腸道菌群在糖尿病合并衰弱的發生發展中起到了重要的作用,因此研究中國中老年糖尿病衰弱人群腸道菌群的結構及功能特征性,具有重要意義。本研究收集30例社區中老年糖尿病患者糞便進行宏基因測序,結合臨床數據,分析糖尿病衰弱及衰弱前期與糖尿病非衰弱兩組人群菌群結構及功能差異性。結果表明:糖尿病衰弱及衰弱前期人群菌群α多樣性有所降低,柯林斯氏菌屬(Collinsella)、丁酸弧菌屬(Butyricimonas)在衰弱人群中相對豐度較高。糖尿病衰弱及衰弱前期人群富集上調的功能基因分布在組氨酸代謝(Histidine metabolism)、EB病毒感染(Epstein-Barr virus infection)、硫代謝(Sulfur metabolism)和Ⅱ型聚酮生物合成(Biosynthesis of type Ⅱ polyketide products)等,而丁酸代謝(Butanoate metabolism)以及苯丙氨酸代謝(Phenylalanine metabolism)等在該人群中下調。本研究為糖尿病衰弱人群的腸道菌群機制探索奠定了理論基礎,同時也為糖尿病衰弱及衰弱前期人群的預防與干預提供了一定的科學依據。
引用本文: 彭旭超, 趙艷莉, 林泰平, 舒嘵宇, 侯利莎, 高浪麗, 王慧, 葛寧, 岳冀蓉. 中老年糖尿病衰弱患者與腸道菌群結構及功能相關性研究. 生物醫學工程學雜志, 2021, 38(6): 1126-1133. doi: 10.7507/1001-5515.202101083 復制
版權信息: ?四川大學華西醫院華西期刊社《生物醫學工程學雜志》版權所有,未經授權不得轉載、改編
引言
全球老齡化問題正日益凸顯,中國老齡化進程明顯快于世界平均水平。2010年我國65歲及以上人群占比8.87%,預計到2050年,這一數字將提高至34%[1]。衰弱是老齡化突出問題之一,是指老年人生理儲備下降導致機體易損性增加和抗應激能力減退的非特異性狀態,其核心是老年人生理儲備下降或異常,外界較小刺激即可引起臨床事件的發生[2]。衰弱人群更容易出現各種不良預后,包括跌倒、失能、認識功能下降、再入院率增加甚至死亡風險等[3]。
糖尿病與衰弱兩者可相互作用。一方面,高血糖狀態會抑制骨骼肌生長,嚴重時會造成肌肉萎縮[4],而胰島素抵抗會抑制骨骼肌的能量代謝造成肌肉再生功能障礙[5],機體衰弱發生風險增加[6]。另一方面,衰弱促使胰島素抵抗增加,血糖呈高水平狀態,增加糖尿病發生風險[7]。我國的流行病學調研顯示,我國65歲以上糖尿病合并衰弱人群占糖尿病人群比例為5%~48%[8],該人群出現低血糖及死亡風險高[9],預后較單純糖尿病老年人群差,極大地增加了家庭照護和社會經濟負擔。
微生物在人體內廣泛分布,其中以胃腸道菌群數量最多。既往研究表明,衰弱與腸道菌群α多樣性呈負相關[10],衰弱程度與Ruminococcacea菌、F. prausnitzii菌及Lachnospiraceae菌豐度呈負相關,與Eubacteriumdolichum菌和Eggerthellalenta菌豐度呈正相關。van Tongeren等[11]采用衰弱指數(frailty index,FI)評價衰弱,發現非衰弱人群,菌群特點表現為普氏菌屬(Prevotella)與擬桿菌屬(Bacteroides)的比值較高;而在衰弱人群中,菌群特征表現為瘤胃球菌屬(Ruminococcus)及奇異菌屬(Atopobium)占比增加,乳酸桿菌屬(Lactobacillus)占比減少。同時,Biagi團隊[12]也發現慢性疾病越多的老年人,腸道內克里斯滕森菌科(Christensenellaceae)、嗜黏蛋白阿克曼菌(Akkermansia)及雙歧桿菌屬(Bifidobacterium)等數量減少,但這些細菌數量級與衰弱并非呈線性關系,個體差異性較大。
腸道菌群可從多個方面影響衰弱。首先,腸道微生物組成失調會影響蛋白質代謝和生物利用度[13-14],影響骨骼肌合成代謝所需的維生素B12、葉酸、核黃酸等物質的產生[14],加速肌細胞線粒體受損累積[15],增加胰島素抵抗風險[16],這些因素會導致骨骼肌數量與質量的下降;其次,隨著年齡增長,微生物組成向著促炎表型發展,總體表現為微生物多樣性減少,個體變異增加[17],該變化與握力降低顯著相關[18];另一方面,腸道菌群結構會隨著地理環境、飲食結構及居住條件、年齡等因素發生改變[19],而目前尚無文獻報道中國中老年糖尿病衰弱人群腸道菌群結構及功能特征。本研究將采用因美納(Illumina)高通量測序等方式,對腸道菌群進行定性定量分析,與糖尿病非衰弱人群對比,探索糖尿病衰弱人群腸道菌群結構及功能特征,揭示菌群與糖尿病衰弱之間的相互關系。
1 材料與方法
1.1 對象
本研究于2017年5月至2017年7月在四川大學華西醫院老年科門診招募患者。納入標準:① 已確診為2型糖尿病的患者;② 愿意參加該研究并配合試驗留取糞便。排除標準:① 終末期疾病,遠期預后不超過半年;② 嚴重肝腎功能不全。衰弱評估采用FRAIL量表,該量表包括了五個條目:疲勞(Fatigue)、抗阻力(Resistance)、步行(Ambulation)、疾病(Illness)、體重下降(Loss weight),其中 ≥ 3項滿足條件為衰弱,1~2項滿足條件為衰弱前期,0項為沒有衰弱。本研究試驗組為糖尿病衰弱及衰弱前期患者,對照組按1∶1比例匹配相同年齡、性別納入糖尿病非衰弱患者,收集臨床信息及患者糞便。
本研究嚴格遵守赫爾辛基宣言,并通過四川大學華西醫院倫理委員會審查,在參與任何實驗活動之前,每位受試者均已簽署知情同意書。
1.2 樣品采集
所有入組患者均正常飲食,采用自然排便方式,于清晨使用糞便取樣器截取樣品中段里部,采取新鮮糞便標本,樣品于2 h內收集完成,放置于 ? 80 ℃冰箱保存,糞便樣本統一由北京量化健康科技有限公司(干冰運輸)進行糞便宏基因測序。
1.3 DNA提取
采用QIAamp PowerFecal Pro DNA Kit試劑對糞便樣本DNA進行提取,并采用瓊脂糖凝膠電泳(agarose gel electrophoresis,AGE)的方式分析DNA純度和完整性;采用Qubit對DNA濃度進行精確定量。
1.4 文庫構建及測序
采用KAPA HyperPlus PCR-free(96rxn)試劑盒進行文庫構建,檢測合格的DNA樣品采用Covaris超聲破碎儀隨機打斷成長度約350 bp的片段,經末端修復、加A尾、加測序接頭、純化、PCR擴增等步驟完成文庫制備,文庫構建完成后,使用Qubit2.0進行初步定量,稀釋文庫至2 ng/μL,隨后使用普瑞凱Qsep 100對文庫的嵌入部分(insert size)進行檢測,符合預期后,再采用Q-PCR方法對文庫有效濃度進行精確定量。把不同文庫按照有效濃度及目標下機數據量需求,合并進行測序。
1.5 測序分析
所有原始宏基因組測序數據將通過MOCAT2軟件進行質量控制[20],采用Cutadapt軟件(v1.14,參數:-M30)對原始序列進行接頭處理,使用SolexaQA package過濾掉長度小于30 bp的序列,質控后得到較為干凈的序列。再使用SOAPaligner軟件(v2.21,參數:-M4 -I30 -V10)比對宿主基因組去除污染的宿主基因序列,得到高質量的干凈數據。采用SOAPdenovo軟件對序列進行從頭組裝(v2.04,參數:all -D1 -M3 -L500),將干凈序列打斷成K-mer構建德布魯因圖(de Bruijin),尋找歐拉途徑(Eulerian Path)組裝成重疊群(contigs),根據雙端測序(pair-end reads)位置關系連接成支架序列(scaffold),并從中挑選出連續的重疊群,得到不含氮端的序列片段(scaftigs)。
基因組裝后獲得的序列采用MetaGeneMark進行基因結構預測[21],預測后用CD-HIT的方式進行聚類去冗余[22],構建基因集,采用DIAMOND軟件,將所獲得的非冗余參考基因對比到京都基因組百科全書基因庫(KEGG Orthology,KO),對所有預測出的基因進行功能注釋。
在物種及其他高級分類層面上,采用MetaPhlan2.0獲得菌群相對豐度,并用來自7 500個物種的100多萬個標記基因做預測,獲得不同分類水平下的相對豐度[23]。同時采用MOCAT2基于宏基因組操作分類單位(metagenomic operational taxonomic units,MetaOTUs)對基因組序列未知物種進行分析,獲得MetaOTUs的相對豐度[24]。
對于KO的相對豐度,采用R/bioconductor的GAGE包進行KEGG Pathway富集分析[25]。
1.6 物種累積曲線及多樣性分析
處理好后的菌群基因,采用R軟件中vegan-specaccum函數計算物種累積曲線,對物種豐度進行預測;在屬水平使用vegan-diversity函數計算香農指數(Shannon Index)以及辛普森指數(Simpson Index),用于評估物種阿爾法(alpha,α)多樣性;采用基于布雷柯蒂斯距離(Bray-Curtis)的主坐標分析(principal co-ordinates analysis,PCoA)法,在二維坐標上顯示不同樣本之間的相似性及差異性,同時采用相似百分比分析法(similar percentage,SIMPER)確定主要差異菌群。
為比較兩組間腸道菌群基因、功能和分類群差異,本研究通過威爾科克森符號秩檢驗(Wilcoxon)比較兩組間相對豐度來得到顯著差異單元,通過本賈米尼-霍克伯格法(Benjamini-Hochberg,HB)方法來矯正P值;并采用線性判別分析(linear discriminant analysis,LDA)對數據進行降維,評估差異顯著物種的影響力(LDA Score),得到線性判別分析效果大小結果圖(Linear discriminant analysis Effect Size,LEfSe)。
2 結果
2.1 納入人群基本特征
本研究共納入糖尿病衰弱或衰弱前期患者15名(A組),按照年齡、性別、衰弱程度等進行1∶1配對后納入糖尿病非衰弱患者15名(B組)。具體情況見表1。
2.2 糞便樣本測序基本統計結果
采用Illumina測序平臺進行測序,共獲得了193.9 GB的原始測序數據,樣本的序列數量在34 956 712~59 919 586之間,平均為46 275 356,所有樣本純凈序列占未處理序列的95%以上,單樣本序列豐度 > 0.1%。
2.3 腸道微生物多樣性
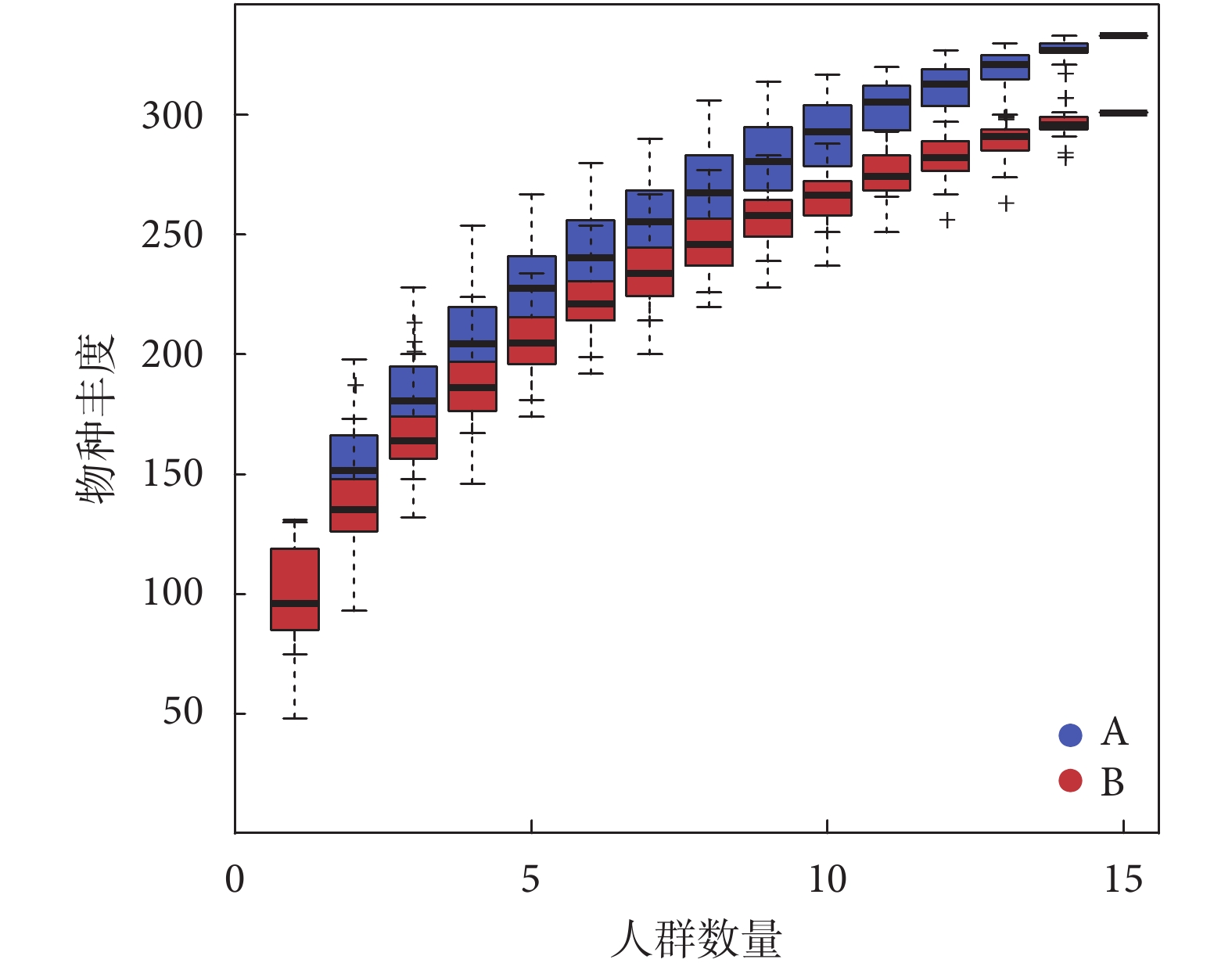
通過對兩組共30例患者的糞便進行宏基因測序,共得到16門、127屬、298種細菌。其中,糖尿病衰弱及衰弱前期組共提取出12門、116屬、264種微生物;糖尿病非衰弱組共提取出16門、100屬、243種微生物。通過物種累積曲線驗證微生物測序數據量在多樣性分析中是否充足,是反映樣本物種多樣性指標之一。橫坐標代表樣本量,縱坐標代表被檢測的物種數,結果顯示末端曲線趨于平緩,表明樣本量足夠(見圖1)。
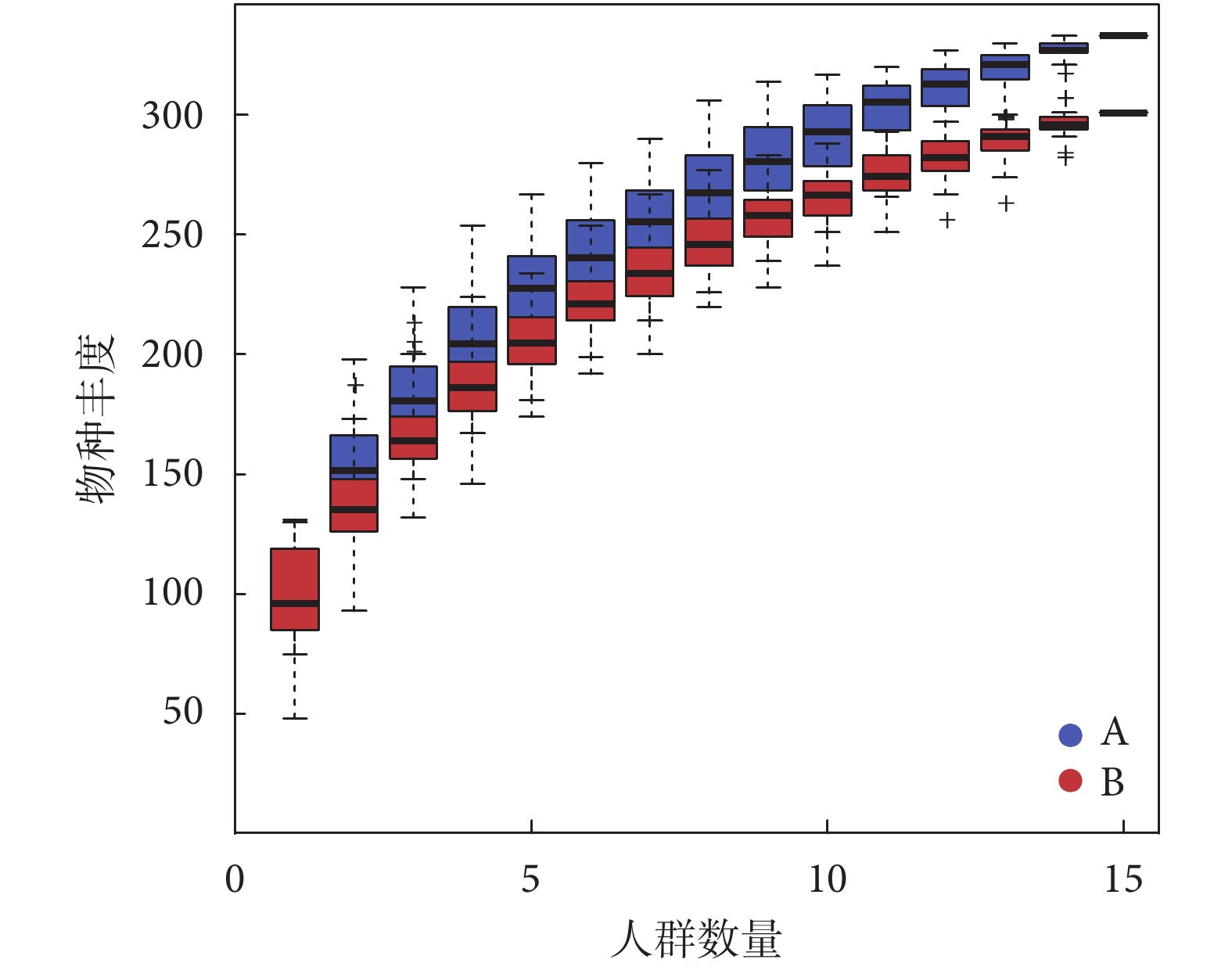 圖1
物種豐度圖
Figure1.
Abundance of microbiota
圖1
物種豐度圖
Figure1.
Abundance of microbiota
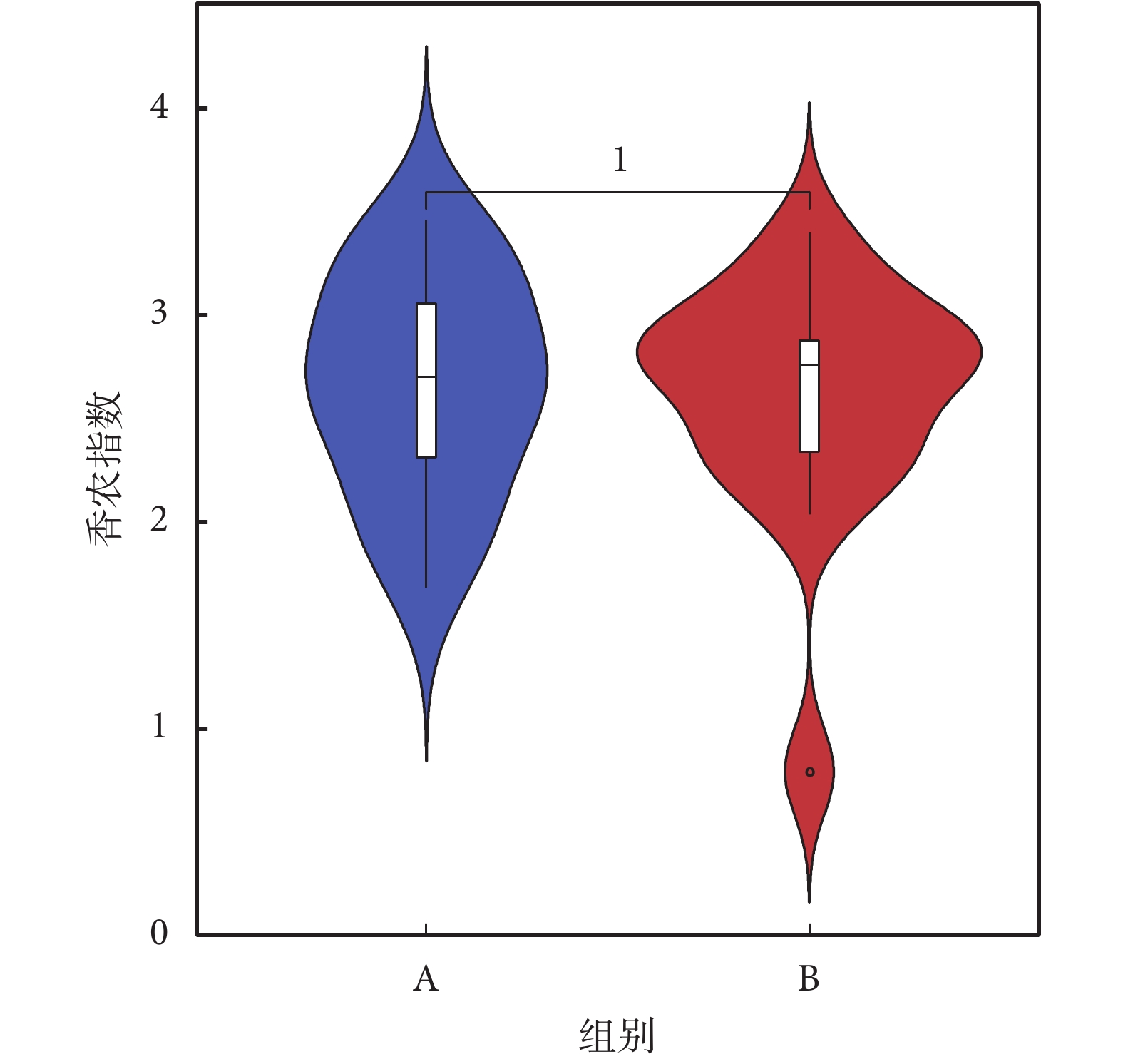
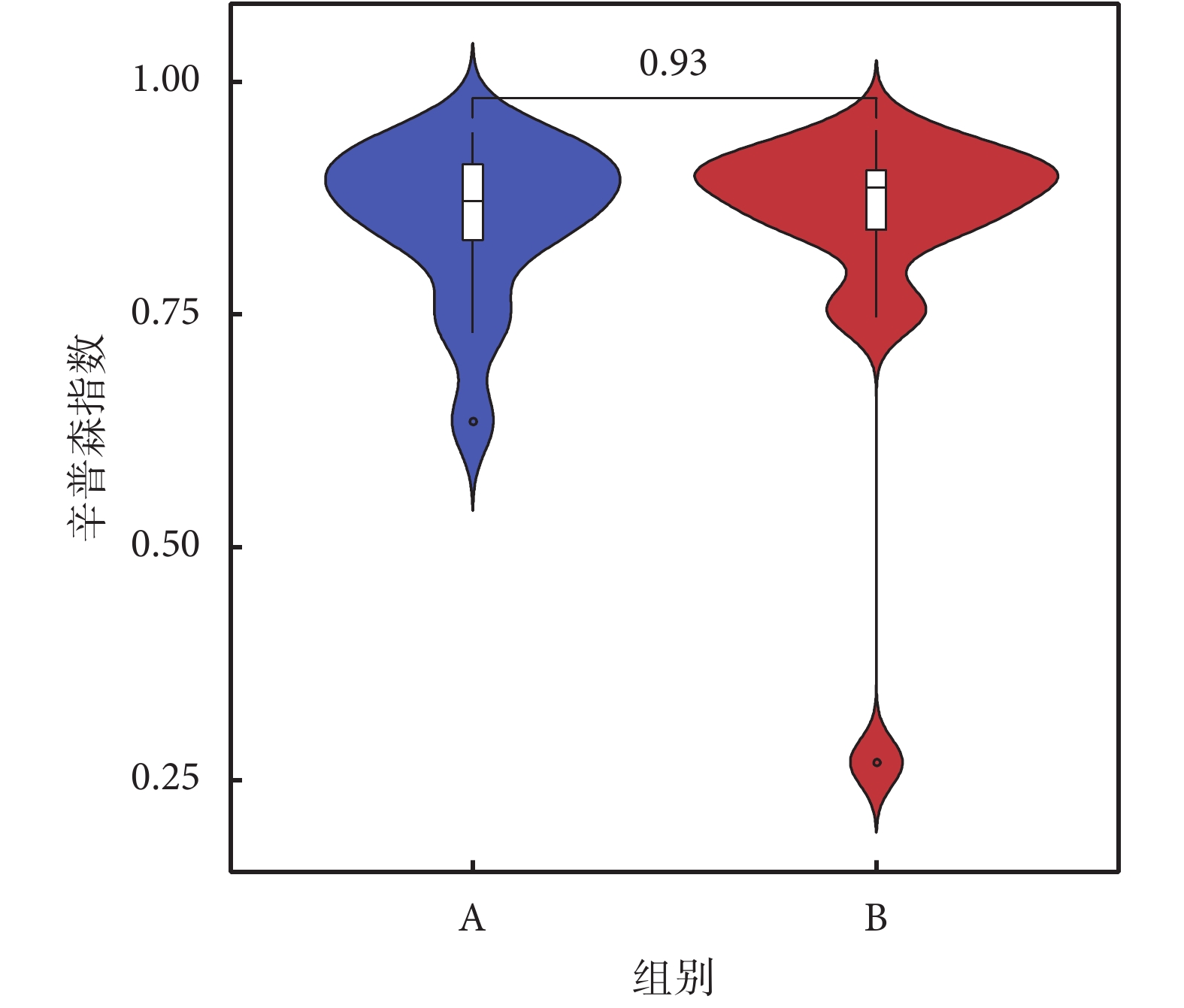
香農指數常用于反映α多樣性指數,香農指數值越大,說明菌落多樣性越高。對兩組人群進行香農指數計算,如圖2所示,A組較B組香農指數略小,但經秩和檢驗分析,兩組α多樣性差異無統計學意義。再采用辛普森指數進行計算,A、B兩組間菌群多樣性差異無統計學意義(見圖3)。
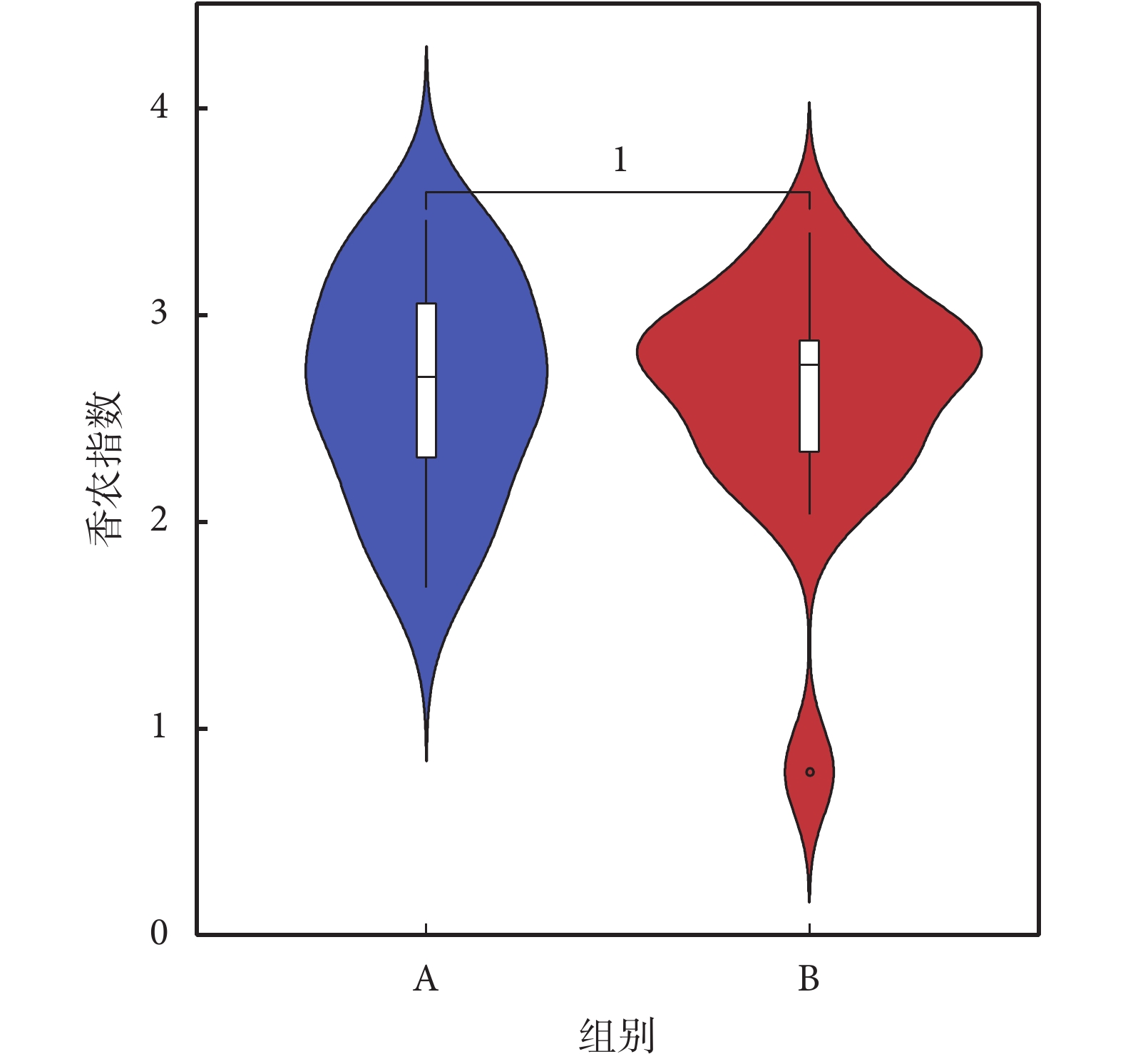 圖2
香農指數
Figure2.
Shannon Index
圖2
香農指數
Figure2.
Shannon Index
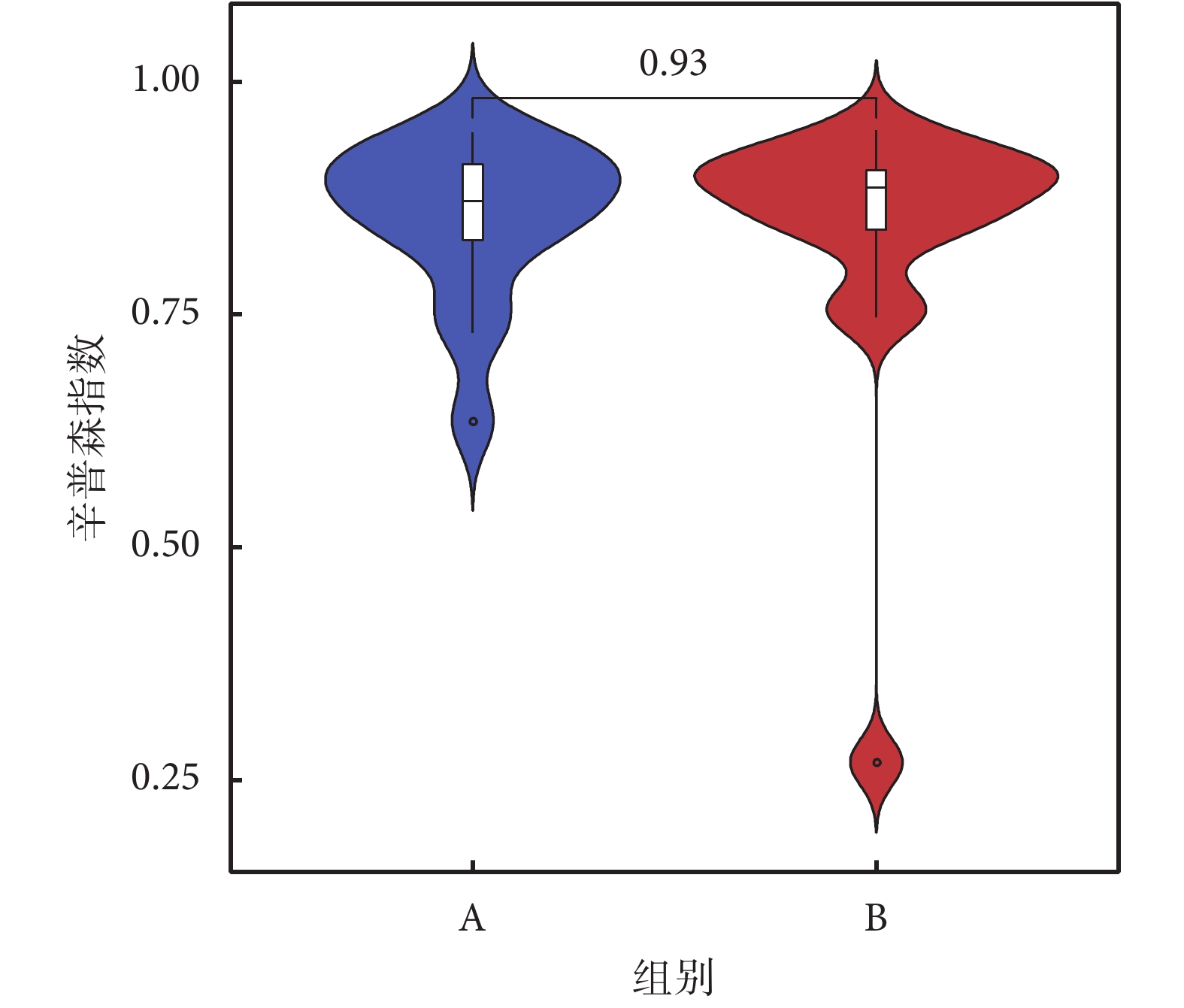 圖3
辛普森指數
Figure3.
Simpson Index
圖3
辛普森指數
Figure3.
Simpson Index
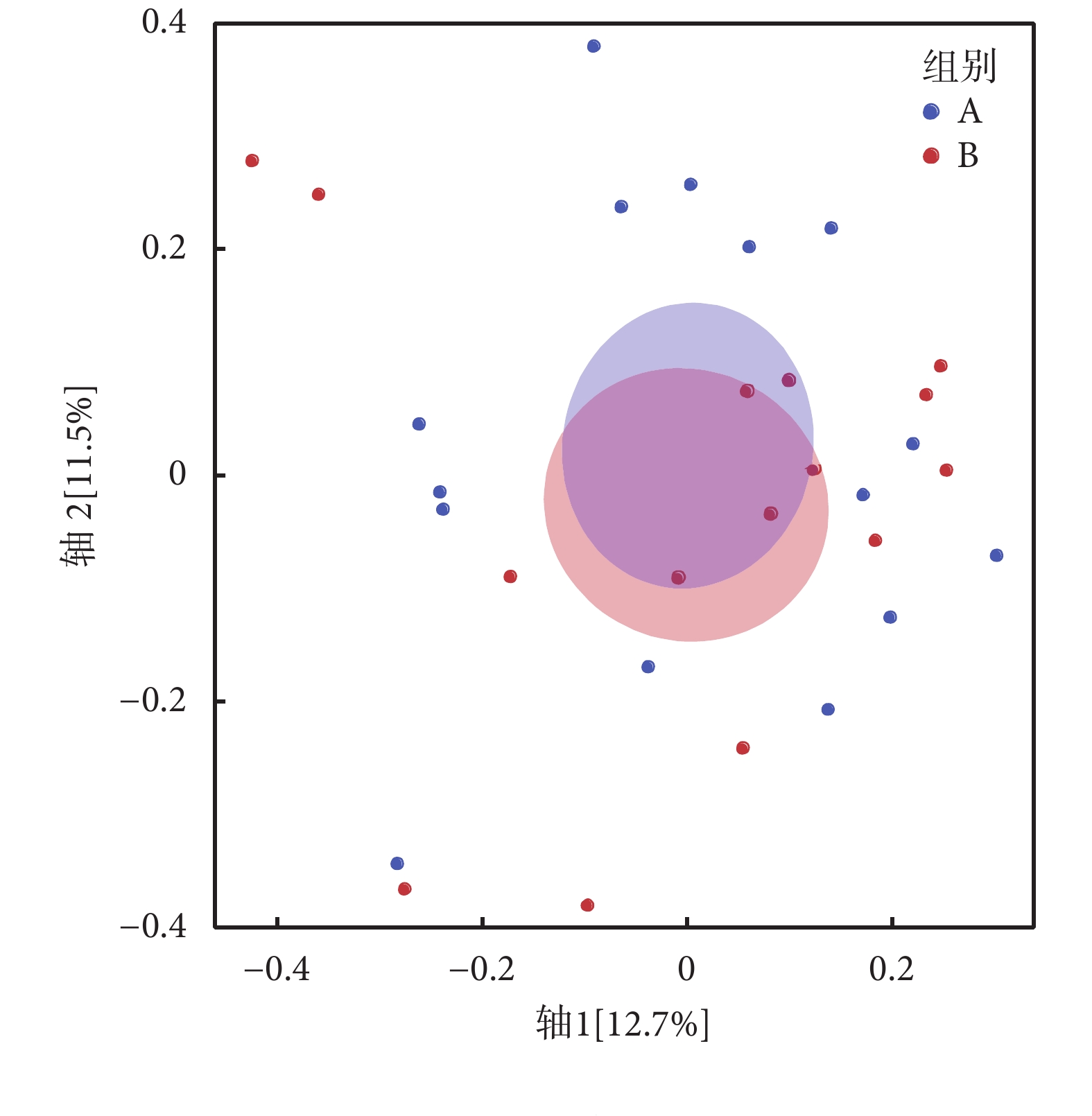
本研究使用PCoA來分析兩組人群糞便微生物β多樣性(見圖4),該方法通過計算Bray-Curtis距離,對菌群進行排序、降維,形成相似度及相異度可視化圖表。結果顯示,兩組人群腸道菌群分布具有差異性,但各自聚集不明顯。使用非參數多元方差分析檢驗分組因素對菌群差異的影響,結果顯示衰弱對糖尿病患者菌群結構總體變化影響不顯著,無統計學意義。
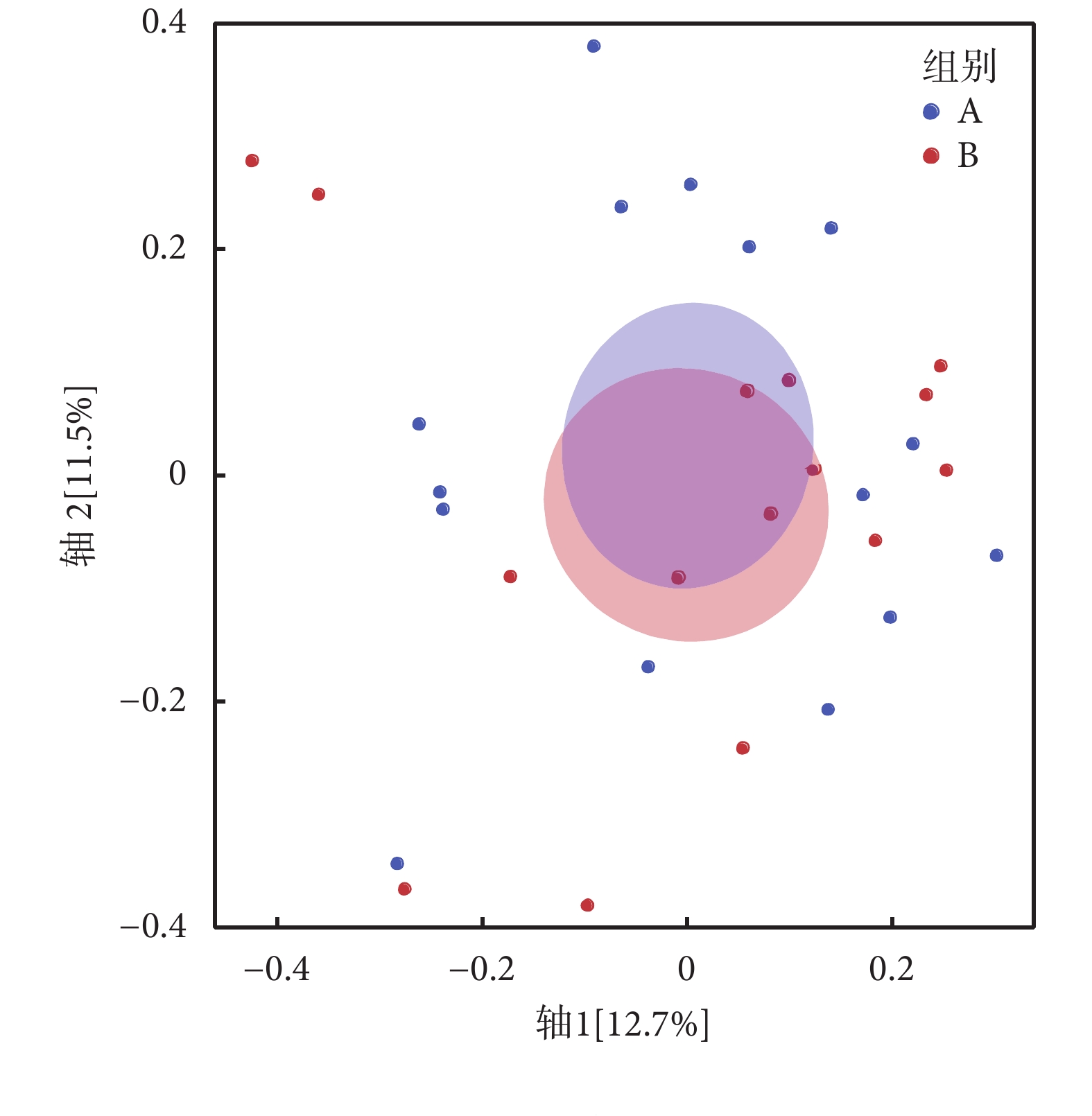 圖4
PCoA結果
Figure4.
The result of PCoA
圖4
PCoA結果
Figure4.
The result of PCoA
2.4 腸道微生物組成特征
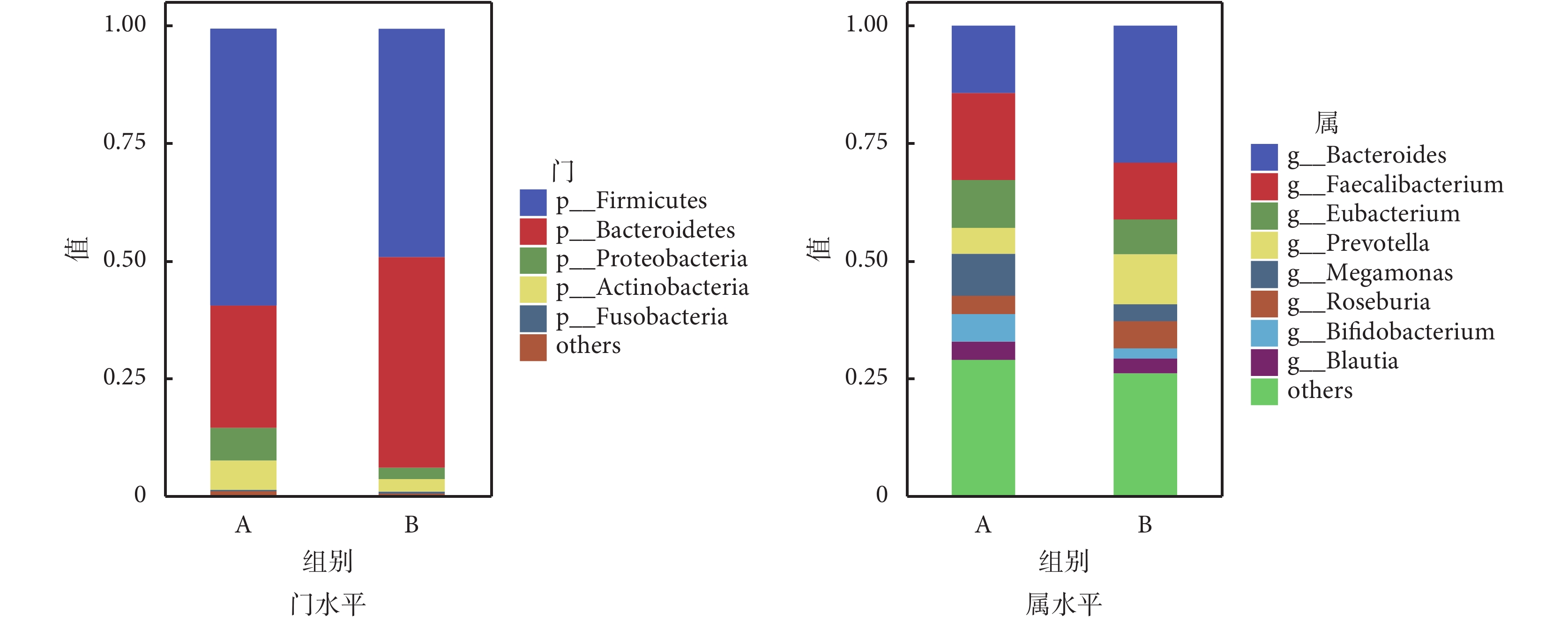
如圖5所示,在門水平上,糖尿病衰弱及衰弱前期組腸道菌群相對豐度較高的為厚壁菌門(Firmicutes)、擬桿菌門(Bacteroidetes)、變形菌門(Proteobacteria)、放線菌門(Actinobacteria);在科水平上,糖尿病衰弱及衰弱前期組腸道菌群相對豐度較高的為瘤胃菌科(Ruminococcaceae)、擬桿菌科(Bacteroidaceae)、韋榮氏球菌科(Veillonellaceae)、毛螺菌科(Lachnospir-aceae)、優桿菌科(Eubacteriaceae)、普雷沃氏菌科(Prevotellaceae)等;在屬水平上,糖尿病衰弱及衰弱前期組患者腸道菌群相對豐度較高的為糞桿菌屬(Faecalibacterium)、擬桿菌屬(Bacteroides)、優桿菌屬(Eubacterium);在種水平上,糖尿病衰弱組患者腸道菌群相對豐度較高的為普拉梭菌(Faecalibacterium prausnitzii)、直腸真桿菌(Eubacterium rectale)、Prevotellacopri菌、長雙歧桿菌(Bifidobacterium longum)以及大腸桿菌(Escherichia coli)。
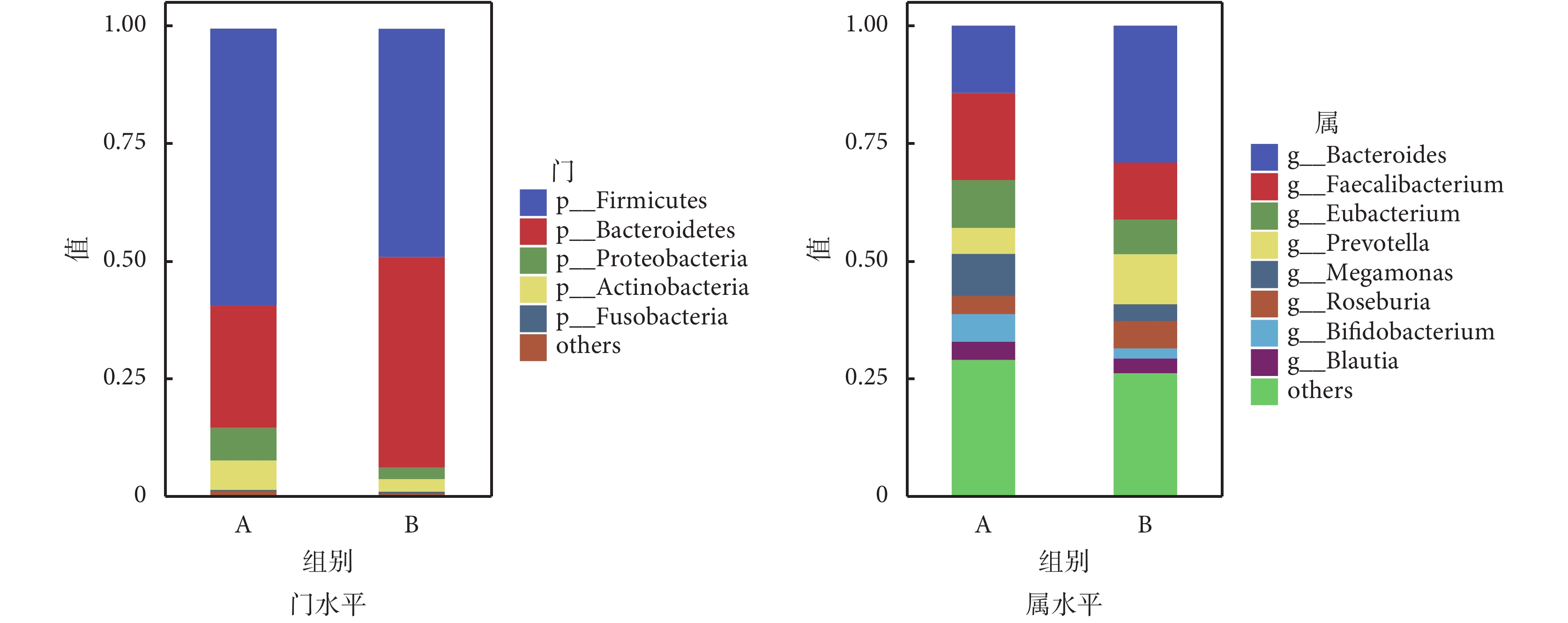 圖5
門水平和屬水平物種分布圖
Figure5.
Diversity in phylum and genus levels
圖5
門水平和屬水平物種分布圖
Figure5.
Diversity in phylum and genus levels
2.5 腸道菌群組間差異性分析
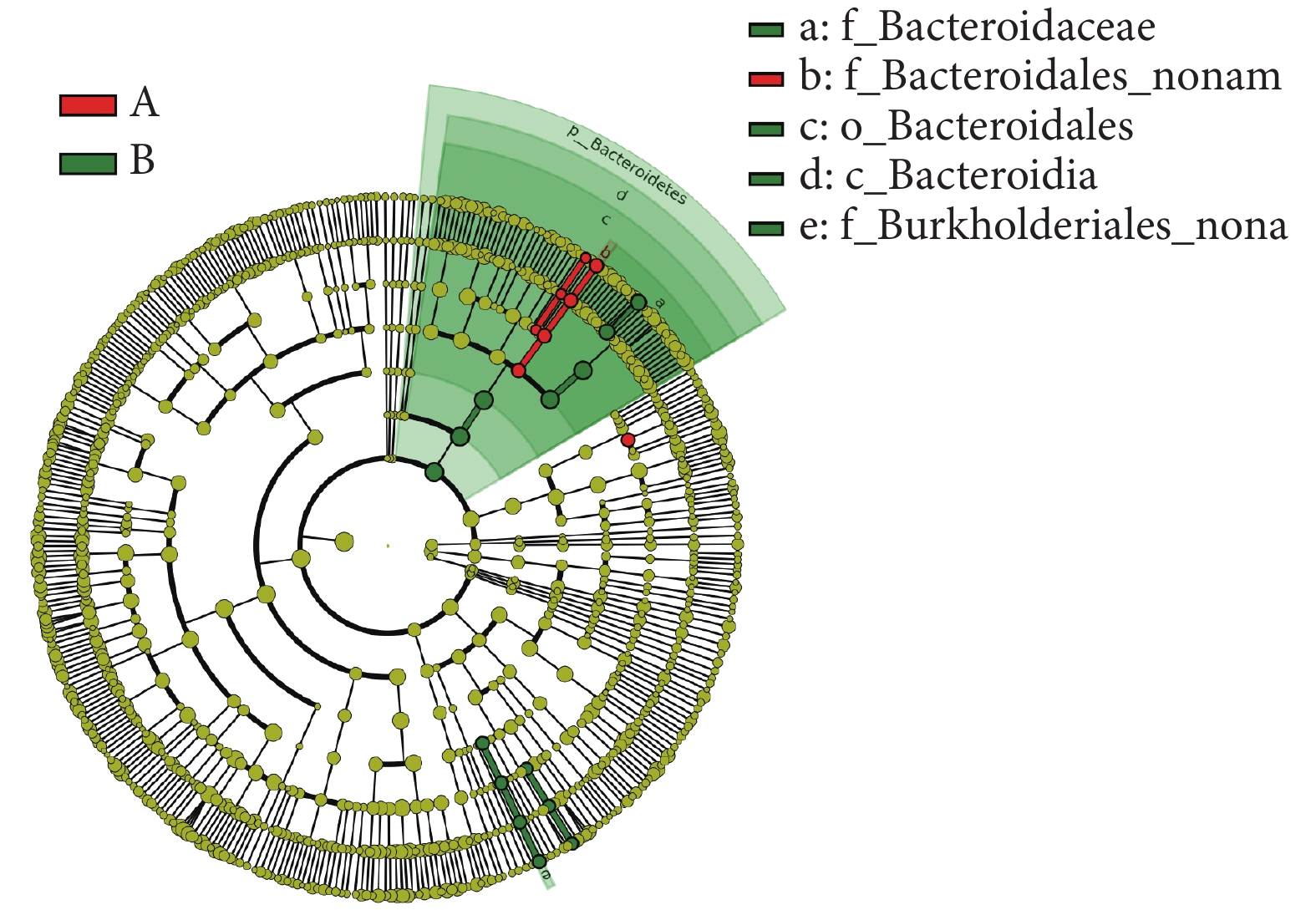
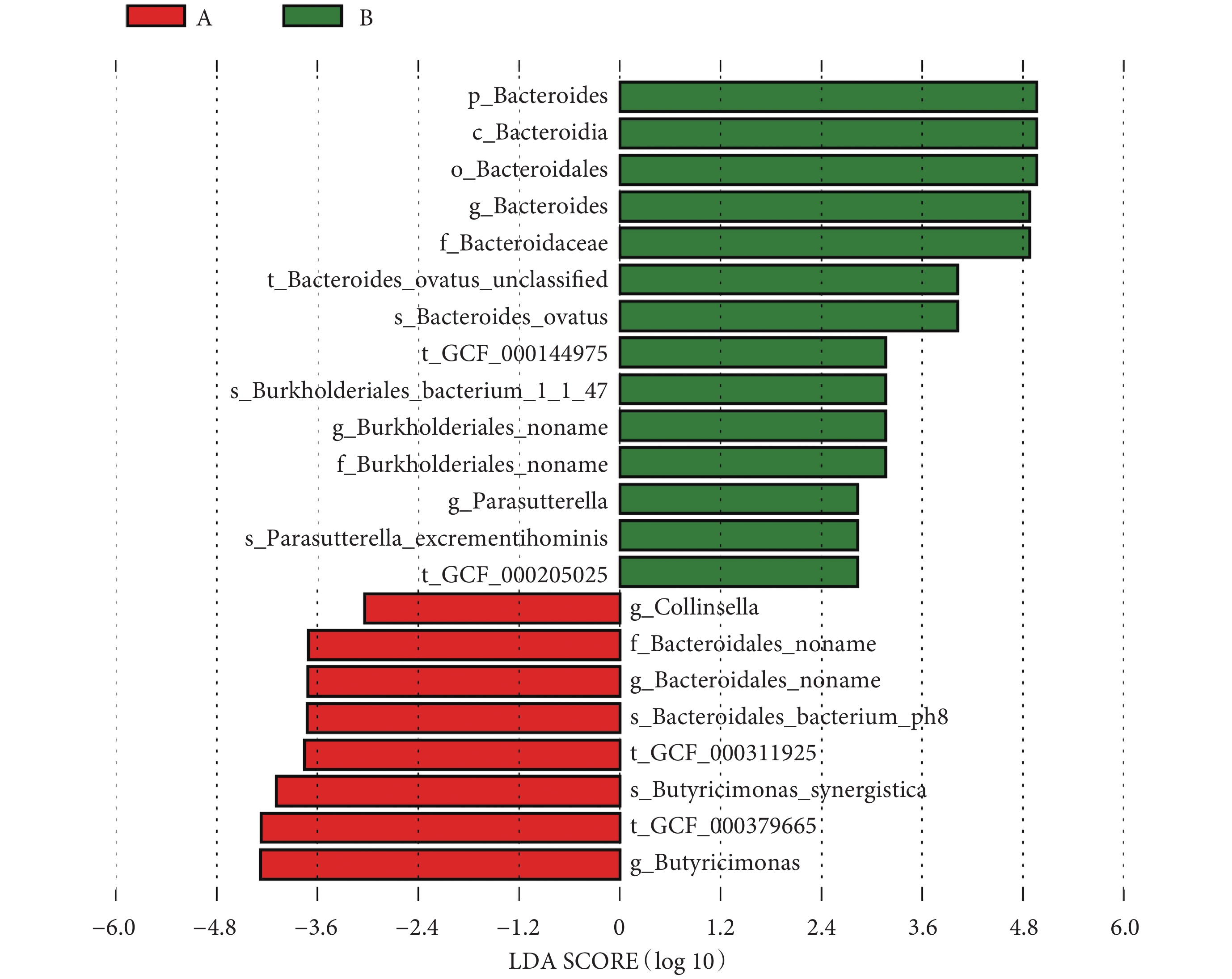
本研究采用秩和檢驗對兩組患者腸道菌群各個水平相對豐度進行統計分析,如見表2所示。在門水平,糖尿病衰弱及衰弱前期組的擬桿菌門(Bacteroidetes,紅色)相對豐度高于糖尿病非衰弱組(P = 0.016)。在綱水平,糖尿病衰弱及衰弱前期組的擬桿菌綱(Bacteroidia)相對豐度低于糖尿病非衰弱組(P = 0.02)。在目水平,糖尿病衰弱及衰弱前期Bacteroidales相對豐度低于糖尿病非衰弱組(P = 0.016)。在科水平,糖尿病衰弱及衰弱前期組Bacteroidales noname高于糖尿病非衰弱組(P < 0.01),而Bacteroidaceae在糖尿病非衰弱組中相對豐度較高(P = 0.03)。在屬水平上,糖尿病衰弱及衰弱前期組Bacteroides_noname(P = 0.01)、Collinsella(P = 0.022)、Butyricimonas(P = 0.03)相對豐度高于糖尿病非衰弱組,而Parasutterella(P = 0.01)、Bacteroides(P = 0.03)在糖尿病非衰弱組中相對豐度較高。在種水平豐度上,衰弱及衰弱前期組人群Bacteroidales-bacterium-ph8(P < 0.01)、Butyricimonas_synergistica(P = 0.03)、Bacteroides_ovatus(P = 0.01)相對豐度顯著高于糖尿病非衰弱組患者,而Parasutterella_excrementihominis(P = 0.02)在糖尿病非衰弱組中相對豐度較高。LEfSe分析結果顯示糖尿病衰弱及衰弱前期患者腸道菌群貢獻最大的菌為Butyricimonas菌屬,而糖尿病非衰弱組人群關鍵菌包括Bacteroidetes門bacteroides屬等(見圖6、圖7)。
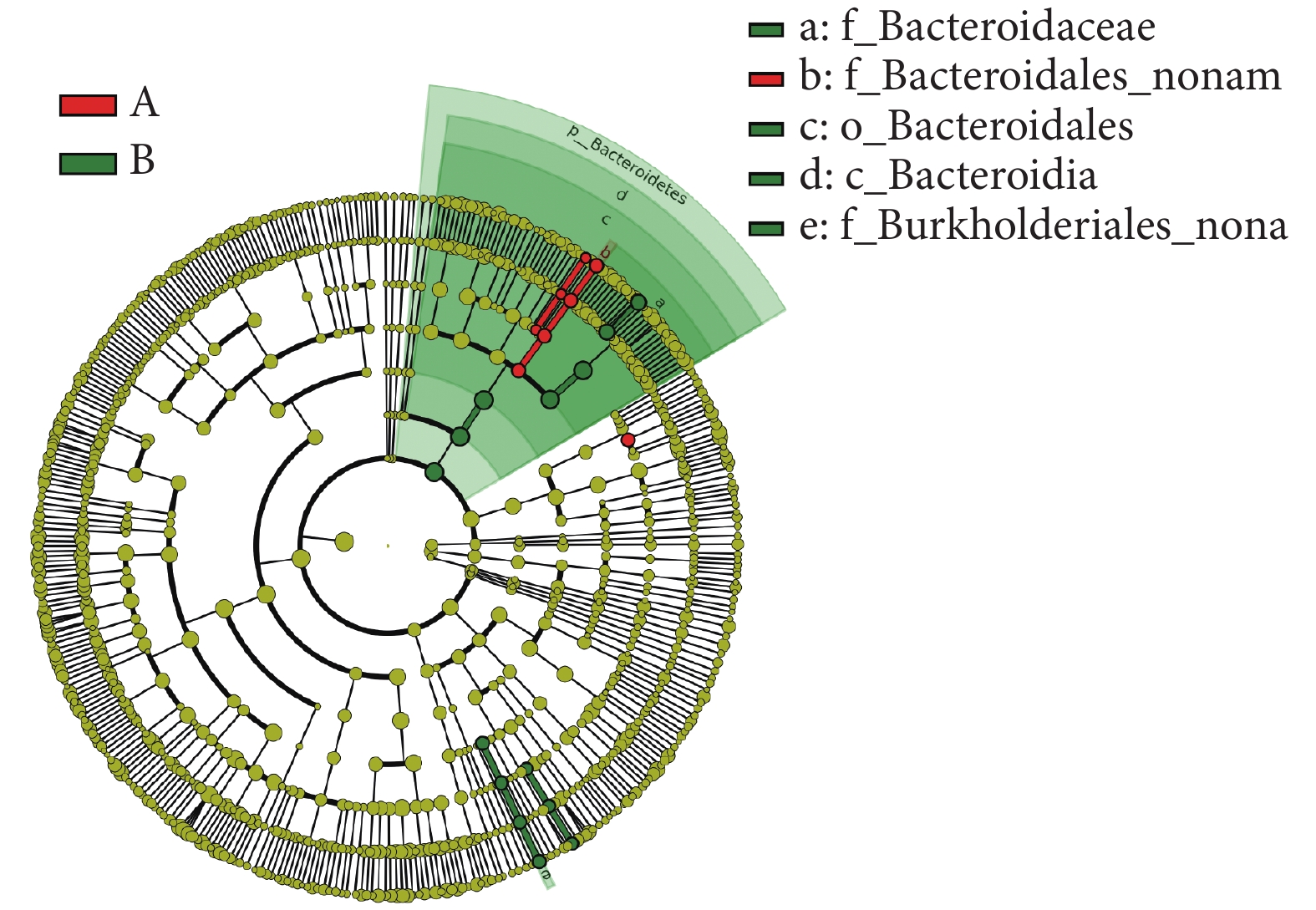 圖6
進化分支圖
Figure6.
Cladogram
圖6
進化分支圖
Figure6.
Cladogram
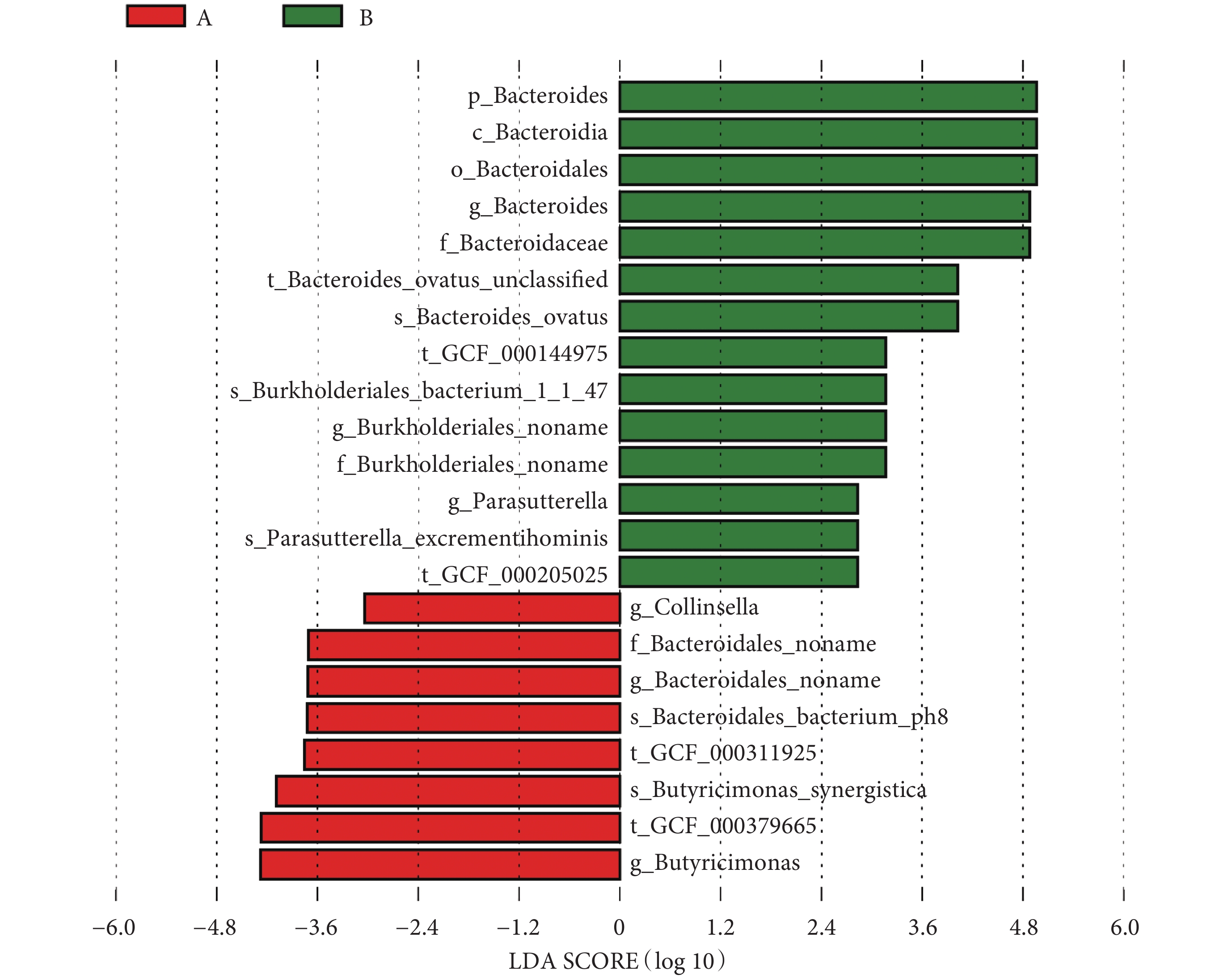 圖7
差異菌群貢獻圖
Figure7.
The most important gut microbiota in different group
圖7
差異菌群貢獻圖
Figure7.
The most important gut microbiota in different group
2.6 差異功能分析
本研究從30例樣本中共提取及構建了1 264 594非冗余基因集。其中,糖尿病衰弱及衰弱前期組人群共有1 147 676個基因集,糖尿病非衰弱組人群共有1 044 831個基因集。其中,共有基因集占有927 913個,占總基因集73.4%。兩組人群微生物功能基因涉及多條功能通路,通過將糖尿病衰弱及衰弱前期和糖尿病非衰弱組人群差異性表達的KO注釋到KEGG模塊水平,發現糖尿病衰弱及衰弱前期的人群富集上調的功能基因分布在組氨酸代謝(histidine metabolism)、EB病毒感染(Epstein-Barr virus infection)、硫代謝(sulfur metabolism)以及Ⅱ型聚酮生物合成(biosynthesis of type Ⅱ polyketide products)等;而磷酸轉移酶系統(phosphotransferase system,pts)、生物膜形成(biofilm formation)、ABC轉運蛋白(ABC transporters)、鞭毛組裝(flagellar assembly)、丁酸代謝(butanoate metabolism)以及苯丙氨酸代謝(phenylalanine metabolism)等在糖尿病衰弱及衰弱前期人群中出現下調(見表3)。
3 討論
本研究調查了糖尿病衰弱人群的腸道微生物,發現糖尿病衰弱與腸道菌群α多樣性并無相關性,兩組人群β多樣性呈現差異性分布,但各自微生物群聚集不明顯。對差異菌群進行功能分析,發現衰弱人群腸道微生物在氨基酸代謝、EB病毒感染等方面上調,而在生物膜形成、丁酸代謝、苯丙氨酸代謝等方面下調。
3.1 菌群結構特征分析
腸道微生物會隨著年齡增長而出現多樣性下降或生態失衡,個體變異性增加,促使寄主生理紊亂顯著[26],而這種變化與腸道通透性增加以及慢性低度炎癥具有相關性[27]。已有證據表明衰弱狀態伴隨著系統性炎性[28]。既往研究中,衰弱人群的腸道微生物最一致的特征是多樣性的喪失[27],本研究發現糖尿病衰弱及衰弱前期組的α多樣性少于糖尿病非衰弱組人群,但兩組并沒有統計學意義,原因可能是糖尿病對腸道菌群的影響[29],導致在沒有衰弱狀態時就缺乏微生物多樣性,β多樣性分析也提示兩組人群差異并沒有統計學意義。
兩組人群的菌群組成具有明顯差異性。在糖尿病衰弱及衰弱前期組中,柯林斯氏菌屬(Collinsella)和丁酸弧菌屬(Butyricimonas)富集。既往研究提示Collinsella豐度與空腹甘油三酯、總膽固醇濃度呈正相關[30],促進體內脂肥胖形成,進一步促進體內炎癥因子形成[31]。在給無菌小鼠腸道喂養Collinsella菌后發現腸細胞緊密連接蛋白表達降低,腸漏及菌血癥的發生風險顯著升高[32]。因此我們推測,Collinsella菌屬可能通過改變體內慢性炎癥環境,導致衰弱發病率上升,是潛在的糖尿病衰弱致病菌群。然而,目前尚不清楚該菌的富集是衰弱的原因還是結果,其潛在機制有待進一步研究。
在糖尿病非衰弱組中,副薩特氏菌屬(Parasutterella)及卵形擬桿菌(Bacteroides ovatus)富集,目前的研究認為副薩特氏菌屬是腸道的核心菌種,在膽汁酸維持和膽固醇代謝中具有重要作用[33]。在一項隨機試驗中,副薩特氏菌屬與低密度脂蛋白(low density lipoprotein,LDL)降低具有相關性,而低水平LDL與較低的衰弱發病率具有相關性[34],因此我們推測副沙特氏菌是糖尿病衰弱的保護菌群。卵形擬桿菌在腸道中參與了多種碳水化合物的消化分解,提供能量[35],目前有研究已證明卵形擬桿菌能夠誘導腸道產生免疫球蛋白A[36],限制細菌與細菌源性毒素進入腸上皮細胞,減少腸道感染風險[37],同時進食含有卵形擬桿菌的益生菌,能夠改善社區人群腹瀉等癥狀[38]。因此我們推測,卵形擬桿菌數量增加可能會降低糖尿病人群衰弱發生的風險,而相關數量級的確定,還需要更多的研究來支持。
3.2 代謝功能特征性分析
通過與KEGG代謝通路數據庫進行比對,發現兩組人群菌群代謝通路具有顯著差異,在糖尿病衰弱及衰弱前期組人群中,上調的通路包括組氨酸代謝通路、EB病毒感染通路、硫代謝通路,以及Ⅱ型聚酮生物合成通路。組氨酸代謝通路中,咪唑丙酸是其重要產物,通過激活雷帕霉素靶蛋白敏感性復合體1通路(mammalian target of rapamycin complex 1,mTORC1),降解胰島素受體底物1(Insulin Receptor Substrate 1,IRS1)和胰島素受體底物2(Insulin Receptor Substrate 2,IRS2),抑制胰島素信號通路,造成胰島素抵抗,可能是2型糖尿病的病因之一[39];另一方面,胰島素抵抗可以通過抑制骨骼肌細胞能量代謝造成肌肉收縮障礙[5]。這提示我們,組氨酸代謝的相關細菌所引起的胰島素抵抗,可能在老年衰弱的發生發展中起到一定的作用。同時EB病毒感染通路能激活Toll樣受體信號通路(Toll-like receptor signaling pathway,TLR pathway),促進體內白細胞介素6(interleukin-6,IL-6)及腫瘤細胞壞死因子α(tumor necrosis factor-α,TNF-α)的產生[40],而這些炎性標志物與衰弱具有相關性,因此推測衰弱老年人與EB病毒感染通路激活相關,而具體機制還需要更多研究支持。
在糖尿病衰弱及衰弱前期人群菌群通路中,下調的通路包括磷酸轉移酶系統、生物膜形成、ABC轉運蛋白、鞭毛組裝及丁酸代謝。其中丁酸代謝在長壽人群當中具有重要意義[41]。同時丁酸通過結腸細胞上的單羧酸轉運蛋白(monocarboxylate transporter,MCT-1)和溶質轉運蛋白(solute carrier family 5 member 8,SLC5A8)轉運,作為組蛋白脫乙酰酶抑制劑或G蛋白偶聯受體分子,在抗炎、維持腸黏膜屏障以及腸黏膜免疫方面發揮重要作用[42]。糖尿病衰弱及衰弱前期人群腸道微生物對丁酸利用減少,上述丁酸作用相對減弱,前期的研究中發現,腸道免疫系統紊亂是衰弱發生發展的重要因素[43],因此我們推測丁酸可能在預防衰弱發生或改善衰弱程度等方面具有一定的作用,具體機制還需要更多研究證明。
3.3 創新性
既往的研究多集中在歐美等地國家,研究地點多選擇在病房以及養老院等[10-11, 44],而關于社區衰弱人群的腸道菌群差異性在國內未見報道。由于腸道菌群受宿主種族、地域及飲食等的密切影響[45],不同地點人群腸道菌群特點值得研究。本研究采用人群年齡、性別完全配比的方式控制了混雜因素,發現柯林斯氏菌以及EB病毒在衰弱發生中起到重要作用,拓寬了以往的研究結果,為衰弱的干預提供了新的理論依據。
4 結論
本研究通過比較糖尿病衰弱及衰弱前期和非衰弱人群的腸道菌群差異,揭示了糖尿病衰弱及衰弱前期人群的腸道菌群結構特征。本研究采用宏基因測序對糖尿病衰弱及衰弱前期患者腸道菌群結構及功能特征性進行研究,結果表明衰弱及衰弱前期對腸道菌群整體結構影響不顯著,但仍具有特異性菌群。通過對菌群基因功能分析,表明腸道菌群可通過胰島素抵抗和腸道免疫等途徑對糖尿病人群的衰弱產生一定影響。本研究的結果為衰弱人群的腸道菌群機制探索奠定了理論基礎,同時也為糖尿病衰弱及衰弱前期人群的預防與干預提供了一定的科學依據。
利益沖突聲明:本文全體作者均聲明不存在利益沖突。
引言
全球老齡化問題正日益凸顯,中國老齡化進程明顯快于世界平均水平。2010年我國65歲及以上人群占比8.87%,預計到2050年,這一數字將提高至34%[1]。衰弱是老齡化突出問題之一,是指老年人生理儲備下降導致機體易損性增加和抗應激能力減退的非特異性狀態,其核心是老年人生理儲備下降或異常,外界較小刺激即可引起臨床事件的發生[2]。衰弱人群更容易出現各種不良預后,包括跌倒、失能、認識功能下降、再入院率增加甚至死亡風險等[3]。
糖尿病與衰弱兩者可相互作用。一方面,高血糖狀態會抑制骨骼肌生長,嚴重時會造成肌肉萎縮[4],而胰島素抵抗會抑制骨骼肌的能量代謝造成肌肉再生功能障礙[5],機體衰弱發生風險增加[6]。另一方面,衰弱促使胰島素抵抗增加,血糖呈高水平狀態,增加糖尿病發生風險[7]。我國的流行病學調研顯示,我國65歲以上糖尿病合并衰弱人群占糖尿病人群比例為5%~48%[8],該人群出現低血糖及死亡風險高[9],預后較單純糖尿病老年人群差,極大地增加了家庭照護和社會經濟負擔。
微生物在人體內廣泛分布,其中以胃腸道菌群數量最多。既往研究表明,衰弱與腸道菌群α多樣性呈負相關[10],衰弱程度與Ruminococcacea菌、F. prausnitzii菌及Lachnospiraceae菌豐度呈負相關,與Eubacteriumdolichum菌和Eggerthellalenta菌豐度呈正相關。van Tongeren等[11]采用衰弱指數(frailty index,FI)評價衰弱,發現非衰弱人群,菌群特點表現為普氏菌屬(Prevotella)與擬桿菌屬(Bacteroides)的比值較高;而在衰弱人群中,菌群特征表現為瘤胃球菌屬(Ruminococcus)及奇異菌屬(Atopobium)占比增加,乳酸桿菌屬(Lactobacillus)占比減少。同時,Biagi團隊[12]也發現慢性疾病越多的老年人,腸道內克里斯滕森菌科(Christensenellaceae)、嗜黏蛋白阿克曼菌(Akkermansia)及雙歧桿菌屬(Bifidobacterium)等數量減少,但這些細菌數量級與衰弱并非呈線性關系,個體差異性較大。
腸道菌群可從多個方面影響衰弱。首先,腸道微生物組成失調會影響蛋白質代謝和生物利用度[13-14],影響骨骼肌合成代謝所需的維生素B12、葉酸、核黃酸等物質的產生[14],加速肌細胞線粒體受損累積[15],增加胰島素抵抗風險[16],這些因素會導致骨骼肌數量與質量的下降;其次,隨著年齡增長,微生物組成向著促炎表型發展,總體表現為微生物多樣性減少,個體變異增加[17],該變化與握力降低顯著相關[18];另一方面,腸道菌群結構會隨著地理環境、飲食結構及居住條件、年齡等因素發生改變[19],而目前尚無文獻報道中國中老年糖尿病衰弱人群腸道菌群結構及功能特征。本研究將采用因美納(Illumina)高通量測序等方式,對腸道菌群進行定性定量分析,與糖尿病非衰弱人群對比,探索糖尿病衰弱人群腸道菌群結構及功能特征,揭示菌群與糖尿病衰弱之間的相互關系。
1 材料與方法
1.1 對象
本研究于2017年5月至2017年7月在四川大學華西醫院老年科門診招募患者。納入標準:① 已確診為2型糖尿病的患者;② 愿意參加該研究并配合試驗留取糞便。排除標準:① 終末期疾病,遠期預后不超過半年;② 嚴重肝腎功能不全。衰弱評估采用FRAIL量表,該量表包括了五個條目:疲勞(Fatigue)、抗阻力(Resistance)、步行(Ambulation)、疾病(Illness)、體重下降(Loss weight),其中 ≥ 3項滿足條件為衰弱,1~2項滿足條件為衰弱前期,0項為沒有衰弱。本研究試驗組為糖尿病衰弱及衰弱前期患者,對照組按1∶1比例匹配相同年齡、性別納入糖尿病非衰弱患者,收集臨床信息及患者糞便。
本研究嚴格遵守赫爾辛基宣言,并通過四川大學華西醫院倫理委員會審查,在參與任何實驗活動之前,每位受試者均已簽署知情同意書。
1.2 樣品采集
所有入組患者均正常飲食,采用自然排便方式,于清晨使用糞便取樣器截取樣品中段里部,采取新鮮糞便標本,樣品于2 h內收集完成,放置于 ? 80 ℃冰箱保存,糞便樣本統一由北京量化健康科技有限公司(干冰運輸)進行糞便宏基因測序。
1.3 DNA提取
采用QIAamp PowerFecal Pro DNA Kit試劑對糞便樣本DNA進行提取,并采用瓊脂糖凝膠電泳(agarose gel electrophoresis,AGE)的方式分析DNA純度和完整性;采用Qubit對DNA濃度進行精確定量。
1.4 文庫構建及測序
采用KAPA HyperPlus PCR-free(96rxn)試劑盒進行文庫構建,檢測合格的DNA樣品采用Covaris超聲破碎儀隨機打斷成長度約350 bp的片段,經末端修復、加A尾、加測序接頭、純化、PCR擴增等步驟完成文庫制備,文庫構建完成后,使用Qubit2.0進行初步定量,稀釋文庫至2 ng/μL,隨后使用普瑞凱Qsep 100對文庫的嵌入部分(insert size)進行檢測,符合預期后,再采用Q-PCR方法對文庫有效濃度進行精確定量。把不同文庫按照有效濃度及目標下機數據量需求,合并進行測序。
1.5 測序分析
所有原始宏基因組測序數據將通過MOCAT2軟件進行質量控制[20],采用Cutadapt軟件(v1.14,參數:-M30)對原始序列進行接頭處理,使用SolexaQA package過濾掉長度小于30 bp的序列,質控后得到較為干凈的序列。再使用SOAPaligner軟件(v2.21,參數:-M4 -I30 -V10)比對宿主基因組去除污染的宿主基因序列,得到高質量的干凈數據。采用SOAPdenovo軟件對序列進行從頭組裝(v2.04,參數:all -D1 -M3 -L500),將干凈序列打斷成K-mer構建德布魯因圖(de Bruijin),尋找歐拉途徑(Eulerian Path)組裝成重疊群(contigs),根據雙端測序(pair-end reads)位置關系連接成支架序列(scaffold),并從中挑選出連續的重疊群,得到不含氮端的序列片段(scaftigs)。
基因組裝后獲得的序列采用MetaGeneMark進行基因結構預測[21],預測后用CD-HIT的方式進行聚類去冗余[22],構建基因集,采用DIAMOND軟件,將所獲得的非冗余參考基因對比到京都基因組百科全書基因庫(KEGG Orthology,KO),對所有預測出的基因進行功能注釋。
在物種及其他高級分類層面上,采用MetaPhlan2.0獲得菌群相對豐度,并用來自7 500個物種的100多萬個標記基因做預測,獲得不同分類水平下的相對豐度[23]。同時采用MOCAT2基于宏基因組操作分類單位(metagenomic operational taxonomic units,MetaOTUs)對基因組序列未知物種進行分析,獲得MetaOTUs的相對豐度[24]。
對于KO的相對豐度,采用R/bioconductor的GAGE包進行KEGG Pathway富集分析[25]。
1.6 物種累積曲線及多樣性分析
處理好后的菌群基因,采用R軟件中vegan-specaccum函數計算物種累積曲線,對物種豐度進行預測;在屬水平使用vegan-diversity函數計算香農指數(Shannon Index)以及辛普森指數(Simpson Index),用于評估物種阿爾法(alpha,α)多樣性;采用基于布雷柯蒂斯距離(Bray-Curtis)的主坐標分析(principal co-ordinates analysis,PCoA)法,在二維坐標上顯示不同樣本之間的相似性及差異性,同時采用相似百分比分析法(similar percentage,SIMPER)確定主要差異菌群。
為比較兩組間腸道菌群基因、功能和分類群差異,本研究通過威爾科克森符號秩檢驗(Wilcoxon)比較兩組間相對豐度來得到顯著差異單元,通過本賈米尼-霍克伯格法(Benjamini-Hochberg,HB)方法來矯正P值;并采用線性判別分析(linear discriminant analysis,LDA)對數據進行降維,評估差異顯著物種的影響力(LDA Score),得到線性判別分析效果大小結果圖(Linear discriminant analysis Effect Size,LEfSe)。
2 結果
2.1 納入人群基本特征
本研究共納入糖尿病衰弱或衰弱前期患者15名(A組),按照年齡、性別、衰弱程度等進行1∶1配對后納入糖尿病非衰弱患者15名(B組)。具體情況見表1。
2.2 糞便樣本測序基本統計結果
采用Illumina測序平臺進行測序,共獲得了193.9 GB的原始測序數據,樣本的序列數量在34 956 712~59 919 586之間,平均為46 275 356,所有樣本純凈序列占未處理序列的95%以上,單樣本序列豐度 > 0.1%。
2.3 腸道微生物多樣性
通過對兩組共30例患者的糞便進行宏基因測序,共得到16門、127屬、298種細菌。其中,糖尿病衰弱及衰弱前期組共提取出12門、116屬、264種微生物;糖尿病非衰弱組共提取出16門、100屬、243種微生物。通過物種累積曲線驗證微生物測序數據量在多樣性分析中是否充足,是反映樣本物種多樣性指標之一。橫坐標代表樣本量,縱坐標代表被檢測的物種數,結果顯示末端曲線趨于平緩,表明樣本量足夠(見圖1)。
圖1
物種豐度圖
Figure1.
Abundance of microbiota
香農指數常用于反映α多樣性指數,香農指數值越大,說明菌落多樣性越高。對兩組人群進行香農指數計算,如圖2所示,A組較B組香農指數略小,但經秩和檢驗分析,兩組α多樣性差異無統計學意義。再采用辛普森指數進行計算,A、B兩組間菌群多樣性差異無統計學意義(見圖3)。
圖2
香農指數
Figure2.
Shannon Index
圖3
辛普森指數
Figure3.
Simpson Index
本研究使用PCoA來分析兩組人群糞便微生物β多樣性(見圖4),該方法通過計算Bray-Curtis距離,對菌群進行排序、降維,形成相似度及相異度可視化圖表。結果顯示,兩組人群腸道菌群分布具有差異性,但各自聚集不明顯。使用非參數多元方差分析檢驗分組因素對菌群差異的影響,結果顯示衰弱對糖尿病患者菌群結構總體變化影響不顯著,無統計學意義。
圖4
PCoA結果
Figure4.
The result of PCoA
2.4 腸道微生物組成特征
如圖5所示,在門水平上,糖尿病衰弱及衰弱前期組腸道菌群相對豐度較高的為厚壁菌門(Firmicutes)、擬桿菌門(Bacteroidetes)、變形菌門(Proteobacteria)、放線菌門(Actinobacteria);在科水平上,糖尿病衰弱及衰弱前期組腸道菌群相對豐度較高的為瘤胃菌科(Ruminococcaceae)、擬桿菌科(Bacteroidaceae)、韋榮氏球菌科(Veillonellaceae)、毛螺菌科(Lachnospir-aceae)、優桿菌科(Eubacteriaceae)、普雷沃氏菌科(Prevotellaceae)等;在屬水平上,糖尿病衰弱及衰弱前期組患者腸道菌群相對豐度較高的為糞桿菌屬(Faecalibacterium)、擬桿菌屬(Bacteroides)、優桿菌屬(Eubacterium);在種水平上,糖尿病衰弱組患者腸道菌群相對豐度較高的為普拉梭菌(Faecalibacterium prausnitzii)、直腸真桿菌(Eubacterium rectale)、Prevotellacopri菌、長雙歧桿菌(Bifidobacterium longum)以及大腸桿菌(Escherichia coli)。
圖5
門水平和屬水平物種分布圖
Figure5.
Diversity in phylum and genus levels
2.5 腸道菌群組間差異性分析
本研究采用秩和檢驗對兩組患者腸道菌群各個水平相對豐度進行統計分析,如見表2所示。在門水平,糖尿病衰弱及衰弱前期組的擬桿菌門(Bacteroidetes,紅色)相對豐度高于糖尿病非衰弱組(P = 0.016)。在綱水平,糖尿病衰弱及衰弱前期組的擬桿菌綱(Bacteroidia)相對豐度低于糖尿病非衰弱組(P = 0.02)。在目水平,糖尿病衰弱及衰弱前期Bacteroidales相對豐度低于糖尿病非衰弱組(P = 0.016)。在科水平,糖尿病衰弱及衰弱前期組Bacteroidales noname高于糖尿病非衰弱組(P < 0.01),而Bacteroidaceae在糖尿病非衰弱組中相對豐度較高(P = 0.03)。在屬水平上,糖尿病衰弱及衰弱前期組Bacteroides_noname(P = 0.01)、Collinsella(P = 0.022)、Butyricimonas(P = 0.03)相對豐度高于糖尿病非衰弱組,而Parasutterella(P = 0.01)、Bacteroides(P = 0.03)在糖尿病非衰弱組中相對豐度較高。在種水平豐度上,衰弱及衰弱前期組人群Bacteroidales-bacterium-ph8(P < 0.01)、Butyricimonas_synergistica(P = 0.03)、Bacteroides_ovatus(P = 0.01)相對豐度顯著高于糖尿病非衰弱組患者,而Parasutterella_excrementihominis(P = 0.02)在糖尿病非衰弱組中相對豐度較高。LEfSe分析結果顯示糖尿病衰弱及衰弱前期患者腸道菌群貢獻最大的菌為Butyricimonas菌屬,而糖尿病非衰弱組人群關鍵菌包括Bacteroidetes門bacteroides屬等(見圖6、圖7)。
圖6
進化分支圖
Figure6.
Cladogram
圖7
差異菌群貢獻圖
Figure7.
The most important gut microbiota in different group
2.6 差異功能分析
本研究從30例樣本中共提取及構建了1 264 594非冗余基因集。其中,糖尿病衰弱及衰弱前期組人群共有1 147 676個基因集,糖尿病非衰弱組人群共有1 044 831個基因集。其中,共有基因集占有927 913個,占總基因集73.4%。兩組人群微生物功能基因涉及多條功能通路,通過將糖尿病衰弱及衰弱前期和糖尿病非衰弱組人群差異性表達的KO注釋到KEGG模塊水平,發現糖尿病衰弱及衰弱前期的人群富集上調的功能基因分布在組氨酸代謝(histidine metabolism)、EB病毒感染(Epstein-Barr virus infection)、硫代謝(sulfur metabolism)以及Ⅱ型聚酮生物合成(biosynthesis of type Ⅱ polyketide products)等;而磷酸轉移酶系統(phosphotransferase system,pts)、生物膜形成(biofilm formation)、ABC轉運蛋白(ABC transporters)、鞭毛組裝(flagellar assembly)、丁酸代謝(butanoate metabolism)以及苯丙氨酸代謝(phenylalanine metabolism)等在糖尿病衰弱及衰弱前期人群中出現下調(見表3)。
3 討論
本研究調查了糖尿病衰弱人群的腸道微生物,發現糖尿病衰弱與腸道菌群α多樣性并無相關性,兩組人群β多樣性呈現差異性分布,但各自微生物群聚集不明顯。對差異菌群進行功能分析,發現衰弱人群腸道微生物在氨基酸代謝、EB病毒感染等方面上調,而在生物膜形成、丁酸代謝、苯丙氨酸代謝等方面下調。
3.1 菌群結構特征分析
腸道微生物會隨著年齡增長而出現多樣性下降或生態失衡,個體變異性增加,促使寄主生理紊亂顯著[26],而這種變化與腸道通透性增加以及慢性低度炎癥具有相關性[27]。已有證據表明衰弱狀態伴隨著系統性炎性[28]。既往研究中,衰弱人群的腸道微生物最一致的特征是多樣性的喪失[27],本研究發現糖尿病衰弱及衰弱前期組的α多樣性少于糖尿病非衰弱組人群,但兩組并沒有統計學意義,原因可能是糖尿病對腸道菌群的影響[29],導致在沒有衰弱狀態時就缺乏微生物多樣性,β多樣性分析也提示兩組人群差異并沒有統計學意義。
兩組人群的菌群組成具有明顯差異性。在糖尿病衰弱及衰弱前期組中,柯林斯氏菌屬(Collinsella)和丁酸弧菌屬(Butyricimonas)富集。既往研究提示Collinsella豐度與空腹甘油三酯、總膽固醇濃度呈正相關[30],促進體內脂肥胖形成,進一步促進體內炎癥因子形成[31]。在給無菌小鼠腸道喂養Collinsella菌后發現腸細胞緊密連接蛋白表達降低,腸漏及菌血癥的發生風險顯著升高[32]。因此我們推測,Collinsella菌屬可能通過改變體內慢性炎癥環境,導致衰弱發病率上升,是潛在的糖尿病衰弱致病菌群。然而,目前尚不清楚該菌的富集是衰弱的原因還是結果,其潛在機制有待進一步研究。
在糖尿病非衰弱組中,副薩特氏菌屬(Parasutterella)及卵形擬桿菌(Bacteroides ovatus)富集,目前的研究認為副薩特氏菌屬是腸道的核心菌種,在膽汁酸維持和膽固醇代謝中具有重要作用[33]。在一項隨機試驗中,副薩特氏菌屬與低密度脂蛋白(low density lipoprotein,LDL)降低具有相關性,而低水平LDL與較低的衰弱發病率具有相關性[34],因此我們推測副沙特氏菌是糖尿病衰弱的保護菌群。卵形擬桿菌在腸道中參與了多種碳水化合物的消化分解,提供能量[35],目前有研究已證明卵形擬桿菌能夠誘導腸道產生免疫球蛋白A[36],限制細菌與細菌源性毒素進入腸上皮細胞,減少腸道感染風險[37],同時進食含有卵形擬桿菌的益生菌,能夠改善社區人群腹瀉等癥狀[38]。因此我們推測,卵形擬桿菌數量增加可能會降低糖尿病人群衰弱發生的風險,而相關數量級的確定,還需要更多的研究來支持。
3.2 代謝功能特征性分析
通過與KEGG代謝通路數據庫進行比對,發現兩組人群菌群代謝通路具有顯著差異,在糖尿病衰弱及衰弱前期組人群中,上調的通路包括組氨酸代謝通路、EB病毒感染通路、硫代謝通路,以及Ⅱ型聚酮生物合成通路。組氨酸代謝通路中,咪唑丙酸是其重要產物,通過激活雷帕霉素靶蛋白敏感性復合體1通路(mammalian target of rapamycin complex 1,mTORC1),降解胰島素受體底物1(Insulin Receptor Substrate 1,IRS1)和胰島素受體底物2(Insulin Receptor Substrate 2,IRS2),抑制胰島素信號通路,造成胰島素抵抗,可能是2型糖尿病的病因之一[39];另一方面,胰島素抵抗可以通過抑制骨骼肌細胞能量代謝造成肌肉收縮障礙[5]。這提示我們,組氨酸代謝的相關細菌所引起的胰島素抵抗,可能在老年衰弱的發生發展中起到一定的作用。同時EB病毒感染通路能激活Toll樣受體信號通路(Toll-like receptor signaling pathway,TLR pathway),促進體內白細胞介素6(interleukin-6,IL-6)及腫瘤細胞壞死因子α(tumor necrosis factor-α,TNF-α)的產生[40],而這些炎性標志物與衰弱具有相關性,因此推測衰弱老年人與EB病毒感染通路激活相關,而具體機制還需要更多研究支持。
在糖尿病衰弱及衰弱前期人群菌群通路中,下調的通路包括磷酸轉移酶系統、生物膜形成、ABC轉運蛋白、鞭毛組裝及丁酸代謝。其中丁酸代謝在長壽人群當中具有重要意義[41]。同時丁酸通過結腸細胞上的單羧酸轉運蛋白(monocarboxylate transporter,MCT-1)和溶質轉運蛋白(solute carrier family 5 member 8,SLC5A8)轉運,作為組蛋白脫乙酰酶抑制劑或G蛋白偶聯受體分子,在抗炎、維持腸黏膜屏障以及腸黏膜免疫方面發揮重要作用[42]。糖尿病衰弱及衰弱前期人群腸道微生物對丁酸利用減少,上述丁酸作用相對減弱,前期的研究中發現,腸道免疫系統紊亂是衰弱發生發展的重要因素[43],因此我們推測丁酸可能在預防衰弱發生或改善衰弱程度等方面具有一定的作用,具體機制還需要更多研究證明。
3.3 創新性
既往的研究多集中在歐美等地國家,研究地點多選擇在病房以及養老院等[10-11, 44],而關于社區衰弱人群的腸道菌群差異性在國內未見報道。由于腸道菌群受宿主種族、地域及飲食等的密切影響[45],不同地點人群腸道菌群特點值得研究。本研究采用人群年齡、性別完全配比的方式控制了混雜因素,發現柯林斯氏菌以及EB病毒在衰弱發生中起到重要作用,拓寬了以往的研究結果,為衰弱的干預提供了新的理論依據。
4 結論
本研究通過比較糖尿病衰弱及衰弱前期和非衰弱人群的腸道菌群差異,揭示了糖尿病衰弱及衰弱前期人群的腸道菌群結構特征。本研究采用宏基因測序對糖尿病衰弱及衰弱前期患者腸道菌群結構及功能特征性進行研究,結果表明衰弱及衰弱前期對腸道菌群整體結構影響不顯著,但仍具有特異性菌群。通過對菌群基因功能分析,表明腸道菌群可通過胰島素抵抗和腸道免疫等途徑對糖尿病人群的衰弱產生一定影響。本研究的結果為衰弱人群的腸道菌群機制探索奠定了理論基礎,同時也為糖尿病衰弱及衰弱前期人群的預防與干預提供了一定的科學依據。
利益沖突聲明:本文全體作者均聲明不存在利益沖突。