本文基于 Z 曲線(z-curve)理論和位置權重矩陣(PWM)提出一種構建核小體 DNA 序列的模型。該模型將核小體 DNA 序列集轉換成三維空間坐標,通過計算該序列集的位置權重矩陣獲得相似性權重得分,將兩者整合得到綜合序列特征模型(CSeqFM),并分別計算候選核小體序列和連接序列到模型 CSeqFM 的歐氏距離作為特征集,投入到支持向量機(SVM)中訓練和檢驗,通過十折交叉驗證進行性能評估。結果顯示,酵母核小體定位的敏感性、特異性、準確率和 Matthews 相關系數(MCC)分別為 97.1%、96.9%、94.2% 和 0.89,受試者操作特征(receiver operating characteristic,ROC)曲線下面積(area under curve,AUC)達到 0.980 1。與其他相關 Z 曲線方法比較,CSeqFM 方法在各項評估指標中均表現出優勢,具有更好的識別效果。同時,將 CSeqFM 方法推廣到線蟲、人類和果蠅的核小體定位識別中,AUC 均高于 0.90,與 iNuc-STNC 和 iNuc-PseKNC 方法比較,CSeqFM 方法也表現出較好的穩定性和有效性,進一步表明該方法具有較好的可靠性和識別效能。
引用本文: 崔穎, 徐澤龍, 李建中. 基于綜合 DNA 序列特征的支持向量機方法識別核小體定位. 生物醫學工程學雜志, 2020, 37(3): 496-501. doi: 10.7507/1001-5515.201911064 復制
版權信息: ?四川大學華西醫院華西期刊社《生物醫學工程學雜志》版權所有,未經授權不得轉載、改編
引言
核小體是真核生物染色質的基本結構單元,每個核小體由核心 DNA 序列和連接區 DNA 序列組成,核心 DNA 序列由 147 bp 的 DNA 纏繞組蛋白八聚體近兩圈形成,也稱為核小體 DNA,而相鄰兩個核小體 DNA 之間的序列稱為連接區 DNA[1-3]。核小體定位是指 DNA 雙螺旋相對于組蛋白八聚體的位置,DNA 序列特征一直被認為是影響核小體定位的重要因素之一。核小體參與很多重要的生物學過程,如染色質形成[4]、拮抗轉錄因子[5]以及抑制基因表達[6]等,核小體 DNA 序列的精確定位影響著基因表達調控[7]、DNA 復制[8]、DNA 修復[9]和重組[10]等。隨著高通量測序技術的快速發展,目前已經獲得了多種真核生物高分辨率的核小體定位實驗圖譜,如酵母、果蠅、人等,但現階段完全依靠實驗方法檢測核小體定位還面臨很多問題,例如耗費大量時間和經費,難以滿足部分研究人員希望即時獲得研究數據的現實需要等,因此,通過計算模型進行核小體識別和預測已經成為生物實驗研究的有力補充。
目前核小體定位識別算法的研究已經成為表觀遺傳學研究的重要領域,國內外有很多研究人員通過信息熵[11]、堿基對偏轉角度[12]等方法來表示核小體 DNA 序列特征[13],進而使用模式識別或深度學習方法進行核小體定位識別,但目前識別方法的精度有待提高,其應用范圍也有待進一步推廣。支持向量機(support vector machine,SVM)作為一種監督學習方法[14],在解決小樣本、非線性及高維模式識別中表現出許多特有的優勢,已經在許多領域取得了成功的應用。本文基于 Z 曲線理論和位置權重矩陣(position weight matrix,PWM),提出一種綜合序列特征模型,以計算樣本與該模型間的歐氏距離作為特征,投入到 SVM 中進行訓練和模型檢驗,用于酵母核小體的定位識別,并將該方法推廣到其他物種中,包括線蟲、人類和果蠅等。
1 實驗數據和方法
1.1 實驗數據
1.1.1 酵母數據集
從文獻中獲得兩套酵母(S. cerevisiae)核小體數據[15-17],第一套數據集包括 5 000 條核小體 DNA 序列與 5 000 條連接區 DNA 序列,記為 S1;第二套數據集包含 1 880 條核小體 DNA 序列和 1 740 條連接區 DNA 序列,記為 S2。兩套數據集的 DNA 序列長度均為 150 bp,S2 經過 CD-HIT 軟件[18],閾值設為 80% 后去冗余得到。
1.1.2 線蟲、人類和果蠅數據集
從文獻中獲得線蟲(C. elegans)、人類(H. sapiens)和果蠅(D. melanogaster)三個物種的核小體數據集[19],C. elegans 數據集包括核小體 DNA 序列 2 567 條和連接區 DNA 序列 2 608 條,H. sapiens 數據集包括核小體 DNA 序列 2 273 條和連接區 DNA 序列 2 300 條,D. melanogaster 數據集包括核小體 DNA 序列 2 900 條和連接區 DNA 序列 2 850 條,三個物種的 DNA 序列長度均為 147 bp,處理方法同 1.1.1 小節。
1.2 實驗方法
1.2.1 DNA 序列集的 Z 曲線模型
將每條 DNA 序列都轉化成歸一化的 Z 曲線坐標[20],如式(1)所示:
 |
當  時,
時, 表示 DNA 序列的子序列長度,
表示 DNA 序列的子序列長度, 、
、 、
、 和
和  表示沿 DNA 序列方向從第 1 位到第
表示沿 DNA 序列方向從第 1 位到第 位的子序列中堿基 A、G、C、T 累積出現的頻率,
位的子序列中堿基 A、G、C、T 累積出現的頻率, 為給定 DNA 序列長度,Z 曲線歸一化公式將長度為 N 的 DNA 序列轉換為空間
為給定 DNA 序列長度,Z 曲線歸一化公式將長度為 N 的 DNA 序列轉換為空間  個有序點,xn、yn、zn 為三維空間坐標分量,范圍為[? 1,1]。
個有序點,xn、yn、zn 為三維空間坐標分量,范圍為[? 1,1]。
將 m 條 DNA 序列,從第 1 個位置到第  個位置,分別計算其對應位置的坐標均值,如式(2)所示,即可得到 Z 曲線模型[21]。
個位置,分別計算其對應位置的坐標均值,如式(2)所示,即可得到 Z 曲線模型[21]。
 |
 、
、 、
、 和
和  表示 m 條序列從第 1 個位置到第
表示 m 條序列從第 1 個位置到第  個位置四種堿基 A、G、C、T 累積出現的平均頻率,xn、yn、zn 是三維空間坐標分量,取值范圍為[?1,1]。
個位置四種堿基 A、G、C、T 累積出現的平均頻率,xn、yn、zn 是三維空間坐標分量,取值范圍為[?1,1]。
1.2.2 DNA 序列集的位置權重矩陣模型
根據 DNA 序列集可以構建位置頻率矩陣(position frequency matrix,PFM),為減少 DNA 序列本身的堿基偏好性對模型的影響,把 PFM 轉換為 PWM[22]。具體方法為:引入堿基 i(A,G,C,T)在全基因組背景下出現的頻率(記為 bi),來消除 DNA 序列本身的堿基偏好性,PWM 模型構建方法如式(3)所示:
 |
Min 為 PWM 元素,表示堿基 i 在 DNA 序列的第 n 位置上的權重值,qin 是堿基 i 在 DNA 序列的第 n 個位置出現的頻率,M 為 4*N 的矩陣。S 為相似性權重得分,將每條候選 DNA 序列與 M 的每個位置的對應元素值進行累加,得到每一條序列的序列得分,最后將 m 條序列的得分加和取平均值,即得到新得分。
1.2.3 綜合序列特征模型構建
將構建 PWM 模型后計算得到的 S 得分與 Z 曲線模型中對應位置的三維坐標值相乘,即得到整合的新模型,如式(4)所示:
 |
Xn、Yn、Zn 表示模型的三維空間坐標分量,由于 Z 曲線模型和 PWM 模型都是基于序列模型,故該模型被稱為綜合序列特征模型(comprehensive sequence feature model,CSeqFM)。
1.2.4 歐氏距離特征提取
對于任意一條 DNA 序列,根據公式(1)將其轉化為 Z 曲線坐標,計算其與 CSeqFM 之間的歐式距離(Euclidean distance,ED)[23],得到歐氏距離向量記為 ED,具體過程為:計算一條長度為 N 的 DNA 序列在第  個位置與 CSeqFM 中對應的第
個位置與 CSeqFM 中對應的第  個位置的歐氏距離記為 Edn,計算
個位置的歐氏距離記為 Edn,計算  個位置,共得到
個位置,共得到  個歐氏距離分量 Ed1,Ed2,
個歐氏距離分量 Ed1,Ed2, ,EdN,組成得到歐氏距離向量記為 ED,如公式(5)所示[21]:
,EdN,組成得到歐氏距離向量記為 ED,如公式(5)所示[21]:
 |
Edn 代表一條 DNA 序列的第  個位置到 CSeqFM 的第
個位置到 CSeqFM 的第  個位置的歐式距離,xn、yn、zn 分別代表 DNA 序列的第
個位置的歐式距離,xn、yn、zn 分別代表 DNA 序列的第  個位置三維坐標值,Xn、Yn、Zn 分別代表 CSeqFM 中第
個位置三維坐標值,Xn、Yn、Zn 分別代表 CSeqFM 中第  個位置三維坐標,最終得到一個 1*
個位置三維坐標,最終得到一個 1* 的歐式距離向量 ED,則 ED 表示該條 DNA 序列與 CSeqFM 之間的距離,距離越小,DNA 序列與 CSeqFM 相似性越大,反之距離越大,DNA 序列與 CSeqFM 相似性越小。
的歐式距離向量 ED,則 ED 表示該條 DNA 序列與 CSeqFM 之間的距離,距離越小,DNA 序列與 CSeqFM 相似性越大,反之距離越大,DNA 序列與 CSeqFM 相似性越小。
1.2.5 支持向量機
本文將歐氏距離向量 ED 作為特征,投入到 SVM 中進行訓練和檢驗。具體方法如下:① 計算每一條核小體 DNA 序列與 CSeqFM 的距離得到歐氏距離向量集,記為歐氏距離陽性集  ;② 計算每一條非核小體 DNA 序列即連接 DNA 序列與 CSeqFM 模型的距離得到歐氏距離向量集,記為歐氏距離陰性集
;② 計算每一條非核小體 DNA 序列即連接 DNA 序列與 CSeqFM 模型的距離得到歐氏距離向量集,記為歐氏距離陰性集  ;③ 通過 SVM 方法,使用十折交叉驗證,隨機分配陽性集和陰性集為 10 份,選擇 9 份距離特征樣本集(陽性集
;③ 通過 SVM 方法,使用十折交叉驗證,隨機分配陽性集和陰性集為 10 份,選擇 9 份距離特征樣本集(陽性集  和陰性集
和陰性集  )進行訓練,剩余 1 份距離特征樣本集(陽性集
)進行訓練,剩余 1 份距離特征樣本集(陽性集  和陰性集
和陰性集  )進行檢驗,共進行 10 次,直到 10 份樣本都完成檢驗為止,取 10 次實驗平均值為一次隨機分配的最后結果;④ 隨機分配過程進行 10 次,實驗使用 R 語言編程及 R 包“e1071”完成。
)進行檢驗,共進行 10 次,直到 10 份樣本都完成檢驗為止,取 10 次實驗平均值為一次隨機分配的最后結果;④ 隨機分配過程進行 10 次,實驗使用 R 語言編程及 R 包“e1071”完成。
1.3 評價指標
本文使用敏感性(sensitivity,Sn)、特異性(specificity,Sp)、準確率(accuracy,Acc)和 Matthews 相關系數(Matthews correlation coefficient,MCC),以及受試者操作特征(receiver operating characteristic,ROC)曲線下面積(area under curve,AUC)作為評價參數。其中 ROC 曲線越靠近圖左側邊界和上側邊緣(曲線與橫軸包含的面積越大)則準確率越高,即 AUC 值越接近 1,說明方法的識別性能越好。前四個指標則通常被用于在統計預測理論中從不同角度衡量預測系統性能的評估指標[16, 19],如式(6)所示:
 |
 表示研究樣本中所有核小體 DNA 序列數目,
表示研究樣本中所有核小體 DNA 序列數目, 表示研究樣本中所有連接區 DNA 序列數目,
表示研究樣本中所有連接區 DNA 序列數目, 表示連接區 DNA 序列被錯誤識別為核小體 DNA 序列的數目,
表示連接區 DNA 序列被錯誤識別為核小體 DNA 序列的數目, 表示核小體 DNA 序列被錯誤識別為連接區 DNA 序列的數目。
表示核小體 DNA 序列被錯誤識別為連接區 DNA 序列的數目。
2 實驗結果與討論
2.1 酵母實驗
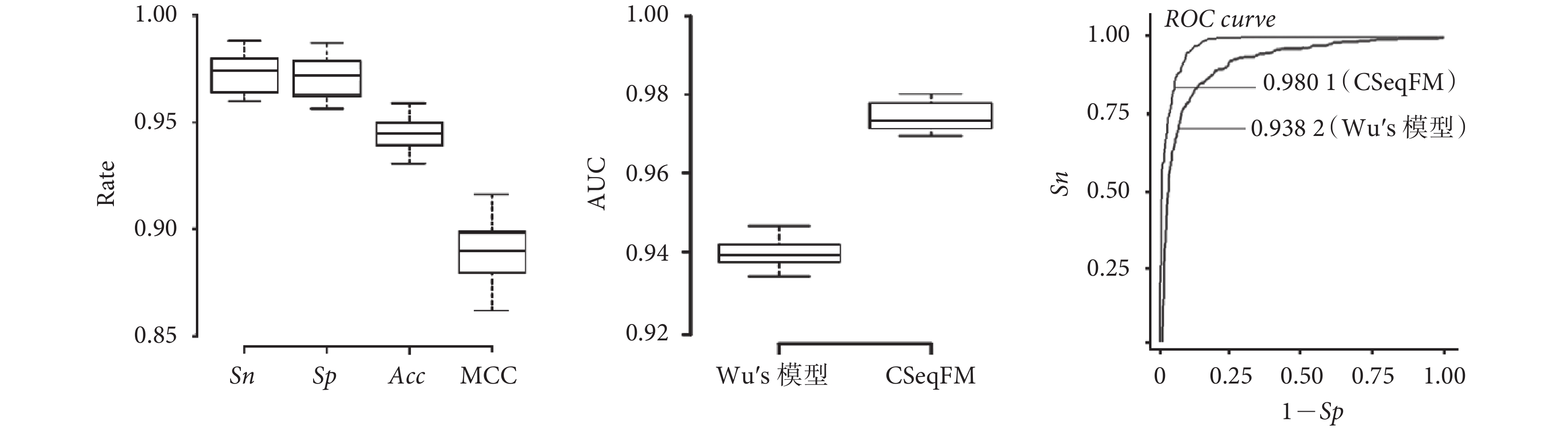
應用 CSeqFM 使用 SVM 方法識別 S. cerevisiae 核小體定位的實驗結果如表 1 和圖 1 所示。在 S. cerevisiae 數據集 S1 結果中,敏感性 Sn、特異性 Sp、準確率 Acc 和 MCC 值分別為 97.1%、96.9%、94.2% 和 0.89,表明該方法性能穩定且效果較好;與基于 Z 曲線理論的 Wu’s 模型[24]結果進行比較,CSeqFM 識別結果的四項評估指標均高于 Wu’s 的結果;同時 AUC 分布箱式圖顯示 CSeqFM 的 AUC 整體分布遠高于 Wu’s 的整體分布,曲線圖顯示 CSeqFM 的 AUC 值為 0.980 1,高于 Wu’s 的 0.938 2,說明 CSeqFM 模型識別性能更好。
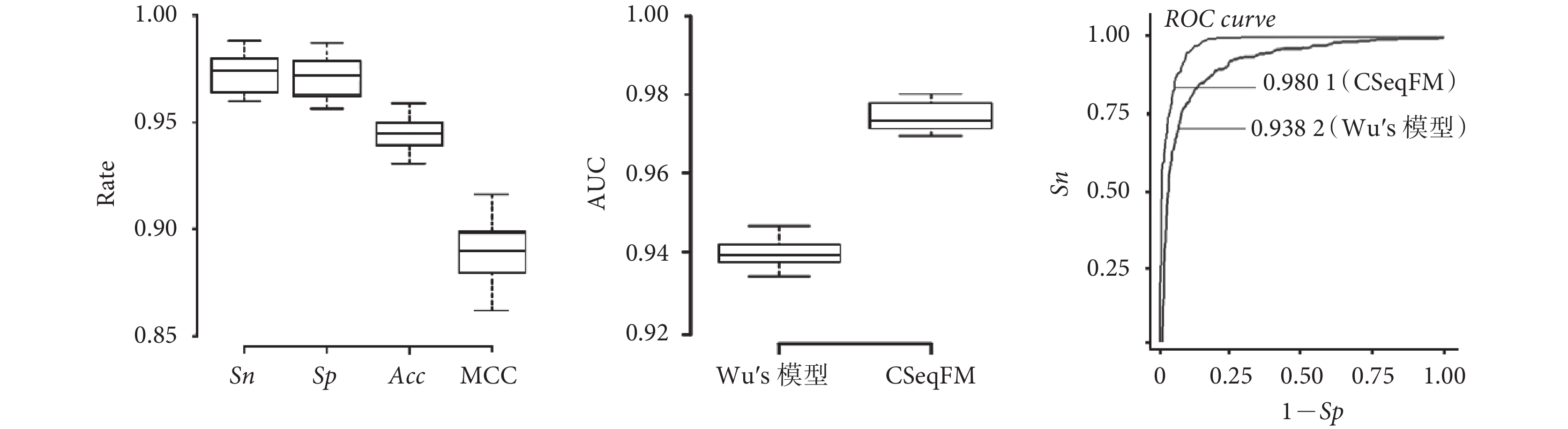 圖1
酵母數據集 S1 結果的四項性能指標、AUC 值分布及 ROC 曲線
Figure1.
Four performances, AUC distribution and ROC curves of dataset S1 for S. cerevisiae
圖1
酵母數據集 S1 結果的四項性能指標、AUC 值分布及 ROC 曲線
Figure1.
Four performances, AUC distribution and ROC curves of dataset S1 for S. cerevisiae
為進一步檢驗性能,用 CSeqFM 識別 S. cerevisiae 數據集 S2,實驗結果如表 1 所示,敏感性 Sn、特異性 Sp、準確率 Acc 和 MCC 值分別為 92.4%、93.9%、93.1% 和 0.86,均高于 Wu’s 模型結果,再次說明 CSeqFM 具有較好的識別效果。
2.2 線蟲、人類和果蠅實驗
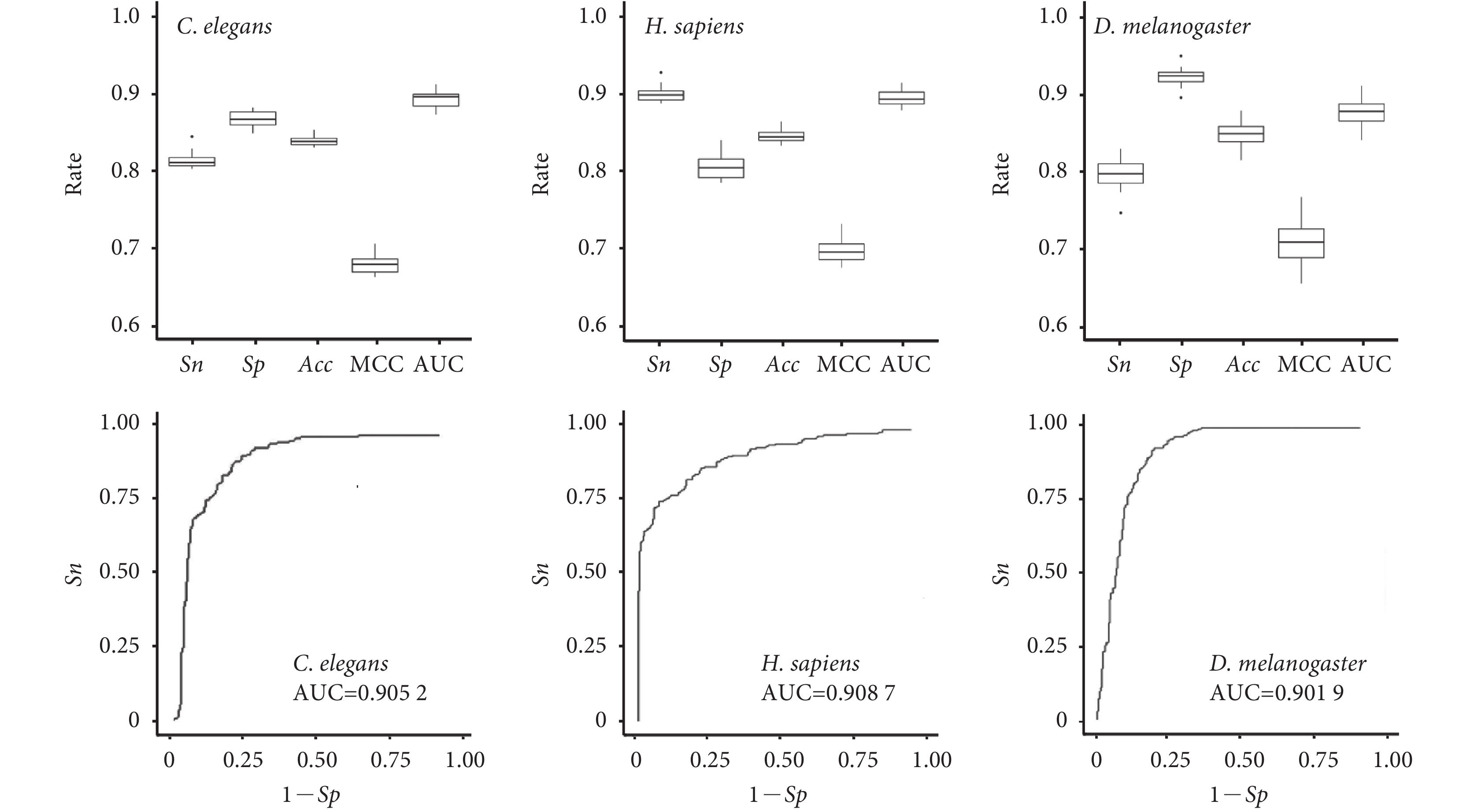
將 CSeqFM 模型推廣到其他物種,包括 C. elegans、H. sapiens 和 D. melanogaster 的核小體定位識別,實驗結果與 iNuc-STNC[16]和 iNuc-PseKNC[19]方法比較,如表 2 和圖 2 所示。
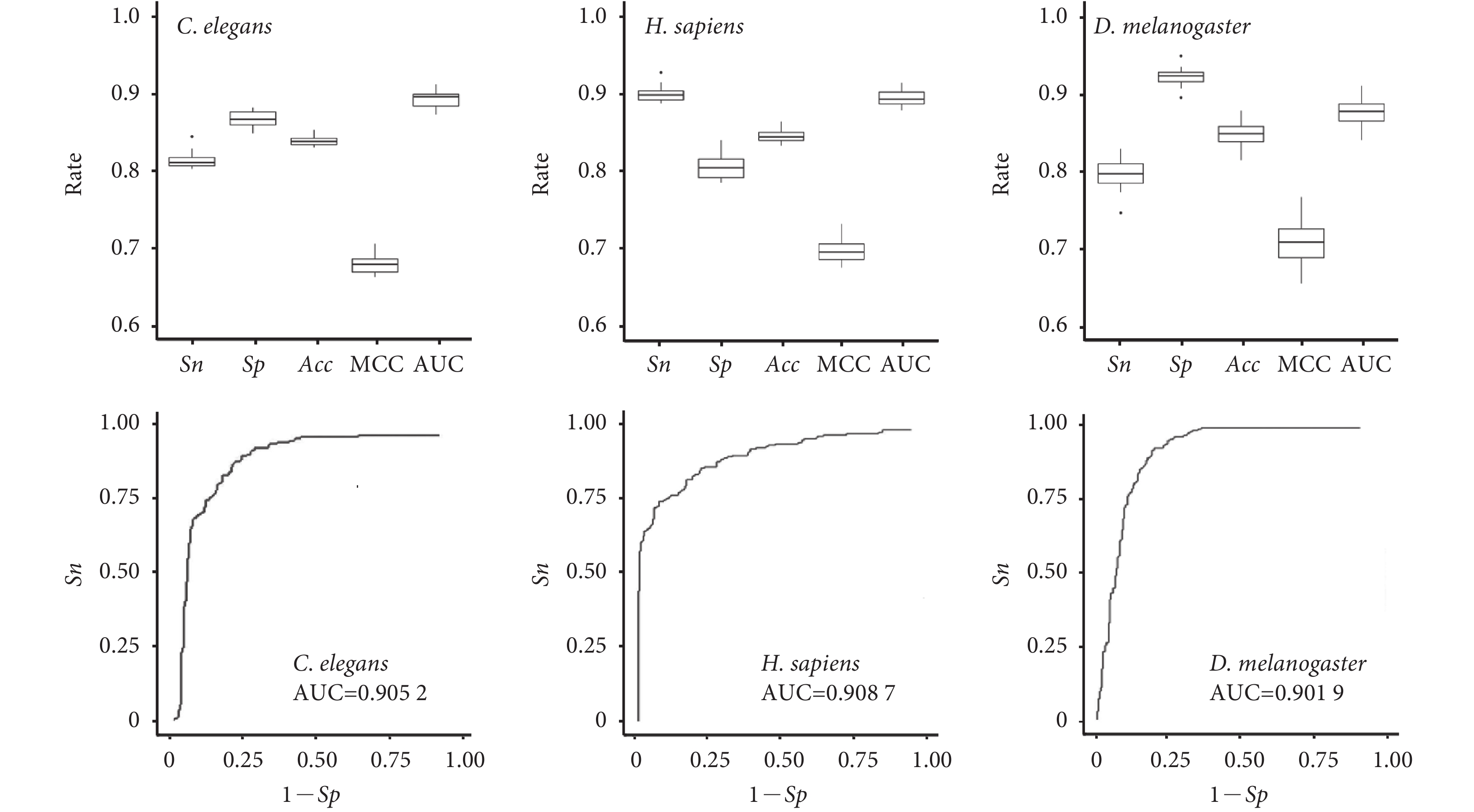 圖2
C. elegans、H. sapiens 和 D. melanogaster 的實驗結果
Figure2.
Experimental results of C. elegans, H. sapiens and D. melanogaster species
圖2
C. elegans、H. sapiens 和 D. melanogaster 的實驗結果
Figure2.
Experimental results of C. elegans, H. sapiens and D. melanogaster species
首先,與 iNuc-STNC 方法比較,在 Sn 方面,CSeqFM 在 H. sapiens 和 D. melanogaster 中均高于 iNuc-STNC 方法;在 Sp、Acc 和 MCC 方面,CSeqFM 方法在 D. melanogaster 中高于 iNuc-STNC 方法;在 AUC 方面,iNuc-STNC 方法沒有給出 AUC 值,而 CSeqFM 在三個物種中的 AUC 值均高于 0.90。如圖 2 所示,總體比較,CSeqFM 與 iNuc-STNC 方法在三個物種中各有優勢,整體性能基本一致。
其次,與 iNuc-PseKNC 方法比較,在 Sn 方面,CSeqFM 在 H. sapiens 和 D. melanogaster 中均高于 iNuc-PseKNC;在 Sp 方面,CSeqFM 在 C. elegans 和 D. melanogaster 中均高于 iNuc-PseKNC 方法;在 Acc、MCC 和 AUC 方面,CSeqFM 在 D. melanogaste 中均高于 iNuc-PseKNC 方法。CSeqFM 與 iNuc-PseKNC 方法在 C. elegans 和 H. sapiens 中各有優勢,但 CSeqFM 在 D. melanogaster 中的五項性能指標均高于 iNuc-PseKNC 方法,識別效果更好。
另外,iNuc-STNC 和 iNuc-PseKNC 方法都沒有 S. cerevisiae 實驗結果,而 CSeqFM 在兩套 S. cerevisiae 數據集中都取得較好的識別效果(如表 1 和圖 1 所示)。
綜上所述,與 iNuc-STNC 和 iNuc-PseKNC 相比,CSeqFM 在 C. elegans、H. sapiens 和 D. melanogaster 的各項性能指標較好,識別效果穩定,說明 CSeqFM 方法具有可靠的物種推廣性和有效性,進一步驗證了 CSeqFM 方法具有好的識別效果。
3 結論
本文提出一種基于綜合序列特征的核小體定位模型 CSeqFM,通過 SVM 進行訓練和檢驗,實驗結果表明,CSeqFM 在 S. cerevisiae 中的 Sn、Sp、Acc 和 MCC 性能指標較好,且 AUC 值達到 0.980 1,各項性能均優于 Wu’s 模型的結果,表明該方法在 S. cerevisiae 中識別性能較好。將 CSeqFM 推廣到 C. elegans、H. sapiens 和 D. melanogaster 物種中,結果顯示 CSeqFM 的各項性能指標較好,三個物種的 AUC 值均高于 0.90,與 iNuc-STNC 和 iNuc-PseKNC 方法比較,CSeqFM 在 D. melanogaster 尤其表現出優勢,進一步驗證了 CSeqFM 方法具有較好的可靠性和有效性。分析原因,可能是由于 CSeqFM 模型是一種綜合序列特征模型,整合了 Z 曲線模型在水平方向上的序列特征和 PWM 在垂直方向上的序列特征,更全面地表示了核小體的序列特征。另外,CSeqFM 也可以用于生物數據有關 DNA 序列或功能元件的分類與識別。總之,CSeqFM 具有較好的識別效果和推廣性,有利于促進核小體定位 DNA 序列特征和功能的研究。
利益沖突聲明:本文全體作者均聲明不存在利益沖突。
引言
核小體是真核生物染色質的基本結構單元,每個核小體由核心 DNA 序列和連接區 DNA 序列組成,核心 DNA 序列由 147 bp 的 DNA 纏繞組蛋白八聚體近兩圈形成,也稱為核小體 DNA,而相鄰兩個核小體 DNA 之間的序列稱為連接區 DNA[1-3]。核小體定位是指 DNA 雙螺旋相對于組蛋白八聚體的位置,DNA 序列特征一直被認為是影響核小體定位的重要因素之一。核小體參與很多重要的生物學過程,如染色質形成[4]、拮抗轉錄因子[5]以及抑制基因表達[6]等,核小體 DNA 序列的精確定位影響著基因表達調控[7]、DNA 復制[8]、DNA 修復[9]和重組[10]等。隨著高通量測序技術的快速發展,目前已經獲得了多種真核生物高分辨率的核小體定位實驗圖譜,如酵母、果蠅、人等,但現階段完全依靠實驗方法檢測核小體定位還面臨很多問題,例如耗費大量時間和經費,難以滿足部分研究人員希望即時獲得研究數據的現實需要等,因此,通過計算模型進行核小體識別和預測已經成為生物實驗研究的有力補充。
目前核小體定位識別算法的研究已經成為表觀遺傳學研究的重要領域,國內外有很多研究人員通過信息熵[11]、堿基對偏轉角度[12]等方法來表示核小體 DNA 序列特征[13],進而使用模式識別或深度學習方法進行核小體定位識別,但目前識別方法的精度有待提高,其應用范圍也有待進一步推廣。支持向量機(support vector machine,SVM)作為一種監督學習方法[14],在解決小樣本、非線性及高維模式識別中表現出許多特有的優勢,已經在許多領域取得了成功的應用。本文基于 Z 曲線理論和位置權重矩陣(position weight matrix,PWM),提出一種綜合序列特征模型,以計算樣本與該模型間的歐氏距離作為特征,投入到 SVM 中進行訓練和模型檢驗,用于酵母核小體的定位識別,并將該方法推廣到其他物種中,包括線蟲、人類和果蠅等。
1 實驗數據和方法
1.1 實驗數據
1.1.1 酵母數據集
從文獻中獲得兩套酵母(S. cerevisiae)核小體數據[15-17],第一套數據集包括 5 000 條核小體 DNA 序列與 5 000 條連接區 DNA 序列,記為 S1;第二套數據集包含 1 880 條核小體 DNA 序列和 1 740 條連接區 DNA 序列,記為 S2。兩套數據集的 DNA 序列長度均為 150 bp,S2 經過 CD-HIT 軟件[18],閾值設為 80% 后去冗余得到。
1.1.2 線蟲、人類和果蠅數據集
從文獻中獲得線蟲(C. elegans)、人類(H. sapiens)和果蠅(D. melanogaster)三個物種的核小體數據集[19],C. elegans 數據集包括核小體 DNA 序列 2 567 條和連接區 DNA 序列 2 608 條,H. sapiens 數據集包括核小體 DNA 序列 2 273 條和連接區 DNA 序列 2 300 條,D. melanogaster 數據集包括核小體 DNA 序列 2 900 條和連接區 DNA 序列 2 850 條,三個物種的 DNA 序列長度均為 147 bp,處理方法同 1.1.1 小節。
1.2 實驗方法
1.2.1 DNA 序列集的 Z 曲線模型
將每條 DNA 序列都轉化成歸一化的 Z 曲線坐標[20],如式(1)所示:
|
當 時, 表示 DNA 序列的子序列長度,、、 和 表示沿 DNA 序列方向從第 1 位到第 位的子序列中堿基 A、G、C、T 累積出現的頻率, 為給定 DNA 序列長度,Z 曲線歸一化公式將長度為 N 的 DNA 序列轉換為空間 個有序點,xn、yn、zn 為三維空間坐標分量,范圍為[? 1,1]。
將 m 條 DNA 序列,從第 1 個位置到第 個位置,分別計算其對應位置的坐標均值,如式(2)所示,即可得到 Z 曲線模型[21]。
|
、、 和 表示 m 條序列從第 1 個位置到第 個位置四種堿基 A、G、C、T 累積出現的平均頻率,xn、yn、zn 是三維空間坐標分量,取值范圍為[?1,1]。
1.2.2 DNA 序列集的位置權重矩陣模型
根據 DNA 序列集可以構建位置頻率矩陣(position frequency matrix,PFM),為減少 DNA 序列本身的堿基偏好性對模型的影響,把 PFM 轉換為 PWM[22]。具體方法為:引入堿基 i(A,G,C,T)在全基因組背景下出現的頻率(記為 bi),來消除 DNA 序列本身的堿基偏好性,PWM 模型構建方法如式(3)所示:
|
Min 為 PWM 元素,表示堿基 i 在 DNA 序列的第 n 位置上的權重值,qin 是堿基 i 在 DNA 序列的第 n 個位置出現的頻率,M 為 4*N 的矩陣。S 為相似性權重得分,將每條候選 DNA 序列與 M 的每個位置的對應元素值進行累加,得到每一條序列的序列得分,最后將 m 條序列的得分加和取平均值,即得到新得分。
1.2.3 綜合序列特征模型構建
將構建 PWM 模型后計算得到的 S 得分與 Z 曲線模型中對應位置的三維坐標值相乘,即得到整合的新模型,如式(4)所示:
|
Xn、Yn、Zn 表示模型的三維空間坐標分量,由于 Z 曲線模型和 PWM 模型都是基于序列模型,故該模型被稱為綜合序列特征模型(comprehensive sequence feature model,CSeqFM)。
1.2.4 歐氏距離特征提取
對于任意一條 DNA 序列,根據公式(1)將其轉化為 Z 曲線坐標,計算其與 CSeqFM 之間的歐式距離(Euclidean distance,ED)[23],得到歐氏距離向量記為 ED,具體過程為:計算一條長度為 N 的 DNA 序列在第 個位置與 CSeqFM 中對應的第 個位置的歐氏距離記為 Edn,計算 個位置,共得到 個歐氏距離分量 Ed1,Ed2,,EdN,組成得到歐氏距離向量記為 ED,如公式(5)所示[21]:
|
Edn 代表一條 DNA 序列的第 個位置到 CSeqFM 的第 個位置的歐式距離,xn、yn、zn 分別代表 DNA 序列的第 個位置三維坐標值,Xn、Yn、Zn 分別代表 CSeqFM 中第 個位置三維坐標,最終得到一個 1* 的歐式距離向量 ED,則 ED 表示該條 DNA 序列與 CSeqFM 之間的距離,距離越小,DNA 序列與 CSeqFM 相似性越大,反之距離越大,DNA 序列與 CSeqFM 相似性越小。
1.2.5 支持向量機
本文將歐氏距離向量 ED 作為特征,投入到 SVM 中進行訓練和檢驗。具體方法如下:① 計算每一條核小體 DNA 序列與 CSeqFM 的距離得到歐氏距離向量集,記為歐氏距離陽性集 ;② 計算每一條非核小體 DNA 序列即連接 DNA 序列與 CSeqFM 模型的距離得到歐氏距離向量集,記為歐氏距離陰性集 ;③ 通過 SVM 方法,使用十折交叉驗證,隨機分配陽性集和陰性集為 10 份,選擇 9 份距離特征樣本集(陽性集 和陰性集 )進行訓練,剩余 1 份距離特征樣本集(陽性集 和陰性集 )進行檢驗,共進行 10 次,直到 10 份樣本都完成檢驗為止,取 10 次實驗平均值為一次隨機分配的最后結果;④ 隨機分配過程進行 10 次,實驗使用 R 語言編程及 R 包“e1071”完成。
1.3 評價指標
本文使用敏感性(sensitivity,Sn)、特異性(specificity,Sp)、準確率(accuracy,Acc)和 Matthews 相關系數(Matthews correlation coefficient,MCC),以及受試者操作特征(receiver operating characteristic,ROC)曲線下面積(area under curve,AUC)作為評價參數。其中 ROC 曲線越靠近圖左側邊界和上側邊緣(曲線與橫軸包含的面積越大)則準確率越高,即 AUC 值越接近 1,說明方法的識別性能越好。前四個指標則通常被用于在統計預測理論中從不同角度衡量預測系統性能的評估指標[16, 19],如式(6)所示:
|
表示研究樣本中所有核小體 DNA 序列數目,表示研究樣本中所有連接區 DNA 序列數目,表示連接區 DNA 序列被錯誤識別為核小體 DNA 序列的數目,表示核小體 DNA 序列被錯誤識別為連接區 DNA 序列的數目。
2 實驗結果與討論
2.1 酵母實驗
應用 CSeqFM 使用 SVM 方法識別 S. cerevisiae 核小體定位的實驗結果如表 1 和圖 1 所示。在 S. cerevisiae 數據集 S1 結果中,敏感性 Sn、特異性 Sp、準確率 Acc 和 MCC 值分別為 97.1%、96.9%、94.2% 和 0.89,表明該方法性能穩定且效果較好;與基于 Z 曲線理論的 Wu’s 模型[24]結果進行比較,CSeqFM 識別結果的四項評估指標均高于 Wu’s 的結果;同時 AUC 分布箱式圖顯示 CSeqFM 的 AUC 整體分布遠高于 Wu’s 的整體分布,曲線圖顯示 CSeqFM 的 AUC 值為 0.980 1,高于 Wu’s 的 0.938 2,說明 CSeqFM 模型識別性能更好。
圖1
酵母數據集 S1 結果的四項性能指標、AUC 值分布及 ROC 曲線
Figure1.
Four performances, AUC distribution and ROC curves of dataset S1 for S. cerevisiae
為進一步檢驗性能,用 CSeqFM 識別 S. cerevisiae 數據集 S2,實驗結果如表 1 所示,敏感性 Sn、特異性 Sp、準確率 Acc 和 MCC 值分別為 92.4%、93.9%、93.1% 和 0.86,均高于 Wu’s 模型結果,再次說明 CSeqFM 具有較好的識別效果。
2.2 線蟲、人類和果蠅實驗
將 CSeqFM 模型推廣到其他物種,包括 C. elegans、H. sapiens 和 D. melanogaster 的核小體定位識別,實驗結果與 iNuc-STNC[16]和 iNuc-PseKNC[19]方法比較,如表 2 和圖 2 所示。
圖2
C. elegans、H. sapiens 和 D. melanogaster 的實驗結果
Figure2.
Experimental results of C. elegans, H. sapiens and D. melanogaster species
首先,與 iNuc-STNC 方法比較,在 Sn 方面,CSeqFM 在 H. sapiens 和 D. melanogaster 中均高于 iNuc-STNC 方法;在 Sp、Acc 和 MCC 方面,CSeqFM 方法在 D. melanogaster 中高于 iNuc-STNC 方法;在 AUC 方面,iNuc-STNC 方法沒有給出 AUC 值,而 CSeqFM 在三個物種中的 AUC 值均高于 0.90。如圖 2 所示,總體比較,CSeqFM 與 iNuc-STNC 方法在三個物種中各有優勢,整體性能基本一致。
其次,與 iNuc-PseKNC 方法比較,在 Sn 方面,CSeqFM 在 H. sapiens 和 D. melanogaster 中均高于 iNuc-PseKNC;在 Sp 方面,CSeqFM 在 C. elegans 和 D. melanogaster 中均高于 iNuc-PseKNC 方法;在 Acc、MCC 和 AUC 方面,CSeqFM 在 D. melanogaste 中均高于 iNuc-PseKNC 方法。CSeqFM 與 iNuc-PseKNC 方法在 C. elegans 和 H. sapiens 中各有優勢,但 CSeqFM 在 D. melanogaster 中的五項性能指標均高于 iNuc-PseKNC 方法,識別效果更好。
另外,iNuc-STNC 和 iNuc-PseKNC 方法都沒有 S. cerevisiae 實驗結果,而 CSeqFM 在兩套 S. cerevisiae 數據集中都取得較好的識別效果(如表 1 和圖 1 所示)。
綜上所述,與 iNuc-STNC 和 iNuc-PseKNC 相比,CSeqFM 在 C. elegans、H. sapiens 和 D. melanogaster 的各項性能指標較好,識別效果穩定,說明 CSeqFM 方法具有可靠的物種推廣性和有效性,進一步驗證了 CSeqFM 方法具有好的識別效果。
3 結論
本文提出一種基于綜合序列特征的核小體定位模型 CSeqFM,通過 SVM 進行訓練和檢驗,實驗結果表明,CSeqFM 在 S. cerevisiae 中的 Sn、Sp、Acc 和 MCC 性能指標較好,且 AUC 值達到 0.980 1,各項性能均優于 Wu’s 模型的結果,表明該方法在 S. cerevisiae 中識別性能較好。將 CSeqFM 推廣到 C. elegans、H. sapiens 和 D. melanogaster 物種中,結果顯示 CSeqFM 的各項性能指標較好,三個物種的 AUC 值均高于 0.90,與 iNuc-STNC 和 iNuc-PseKNC 方法比較,CSeqFM 在 D. melanogaster 尤其表現出優勢,進一步驗證了 CSeqFM 方法具有較好的可靠性和有效性。分析原因,可能是由于 CSeqFM 模型是一種綜合序列特征模型,整合了 Z 曲線模型在水平方向上的序列特征和 PWM 在垂直方向上的序列特征,更全面地表示了核小體的序列特征。另外,CSeqFM 也可以用于生物數據有關 DNA 序列或功能元件的分類與識別。總之,CSeqFM 具有較好的識別效果和推廣性,有利于促進核小體定位 DNA 序列特征和功能的研究。
利益沖突聲明:本文全體作者均聲明不存在利益沖突。