如圖 2所示。
圖2
蛋白質點粗檢測
Figure2.
Protein spots coarse detection
像元數據并行排序:主要是對二維數組進行排序,并行排序方法是建立在雙調排序的基礎上,通過對二維數組的行列交替進行排序。首先對線程進行分配,每個線程塊使用512個線程,需要一個共享寄存器同步所有的線程數據,而共享寄存器只能同步屬于同一個線程塊中線程的數據,每次最多只能處理1 024×1 024的二維數組,如數據較多則可分多次進行。首先將待排序的數據進行格式轉換,以便于排序;其次,啟用線程,每個線程處理一列數據,對列數據進行并行的升序排序,得出排序結果存入共享寄存器中并調用_synchreads()函數同步各個線程;待所有線程執行至_synchreads()函數時,開始對行數據進行并行排序,其中偶數行進行升序排序,奇數行進行降序排序,得出排序結果存入共享寄存器中并調用_synchreads()函數線程同步;重復上述步驟循環,共循環log(m),其中m為二維數組的行數;最后,處理得到的數據是一個蛇形一樣的二維數組(像元并行排序示意圖如圖 3所示)。
圖3
像元排序示意圖
Figure3.
Process of pixel sort
1.4 重疊蛋白質點檢測
蛋白質點粗檢測后,大部分蛋白質點已被檢測出來,但存在重疊蛋白質點檢測效果不佳的問題,如圖 2粗檢測結果中所示的1和2處。針對此問題,采用文獻[8]方法進行重疊蛋白質點分割,主要包括形狀標記點提取和重疊蛋白質點分割兩個過程。并行化設計的主要步驟如下:
步驟1:區域生長法確定凝膠圖像二值圖像重建[12-13]。首先確定種子點,利用預檢測蛋白質點區域的幾何中心做為種子點,以蛋白質點外圍輪廓作為增長的終止規則。每個像素對應一個線程,定義兩個bool類型的變量A、B(原始值都為0),將種子點(即區域極小值)所對應位置的A中值置為1。調用內核函數,對A中為1所對應的像素點取其3×3鄰域進行判斷,并將對應的A值置為0,若其為預檢測蛋白質點邊緣輪廓則將其對應的B中值置為1,反之將A置為1。之后判斷B中數據有無變化,若B中數據無變化,則代表區域生長完成,若有變化,則重復上述步驟。
步驟2:形狀標記點提取。本文采用自適應閾值的H-極小值變換方法(如式2所示),采用一維線程分配方法,每個線程塊分配256個,則其對應的網格線程塊的分配為(蛋白質點連通域個數+線程塊在x方向上的維度+1)/線程塊在x方向上的維度。采用一個線程塊處理一個蛋白質點區域,因為每個蛋白質點區域的像素點數據需相互通信。每個線程塊處理一個單獨的蛋白質點連通區域,以第i個連通域(線程塊)為例,首先從閾值h=1進行極小值變換,計算對應的極小值個數Ni(h),若其小于或等于1,則該連通域中沒有重疊蛋白質點;反之,含有重疊點。重復增長h,增長步長為1,計算Ni(h),直至其小于1。根據各個線程求得的閾值h計算該線程處理蛋白質點區域的形狀標記點。
| $H\left( c,h \right)={{R}^{\delta }}_{f}\left( c+h \right)-c,$ |
式中,c為經過粗檢測后的凝膠圖像C(x,y),h為極小值深度閾值,Rfδ(c+h)為對圖像c進行極小值強加得到的圖像數據,H(c,h)為圖像c的區域極小值。
步驟3:距離變換。線程分配仍用上一步的方法,每個線程處理一個蛋白質點連通區域,形狀標記距離變換是計算形狀標記點到該連通區域中任意像素點的最短路徑。采用8-鄰域的距離變換方法[14],首先對蛋白質點連通區域的位置進行統計,CUDA架構可以利用線程的唯一ID來標識統計,將統計后的數據利用均值法求出該區域的中心。對蛋白質點區域的所有像素點進行位置統計,利用均值法求出其幾何中心;判斷蛋白質點區域中任意像素點相對幾何中心的位置,如A點在其左上,則計算距離的方向順序是:左、左上、上,將這3個方向上的最小值Lmin保存;計算A點的其他5個方向到邊界的距離L,以Lmin為最高的循環次數,在該循環次數內,如果在某方向上計算的距離L小于Lmin,則用L的值替換Lmin的值,否則,停止該方向上的距離計算。重復上述步驟,完成所有像素計算。
步驟4:對形狀距離圖像進行分水嶺變換[15]。由于每個蛋白質點區域的分水嶺算法互不相干,則可以采用每個線程塊負責一個蛋白質點區域,并將各個蛋白質點區域進行數值標記,用線程標識號進行數值標記。在此以第i個蛋白質點連通區域為例,其他像素類似。先以該區域的中心點進行3×3鄰域像素的擴展,將這些數據存入全局寄存器中,再采用設備端的Shear方法對二維數組中的數據進行排序(這是由于分水嶺算法在進行數據“泛洪”時只能先處理像素值小的點),并取出最小的像素值,繼續對這些像素進行3×3數據擴展,對擴展的數據進行判斷,若發現有些點的標記值與該點區域的標記數值不同,則該點為區域的邊緣輪廓點;重復上述步驟,直至遍歷該區域內的所有蛋白質點。將數據存入全局寄存器中,并將其拷貝至主機內存,顯示圖像。
利用上述重疊點分割方法對圖 2粗檢測所示凝膠圖像進行重疊蛋白質點分割。以圖 2結果圖中的重疊點1為檢測對象,其效果如圖 4所示。
圖4
重疊蛋白質點分割
Figure4.
Overlapping protein spots segmentation
2 實驗結果及分析
為驗證本文方法的有效性,在硬件配置為NVIDIA GeForce 605圖形處理器、主機內存為4.0G、Pentium(R) Dual-Core CPU E5800 @ 3.20 GHz 雙核下,以及軟件環境為WIN7操作系統、CUDA6.0 32 bit版本、Visual Studio 2010 32 bit和GEFORCE 344.11顯卡驅動版本下,分別實現蛋白質點檢測算法在CPU上運行的串行方法和在CUDA上運行的并行方法。
為驗證圖像大小對算法效率提高的影響,選取6組不同分辨率的凝膠圖像進行檢測對比實驗,算法耗時取多次運行時間的平均值(取3次)。檢測算法在CPU和GPU上實現的性能比較如表 1所示,其中加速比為CPU耗時與GPU耗時之比。
可以看出,蛋白質點檢測算法在GPU上運行并行方法的效率明顯高于在CPU上運行的串行方法,并且隨著圖像像素的增大,檢測效率提高越明顯。這是因為采用CPU串行實現時,需用循環對每個像素進行遍歷,像素越多,耗時越多,而采用GPU并行實現時,為每一像素開辟一個線程數并行實現每個像素的遍歷,這樣明顯提高了算法的效率。
3 結論
本文提出了基于CUDA的蛋白質點檢測快速實現方法。該方法根據凝膠圖像的特點,利用CUDA單指令多線程指令確定了適合凝膠圖像的線程分配方式,加快了凝膠圖像預處理算法、蛋白質點預檢測及重疊蛋白質點檢測算法的實現,提高了檢測算法的運行效率,同時使用CUDA共享內存和紋理內存的內存管理方式,減少了重復訪問,進一步提高檢測效率。通過不同像素大小的真實凝膠圖像檢測實驗,驗證了本文快速實現方法的有效性。