引用本文: 楊呈浩, 王偉, 羅小彬, 鄒勇, 張明輝. 晝夜節律和代謝途徑在抑郁癥中的角色:生物標志物的發現與新藥物的預測. 華西醫學, 2024, 39(9): 1457-1464. doi: 10.7507/1002-0179.202310239 復制
版權信息: ?四川大學華西醫院華西期刊社《華西醫學》版權所有,未經授權不得轉載、改編
抑郁癥最早記載于我國經典醫學著作《黃帝內經》,其對“郁”解釋為心情抑郁、情緒不寧。近現代對于其病因、發病機制仍不清楚,但目前大多數人一致認為其和體內的生物化學、遺傳、社會因素有關,通過對多數病例進行分析,研究發現其具有發病率高、復發率高、致殘率高、自殺率高等特點[1]。據世界衛生組織估計,2017 年全世界約有 3.22 億抑郁癥患者[1]。預計至 2030 年抑郁癥將成為全球疾病負擔的首位[2]。有研究發現,有 1/5 的中國人具有抑郁相關癥狀,但抑郁癥患者中系統治療者僅占 1/10[3]。目前抑郁癥的治療方式有藥物、心理、物理、替代與補充治療,但治療效果仍不理想。近年來,隨著生物信息學的發展,利用生物信息學對疾病的診斷和治療生物標志物的篩選成為一種新方法[4]。生物信息學特別在探索疾病的分子機制和識別新的治療靶點方面顯示出巨大潛力。盡管如此,將這些生物標志物轉化為實際治療手段仍面臨重大挑戰。此外,晝夜節律失衡與抑郁癥之間的聯系逐漸成為研究焦點,暗示著調節生物鐘可能成為未來治療策略的關鍵。本研究旨在通過生物信息學方法探索抑郁癥的潛在致病機制,并探索可能用于抑郁癥治療的潛在藥物,為抑郁癥的診斷和治療提供新的線索和思路。
1 資料與方法
1.1 數據收集與質量評估
本研究于 2022 年 11 月—2024 年 1 月期間,持續跟蹤 Gene Expression Omnibus 數據庫中的相關數據,并多次檢索關鍵詞“Depression”以獲取符合研究目的的數據集。最終發現 GSE182193 數據集與本研究目的契合,因此選擇其進行后續分析。該數據集包括 6 名抑郁癥患者和 6 名健康志愿者,抑郁癥患者年齡 24~59 歲,健康志愿者年齡 25~64 歲,每組男女性各 3 例,箱線圖提示基線穩定(圖1),可以進行后續分析。
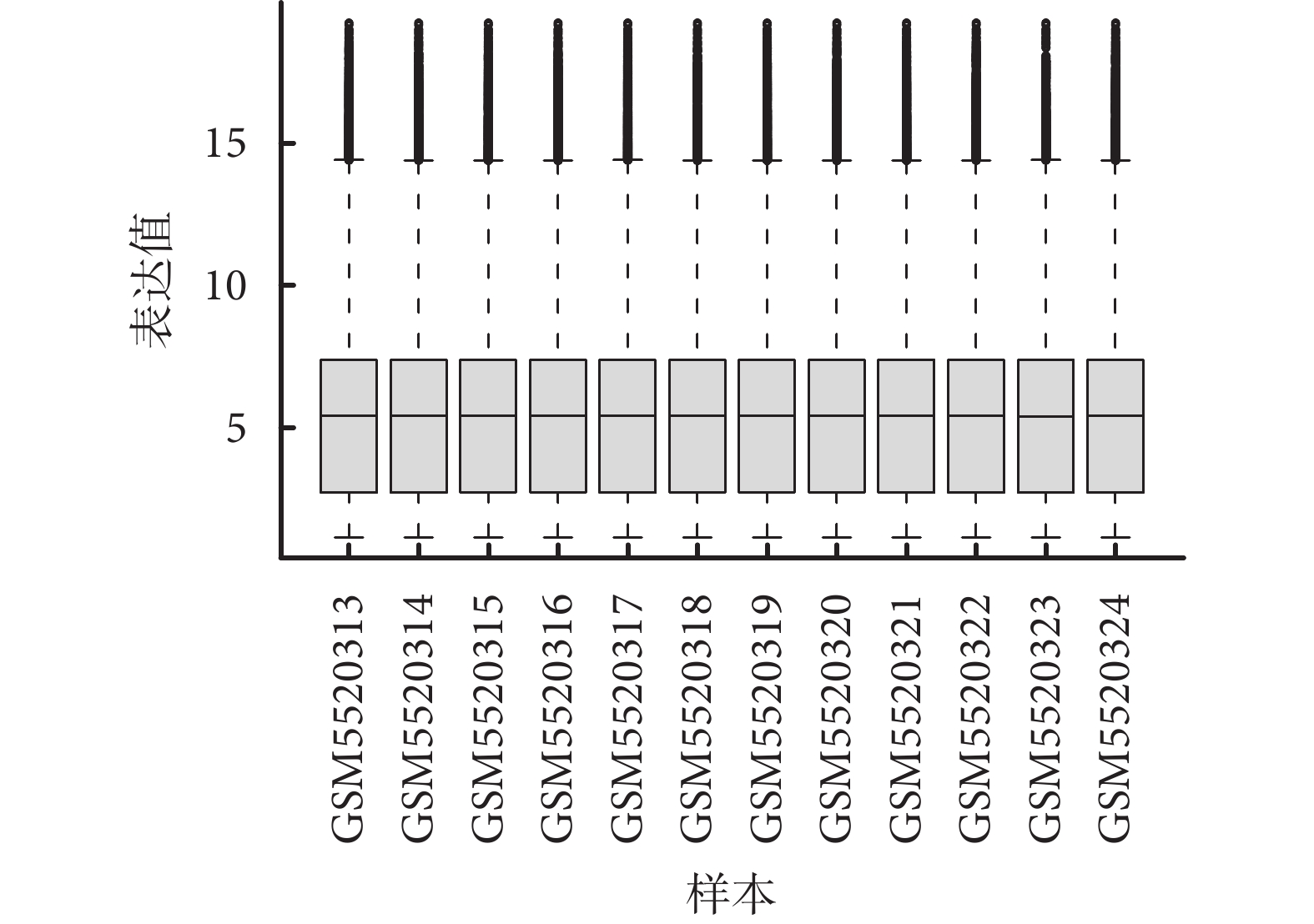 圖1
GSE182193 所有樣本的箱線圖
圖1
GSE182193 所有樣本的箱線圖
本研究還檢索并下載了 GSE217811 數據集。該數據集包含 10 名未用藥的青少年重度抑郁癥患者和 10 名年齡、性別匹配的健康對照樣本,用于血漿外泌體來源的轉錄組表達分析,所有參與者的年齡在 15~19 歲,其中男性患者 4 名,抑郁癥患者均符合《精神障礙診斷與統計手冊(第 5 版)》抑郁癥診斷標準[5]。此外,本研究還選取了 GSE144136 數據集,該數據集通過 10X Genomics Chromium 平臺對大腦背外側前額葉皮質(BA9)中的細胞核進行單核 RNA 測序,分析了 17 名抑郁癥自殺患者與 17 名健康對照者的差異表達基因,所有樣本均為男性,患者平均年齡為 41 歲。通過這兩個數據集的聯合分析,以進一步揭示抑郁癥的潛在分子機制。
1.2 差異表達基因的篩選與驗證
下載 GSE182193 的表達矩陣數據后,使用 GPL22120 平臺對其進行探針注釋,以得到基因名稱;下載 Gencode 的基因注釋文件第 38 版對獲得基因進行區分,以篩選出編碼 mRNA 的基因,并以 R 包對其進行差異分析。為確保分析的準確性和可靠性,本研究設置了明確的篩選標準:差異表達基因的選擇基于絕對對數變化倍數(|log2FC|)≥1 且 P<0.05。其后,利用 GSE217811 的表達矩陣數據,采用同樣的分析流程對上述差異表達基因進行驗證。
1.3 差異表達基因的蛋白質相互作用(protein-protein interaction, PPI)網絡的構建
將得到的差異表達基因通過在線數據庫 STRING(版本 11.5)進行共表達網絡的構建,構建過程中隱藏網絡中斷開連接的節點后得到 PPI 網絡,使用 Cytoscape 軟件(版本 3.8.2)的插件 Cytohubba(版本 0.1)對網絡圖進行連接度(Degree)計算,連接度越高,該基因在抑郁癥中就越重要[6]。
1.4 差異表達基因的富集分析
使用“clusterProfiler”“org.Hs.eg.db”“enrichplot”“ggplot2”包對上述差異表達基因進行基因本體論(Gene Ontology, GO)、京都基因和基因組數據庫(Kyoto Encyclopedia of Genes and Genomes, KEGG)富集分析[7],篩選出其中上調和下調的基因分別進行富集分析,其中篩選術語和通路標準為 P<0.05,最后可視化相關術語和通路。
1.5 單細胞測序分析
為深入探究抑郁癥的分子機制,并探索在抑郁癥發病過程中可能涉及的關鍵細胞類型和分子通路,本研究利用單細胞測序分析數據在細胞層面上探究抑郁癥的分子機制,揭示特定細胞類型中的病理過程。利用 Seurat 軟件包對 GSE144136 數據集中的單細胞 RNA 測序數據進行預處理,包括數據的標準化、細胞周期效應校正、數據降維和細胞群體的聚類分析。為了提高細胞類型識別的準確性,使用 SingleR 軟件對單細胞測序數據進行進一步的標注,對差異表達基因在不同細胞類型中的分布情況進行詳細的映射。
1.6 孟德爾隨機化分析
為理解差異表達基因與抑郁癥易感性之間的關聯性,以及這些基因如何在遺傳層面上影響抑郁癥的風險,本研究采用孟德爾隨機化分析評估差異表達基因與抑郁癥易感性之間的因果關系。首先,利用 R 包 gwasrapidd,從已知的差異表達基因中提取與抑郁癥相關的單核苷酸多態性(single nucleotide polymorphism, SNP)。將這些 SNP 作為工具變量,用于分析基因在抑郁癥易感性中的作用。隨后,使用基于總結數據的孟德爾隨機化分析工具根據所提取的 SNP 列表,獲取進行孟德爾隨機化分析所需的相關數據。孟德爾隨機化分析是通過 TwoSampleMR 進行的,其中暴露組的數據來自于全基因組關聯研究數據庫 UK Biobank 項目的 ukb-b-12064 數據集。采用多種統計方法進行分析,包括 MR Egger 分析、加權中位數法、逆方差加權法、簡單模式和加權模式,以確保結果具有穩健性。
1.7 藥物預測
使用 Cmap 在線數據庫對抑郁癥中的關鍵基因進行藥物預測,Cmap 是由美國麻省理工學院和美國哈佛大學共同創建并維護用于小分子藥物預測的在線數據庫[8]。當富集分數為正時,其表示藥物會誘導相關生物過程,反之,則表明藥物可以逆轉相關生物過程,發揮治療作用。因此,我們的篩選標準為富集分數<–0.7、P<0.05 代表結果具有統計學意義。
1.8 統計學方法
所有分析均使用 R 4.3.2 軟件完成,差異分析使用 Wilcoxon 秩和檢驗,檢驗水準 α=0.05。
2 結果
2.1 差異分析結果
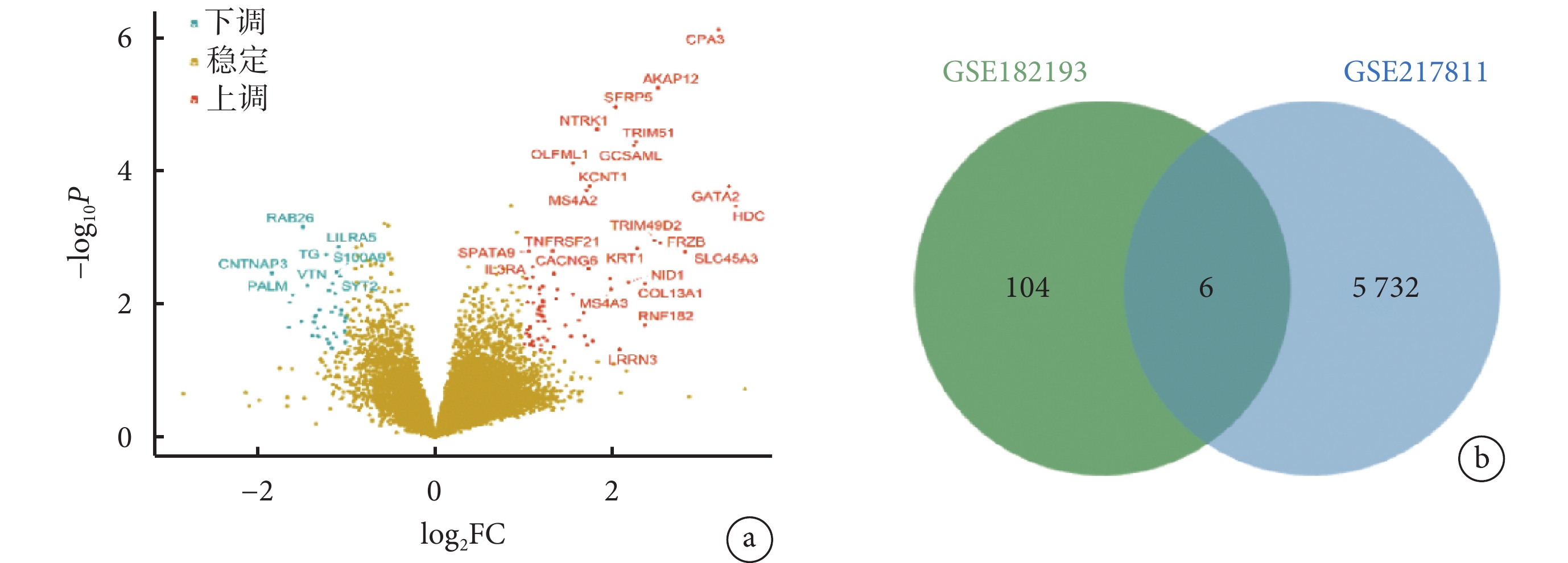
GSE182193 數據集中共有差異表達基因 110 個(上調 74 個,下調 36 個)。以數據集 GSE217811 對 110 個差異表達基因進行驗證,6 個基因(SALL2、AKAP12、GCSAML、CPA3、FCRL3 和 MS4A3)仍滿足篩選標準(|log2FC|≥1,P<0.05)。見圖2。
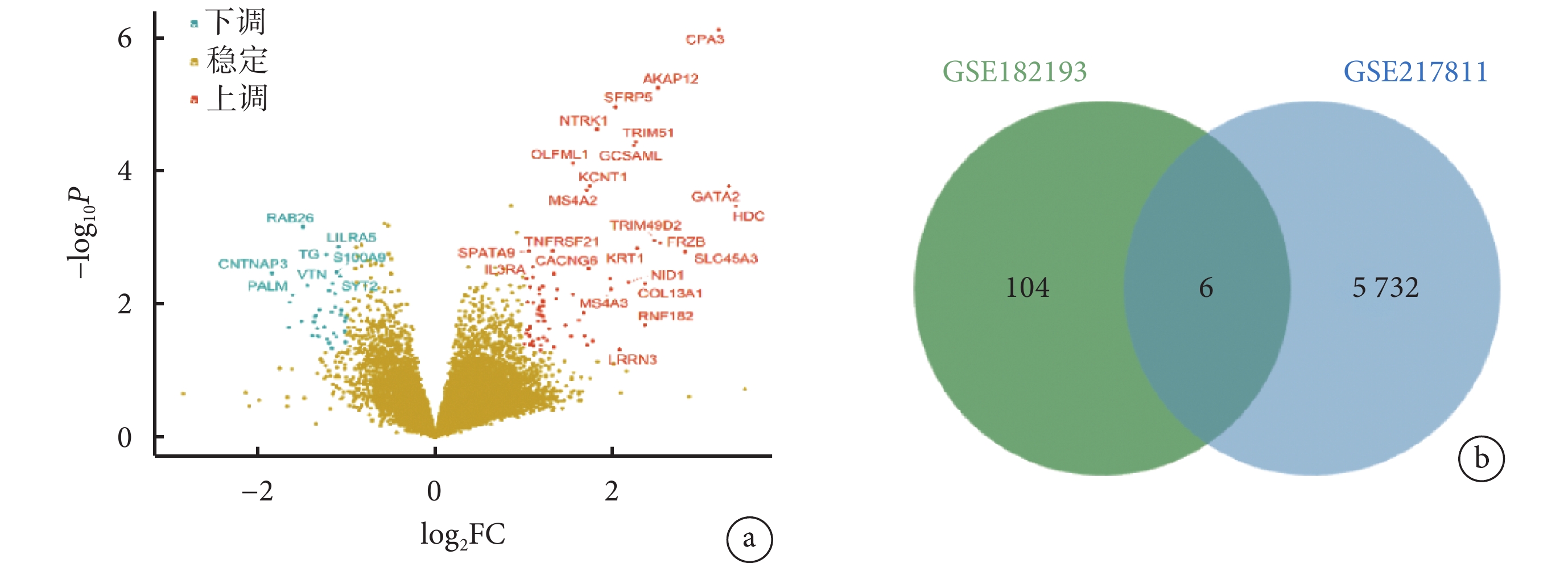 圖2
抑郁癥患者和健康志愿者之間差異表達基因的火山圖和韋恩圖
圖2
抑郁癥患者和健康志愿者之間差異表達基因的火山圖和韋恩圖
a. GSE182193 數據集中差異表達基因的火山圖,FC:差異倍數;b. GSE182193 數據集與 GSE217811 數據集差異表達基因的韋恩圖
2.2 PPI 網絡的構建
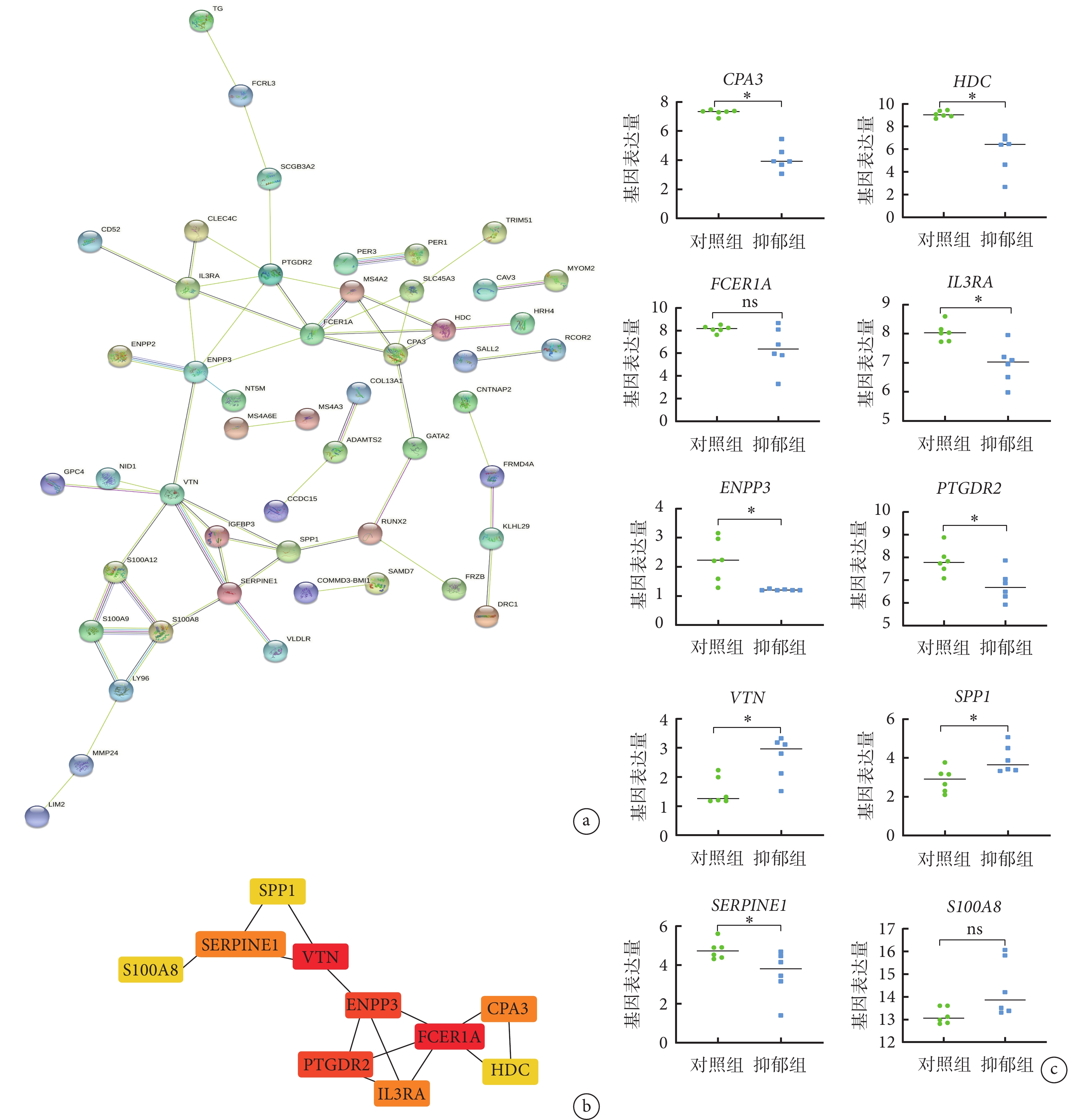
對 GSE182193 數據集中的 110 個差異表達基因進行 PPI 網絡構建,通過 STRING 在線網絡工具,去除沒有連接的點后,獲得含 50 個點、58 條邊的 PPI 網絡,通過 Cytohubba 對連接點進行篩選選出了 10 個連接度最高的基因(CPA3、HDC、FCER1A、IL3RA、ENPP3、PTGDR2、VTN、SPP1、SERPINE1 和 S100A8),并進行差異分析,結果顯示 10 個基因中,有 8 個基因(CPA3、HDC、IL3RA、ENPP3、PTGDR2、VTN、SPP1 和 SERPINE1)在健康志愿者和抑郁癥患者中表達具有差異(P<0.05)。見圖3。
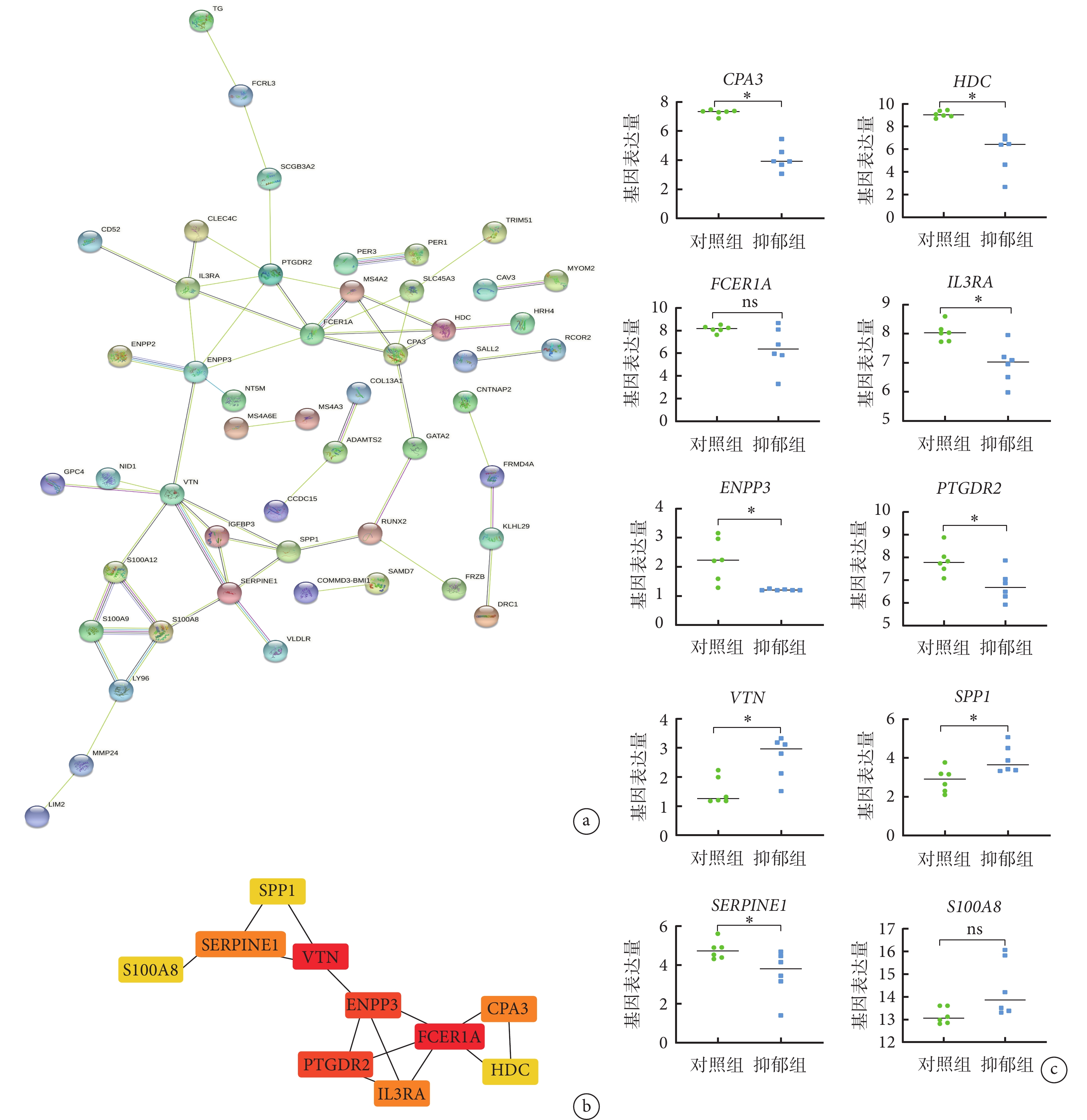 圖3
關鍵基因的篩選
圖3
關鍵基因的篩選
a. 通過差異表達基因構建的蛋白質相互作用網絡;b. 前 10 個關鍵基因的蛋白質相互作用網絡;c. 前 10 個關鍵基因的差異分析結果,對照組為健康志愿者樣本,抑郁組為抑郁癥患者樣本,*代表
2.3 差異基因的富集分析
2.3.1 GO 富集分析
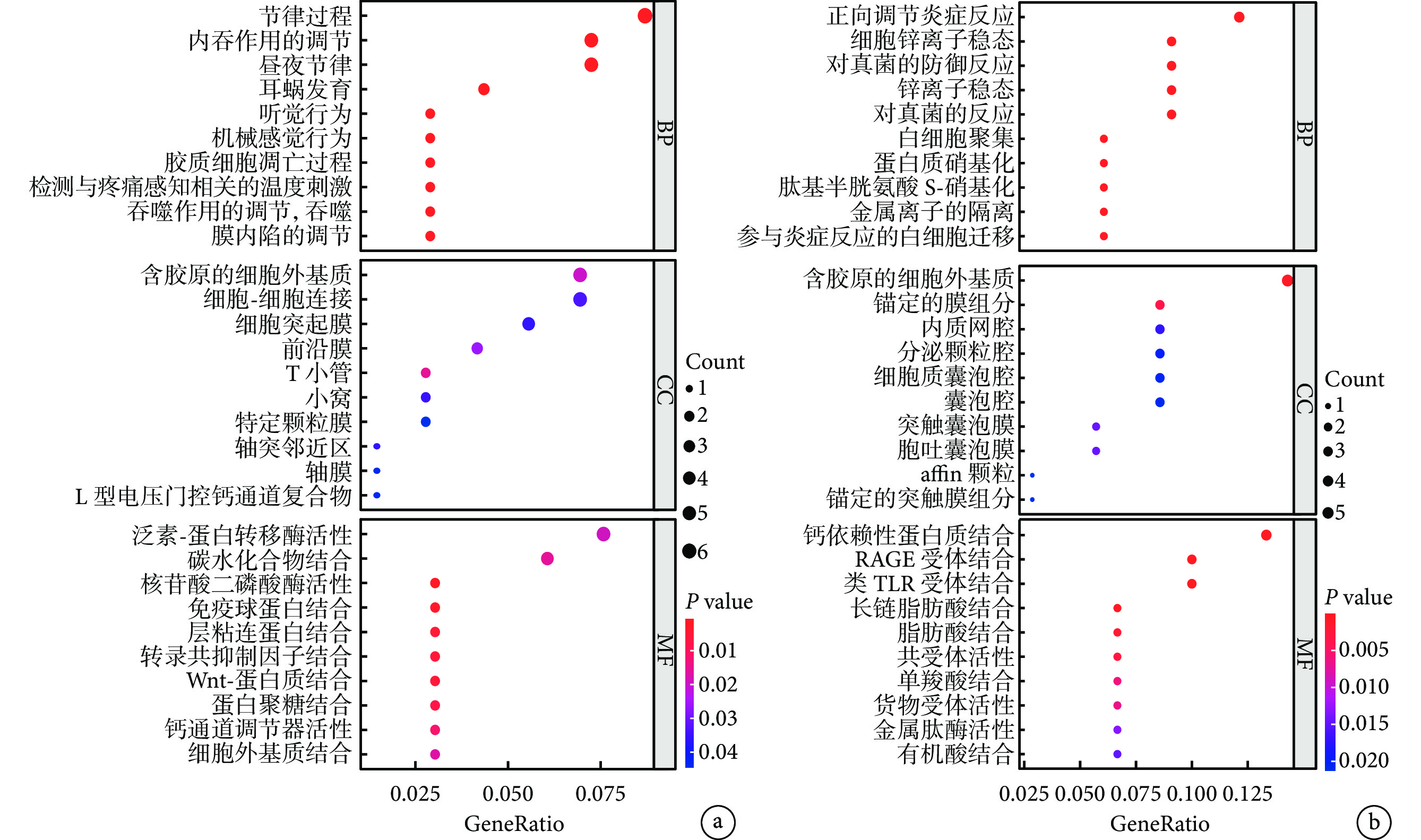
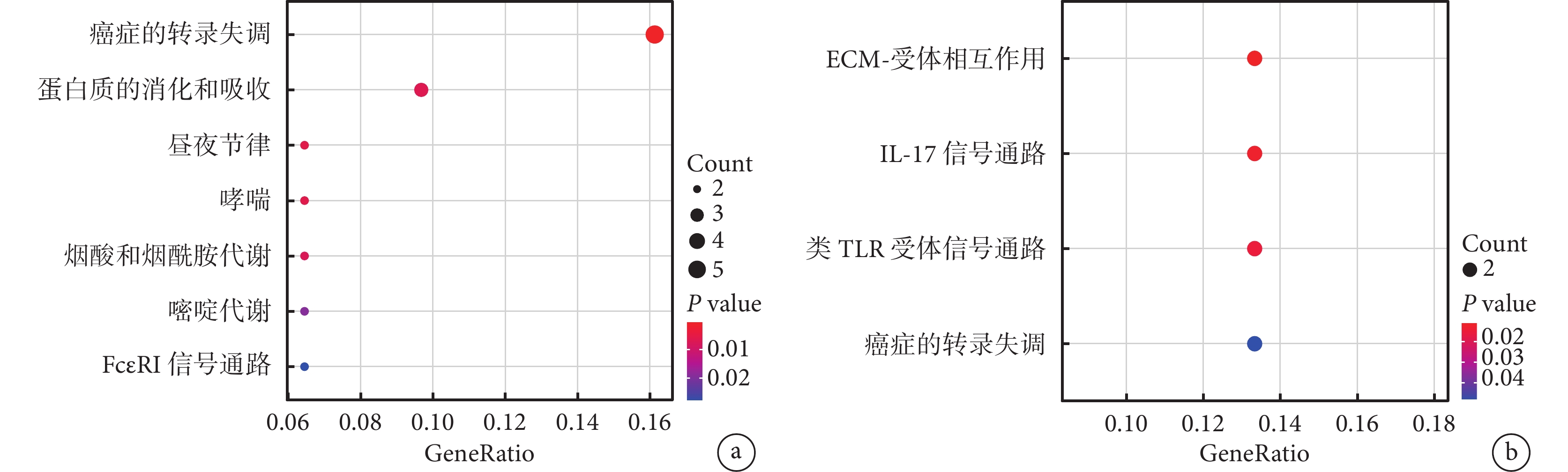
GO 富集分析結果顯示,上調的基因在生物學過程(biological process, BP)主要富集在生理節律的維持中,如節律過程、晝夜節律、機械感覺行為;細胞組分(cellular component, CC)主要富集在各種細胞器上;分子功能(molecular function, MF)主要富集在各種蛋白結合,尤其是 Wnt-蛋白質結合。下調的基因在 BP 主要集中在炎癥反應中,如正向調節炎癥反應、對真菌的反應等;同上調的 CC,下調的 CC 也主要富集在各種細胞器中;在 MF 中,其主要富集在各種受體的結合,尤其是鈣依賴蛋白結合、晚期糖基化終產物受體結合、Toll 樣受體結合。見圖4。
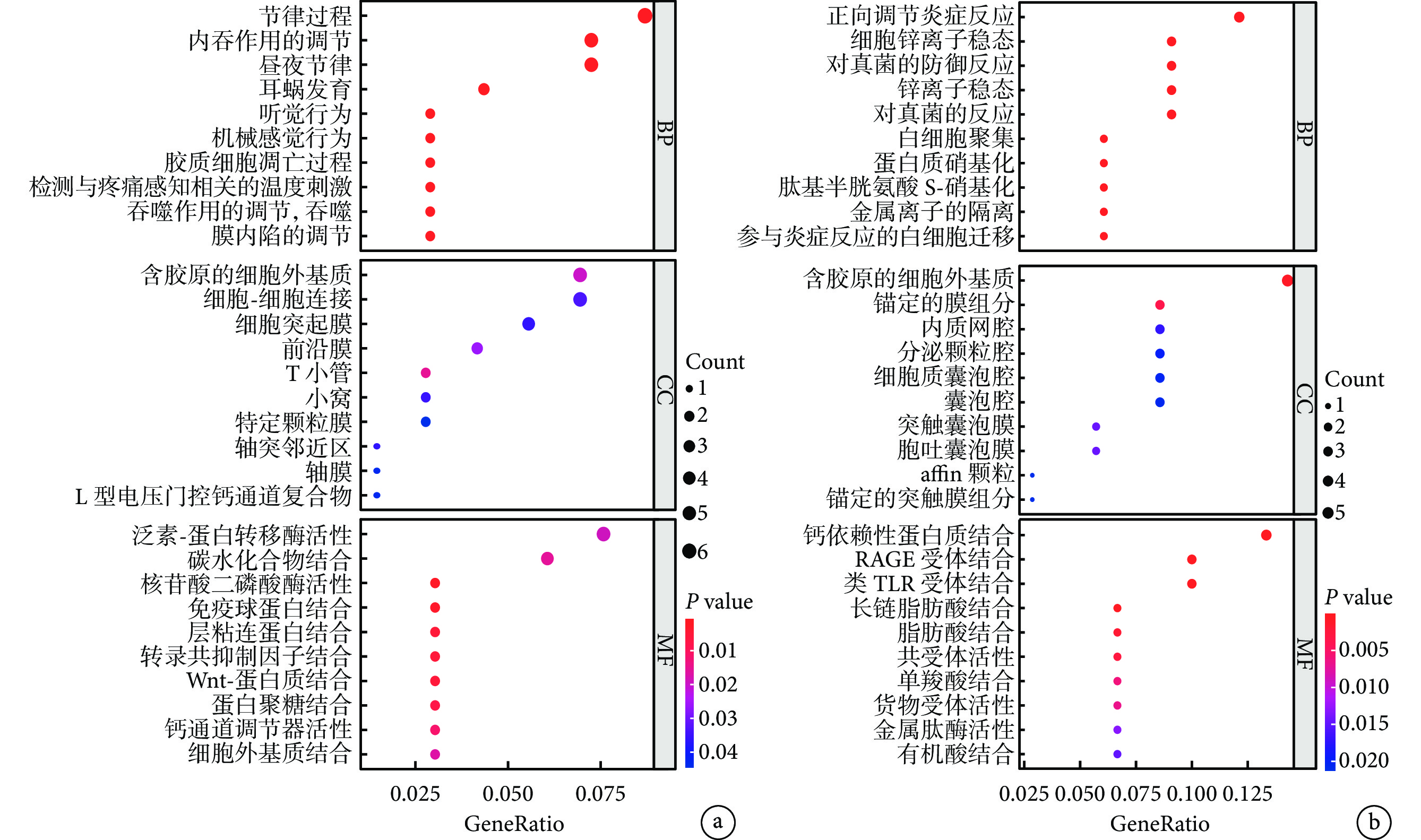 圖4
差異表達基因的 GO 富集分析
圖4
差異表達基因的 GO 富集分析
a. 在抑郁癥患者中上調的差異表達基因的富集分析結果;b. 在抑郁癥患者中下調的差異表達基因的富集分析結果。GO:基因本體論;BP:生物學過程;CC:細胞組分;MF:分子功能;RAGE:晚期糖基化終產物受體;TLR:Toll 樣受體;GeneRatio:基因比率
2.3.2 KEGG 富集分析
KEGG 通路富集分析結果顯示,上調的基因主要富集在多通路的代謝失調,尤其是癌癥中的轉錄失調、晝夜節律、煙酸和煙酰胺代謝、嘧啶代謝、FcεRI 信號通路。下調的基因主要富集在細胞外基質-受體相互作用、白細胞介素-17 信號通路、Toll 樣受體信號通路以及癌癥中的轉錄失調通路。見圖5。
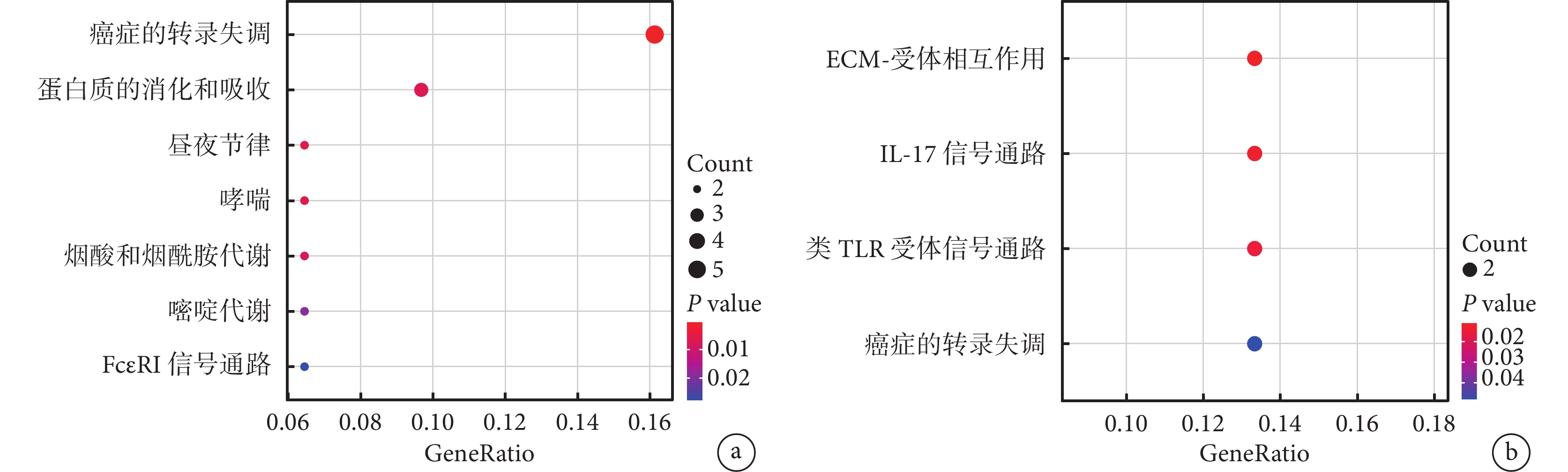 圖5
差異表達基因的 KEGG 富集分析
圖5
差異表達基因的 KEGG 富集分析
a. 在抑郁癥患者中上調的差異表達基因的富集分析結果;b. 在抑郁癥患者中下調的差異表達基因的富集分析結果。KEGG:京都基因與基因組數據庫;ECM:細胞外基質;IL-17:白細胞介素-17;TLR:Toll 樣受體;GeneRatio:基因比率
2.4 單細胞測序分析
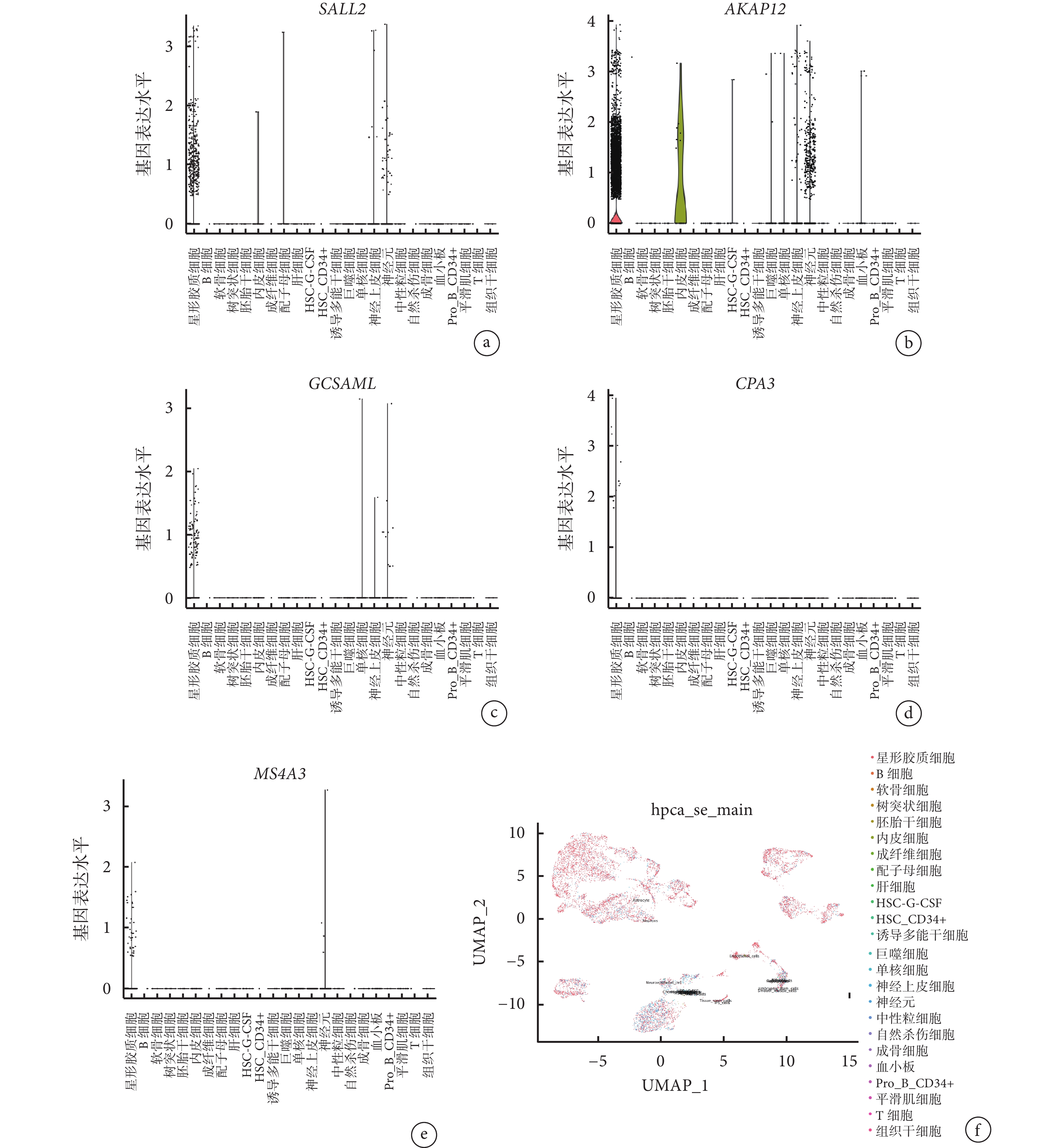
對 6 個獲驗證的差異表達基因(SALL2、AKAP12、GCSAML、CPA3、FCRL3 和 MS4A3)在不同細胞類型中的分布情況進行詳細的映射,除了 FCRL3 未在映射結果中觀察到外,其他 5 個差異表達基因主要在星形膠質細胞和神經細胞中顯著表達。見圖6。
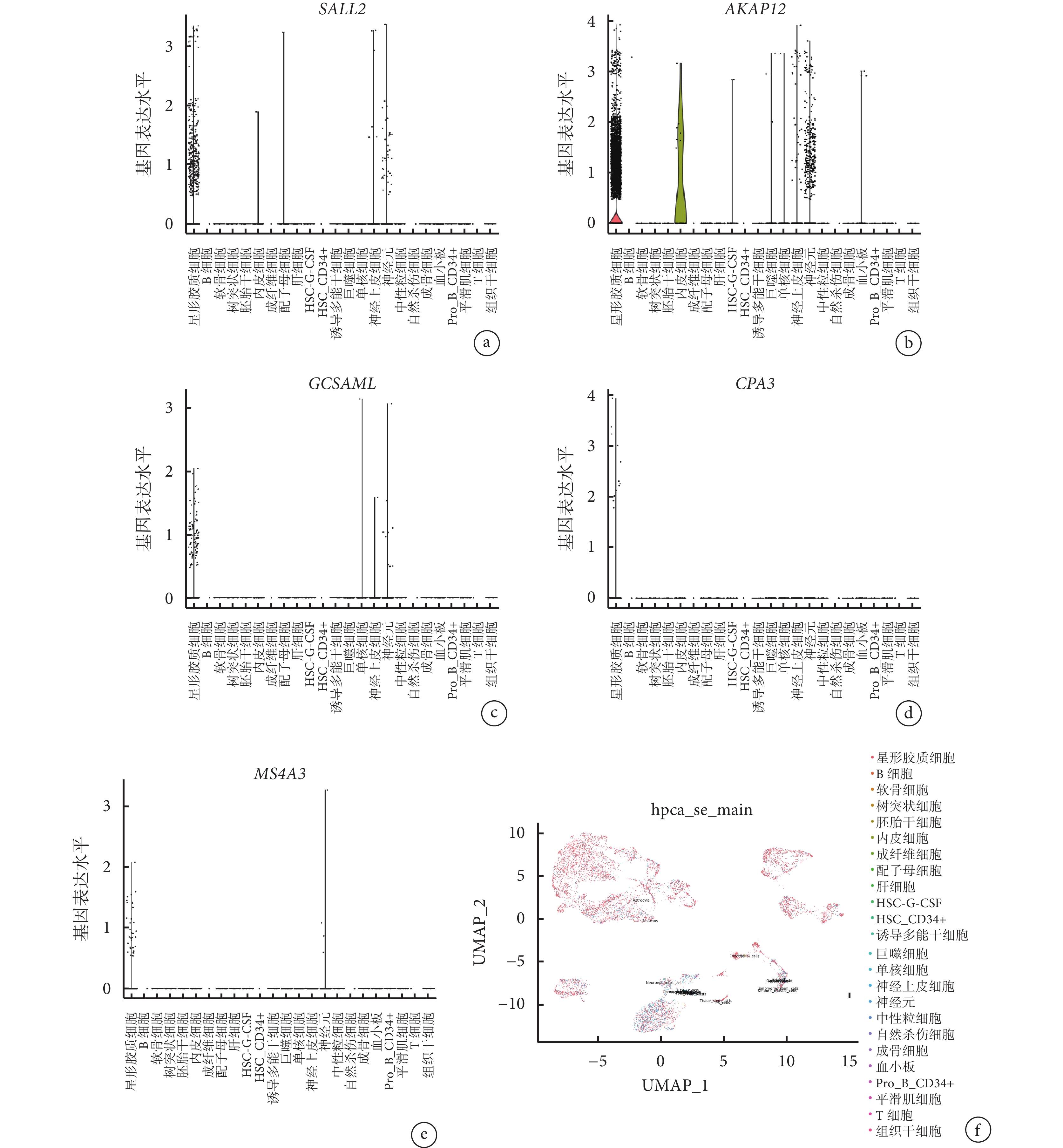 圖6
GSE144136 數據集中重度抑郁癥患者差異表達基因的單細胞測序分析
圖6
GSE144136 數據集中重度抑郁癥患者差異表達基因的單細胞測序分析
a~e. 分別為
2.5 孟德爾隨機化分析
全部差異表達基因相關 SNP 的孟德爾隨機化分析結果表明,不同方法得到的 β 系數和標準誤差各異。MR Egger 方法的 β 系數為
考慮到較高的異質性,我們對 6 個與抑郁癥差異表達基因(SALL2、AKAP12、GCSAML、CPA3、FCRL3 和 MS4A3)相關的 SNP 進行了孟德爾隨機化分析,以探討這些基因在抑郁癥遺傳易感性中的作用。分析結果顯示,大多數基因與抑郁癥之間的關聯無統計學意義(P>0.05)。然而,FCRL3 基因的兩種分析方法(加權中位數法和逆方差加權法)表明該基因與抑郁癥之間的關聯有統計學意義(P=
2.6 藥物預測
8 個關鍵基因中,因 ENPP3 基因進行探針轉換時沒有找到其對應探針,因此只納入了 7 個基因進行 Cmap 藥物預測,通過對 Cmap 數據庫的結果進行篩選,前 10 個潛在的抑郁癥治療藥物分別是頭孢替安、駱駝蓬醇、坎那定、CP-863187、2-氨基苯磺酰胺、林可霉素、病毒唑、苯噻林、α-育亨賓、桿菌肽(表1)。
3 討論
本研究通過對抑郁癥患者和健康志愿者的基因表達進行差異分析,找到其潛在的致病基因,分別是 CPA3、HDC、FCER1A、IL3RA、ENPP3、PTGDR2、VTN、SPP1、SERPINE1 和 S100A8,其中 S100A8、FCER1A 在健康志愿者和抑郁癥患者中差異無統計學意義(P>0.05)。綜合目前的研究,Han 等[9]研究發現在肝癌和抑郁癥中 SERPINE1 基因表達具有差異,可能與細胞死亡或凋亡有關。有趣的是,本研究未發現 S100A8 基因在健康志愿者和抑郁癥患者之間存在差異表達,但現有研究顯示,S100A8 基因可能與抑郁癥患者刺激性行為有關[10]。S100A8 和 FCER1A 在本研究中未顯示出有統計學意義的差異,這可能指向抑郁癥病理機制的復雜性及個體間的異質性。此外,考慮到抑郁癥的多因素性質,未來研究中探索與這些基因相互作用的其他生物標志物,可能揭示更加細致的病理過程。
通過對抑郁癥差異表達的基因進行 GO、KEGG 富集分析,我們發現上調的基因在 GO 術語的 BP 中主要富集在生理節律的維持,CC 主要富集在各種細胞器上,MF 主要富集在各種蛋白結合,下調的基因在 GO 術語的 BP 中主要集中在炎癥反應中,CC 主要富集在各種細胞器中,MF 主要富集在各種受體的結合,由于抑郁癥與體內的生物化學反應相關,我們推測其可能與生理節律失調、炎癥反應有關。另外,KEGG 通路富集分析顯示晝夜節律紊亂明顯。另外,相關研究發現晝夜節律紊亂是抑郁癥的病理生理學基礎[11-12],這與我們的研究相一致。重要的是,在煙酸和煙酰胺代謝通路中,Ma 等[13]研究發現綠原酸可以調節該通路而發揮其抗抑郁作用。此外,Zhao 等[14]研究發現抑郁癥大學生的心率變異性與嘧啶代謝途徑有關。并且,中藥大黃素可能通過白細胞介素-17 信號通路發揮其抗抑郁的作用[15]。另外,Toll 樣受體信號通路可能與針灸發揮抗抑郁作用有關[16]。以上通路均與抑郁癥有關,因此,研究相關的靶向藥物可能對改善抑郁癥患者的癥狀提供依據。
在本研究中,FCRL3 基因在抑郁癥患者與健康對照組之間表現出差異性表達。鑒于近年來研究發現抑郁癥可能與身體炎癥狀態相關,我們猜想 FCRL3 可能通過影響炎癥反應途徑,間接參與到抑郁癥的發病機制中[17]。雖然在單細胞測序數據分析中未檢測到 FCRL3 的特定表達模式,這可能由于樣本的選擇或細胞類型的限制。然而,通過孟德爾隨機化分析,我們發現與 FCRL3 相關的特定 SNP 在抑郁癥的遺傳易感性中顯示出明顯的相關性,為 FCRL3 在抑郁癥中可能扮演的角色提供了初步證據。這一點強調了 FCRL3 作為一個潛在的雙重功能靶點,在免疫調節和精神障礙發病機制中的重要性。
有研究通過使用 Cmap 數據庫對潛在治療抑郁癥藥物進行預測,發現駱駝蓬醇中的吲哚生物堿與神經遞質血清素有關[18],它可以通過 γ-氨基丁酸受體發揮抗抑郁的作用[19]。令人詫異的是,結合當下的研究,我們發現在丙型病毒性肝炎患者中利巴韋林和聚乙二醇干擾素可誘發抑郁發作[20]。其余藥物均未發現其用于治療抑郁癥。因此,本研究可為藥物生產企業研發控制抑郁癥藥物提供參考。
綜上所述,本研究鑒定了與抑郁癥相關的差異表達基因,并通過富集分析揭示了其可能涉及的致病通路。進一步分析中,我們識別出抑郁癥的 10 個潛在關鍵基因,其中 8 個基因(CPA3、HDC、IL3RA、ENPP3、PTGDR2、VTN、SPP1 和 SERPINE1)在健康志愿者和抑郁癥患者之間的表達水平存在差異,并基于這些基因進行了藥物預測,得到了若干可能用于抑郁癥治療的候選藥物。此外,本研究結果提示晝夜節律紊亂可能是抑郁癥發病的重要機制之一。整體而言,本研究為抑郁癥的病理機制和潛在治療靶點提供了新的見解,有望改善抑郁癥患者的預后,提高生活質量,并減輕家庭和社會的負擔。
利益沖突:所有作者聲明不存在利益沖突。
請掃描本文首頁二維碼后在“補充材料”中下載查看附件。
抑郁癥最早記載于我國經典醫學著作《黃帝內經》,其對“郁”解釋為心情抑郁、情緒不寧。近現代對于其病因、發病機制仍不清楚,但目前大多數人一致認為其和體內的生物化學、遺傳、社會因素有關,通過對多數病例進行分析,研究發現其具有發病率高、復發率高、致殘率高、自殺率高等特點[1]。據世界衛生組織估計,2017 年全世界約有 3.22 億抑郁癥患者[1]。預計至 2030 年抑郁癥將成為全球疾病負擔的首位[2]。有研究發現,有 1/5 的中國人具有抑郁相關癥狀,但抑郁癥患者中系統治療者僅占 1/10[3]。目前抑郁癥的治療方式有藥物、心理、物理、替代與補充治療,但治療效果仍不理想。近年來,隨著生物信息學的發展,利用生物信息學對疾病的診斷和治療生物標志物的篩選成為一種新方法[4]。生物信息學特別在探索疾病的分子機制和識別新的治療靶點方面顯示出巨大潛力。盡管如此,將這些生物標志物轉化為實際治療手段仍面臨重大挑戰。此外,晝夜節律失衡與抑郁癥之間的聯系逐漸成為研究焦點,暗示著調節生物鐘可能成為未來治療策略的關鍵。本研究旨在通過生物信息學方法探索抑郁癥的潛在致病機制,并探索可能用于抑郁癥治療的潛在藥物,為抑郁癥的診斷和治療提供新的線索和思路。
1 資料與方法
1.1 數據收集與質量評估
本研究于 2022 年 11 月—2024 年 1 月期間,持續跟蹤 Gene Expression Omnibus 數據庫中的相關數據,并多次檢索關鍵詞“Depression”以獲取符合研究目的的數據集。最終發現 GSE182193 數據集與本研究目的契合,因此選擇其進行后續分析。該數據集包括 6 名抑郁癥患者和 6 名健康志愿者,抑郁癥患者年齡 24~59 歲,健康志愿者年齡 25~64 歲,每組男女性各 3 例,箱線圖提示基線穩定(圖1),可以進行后續分析。
圖1
GSE182193 所有樣本的箱線圖
本研究還檢索并下載了 GSE217811 數據集。該數據集包含 10 名未用藥的青少年重度抑郁癥患者和 10 名年齡、性別匹配的健康對照樣本,用于血漿外泌體來源的轉錄組表達分析,所有參與者的年齡在 15~19 歲,其中男性患者 4 名,抑郁癥患者均符合《精神障礙診斷與統計手冊(第 5 版)》抑郁癥診斷標準[5]。此外,本研究還選取了 GSE144136 數據集,該數據集通過 10X Genomics Chromium 平臺對大腦背外側前額葉皮質(BA9)中的細胞核進行單核 RNA 測序,分析了 17 名抑郁癥自殺患者與 17 名健康對照者的差異表達基因,所有樣本均為男性,患者平均年齡為 41 歲。通過這兩個數據集的聯合分析,以進一步揭示抑郁癥的潛在分子機制。
1.2 差異表達基因的篩選與驗證
下載 GSE182193 的表達矩陣數據后,使用 GPL22120 平臺對其進行探針注釋,以得到基因名稱;下載 Gencode 的基因注釋文件第 38 版對獲得基因進行區分,以篩選出編碼 mRNA 的基因,并以 R 包對其進行差異分析。為確保分析的準確性和可靠性,本研究設置了明確的篩選標準:差異表達基因的選擇基于絕對對數變化倍數(|log2FC|)≥1 且 P<0.05。其后,利用 GSE217811 的表達矩陣數據,采用同樣的分析流程對上述差異表達基因進行驗證。
1.3 差異表達基因的蛋白質相互作用(protein-protein interaction, PPI)網絡的構建
將得到的差異表達基因通過在線數據庫 STRING(版本 11.5)進行共表達網絡的構建,構建過程中隱藏網絡中斷開連接的節點后得到 PPI 網絡,使用 Cytoscape 軟件(版本 3.8.2)的插件 Cytohubba(版本 0.1)對網絡圖進行連接度(Degree)計算,連接度越高,該基因在抑郁癥中就越重要[6]。
1.4 差異表達基因的富集分析
使用“clusterProfiler”“org.Hs.eg.db”“enrichplot”“ggplot2”包對上述差異表達基因進行基因本體論(Gene Ontology, GO)、京都基因和基因組數據庫(Kyoto Encyclopedia of Genes and Genomes, KEGG)富集分析[7],篩選出其中上調和下調的基因分別進行富集分析,其中篩選術語和通路標準為 P<0.05,最后可視化相關術語和通路。
1.5 單細胞測序分析
為深入探究抑郁癥的分子機制,并探索在抑郁癥發病過程中可能涉及的關鍵細胞類型和分子通路,本研究利用單細胞測序分析數據在細胞層面上探究抑郁癥的分子機制,揭示特定細胞類型中的病理過程。利用 Seurat 軟件包對 GSE144136 數據集中的單細胞 RNA 測序數據進行預處理,包括數據的標準化、細胞周期效應校正、數據降維和細胞群體的聚類分析。為了提高細胞類型識別的準確性,使用 SingleR 軟件對單細胞測序數據進行進一步的標注,對差異表達基因在不同細胞類型中的分布情況進行詳細的映射。
1.6 孟德爾隨機化分析
為理解差異表達基因與抑郁癥易感性之間的關聯性,以及這些基因如何在遺傳層面上影響抑郁癥的風險,本研究采用孟德爾隨機化分析評估差異表達基因與抑郁癥易感性之間的因果關系。首先,利用 R 包 gwasrapidd,從已知的差異表達基因中提取與抑郁癥相關的單核苷酸多態性(single nucleotide polymorphism, SNP)。將這些 SNP 作為工具變量,用于分析基因在抑郁癥易感性中的作用。隨后,使用基于總結數據的孟德爾隨機化分析工具根據所提取的 SNP 列表,獲取進行孟德爾隨機化分析所需的相關數據。孟德爾隨機化分析是通過 TwoSampleMR 進行的,其中暴露組的數據來自于全基因組關聯研究數據庫 UK Biobank 項目的 ukb-b-12064 數據集。采用多種統計方法進行分析,包括 MR Egger 分析、加權中位數法、逆方差加權法、簡單模式和加權模式,以確保結果具有穩健性。
1.7 藥物預測
使用 Cmap 在線數據庫對抑郁癥中的關鍵基因進行藥物預測,Cmap 是由美國麻省理工學院和美國哈佛大學共同創建并維護用于小分子藥物預測的在線數據庫[8]。當富集分數為正時,其表示藥物會誘導相關生物過程,反之,則表明藥物可以逆轉相關生物過程,發揮治療作用。因此,我們的篩選標準為富集分數<–0.7、P<0.05 代表結果具有統計學意義。
1.8 統計學方法
所有分析均使用 R 4.3.2 軟件完成,差異分析使用 Wilcoxon 秩和檢驗,檢驗水準 α=0.05。
2 結果
2.1 差異分析結果
GSE182193 數據集中共有差異表達基因 110 個(上調 74 個,下調 36 個)。以數據集 GSE217811 對 110 個差異表達基因進行驗證,6 個基因(SALL2、AKAP12、GCSAML、CPA3、FCRL3 和 MS4A3)仍滿足篩選標準(|log2FC|≥1,P<0.05)。見圖2。
圖2
抑郁癥患者和健康志愿者之間差異表達基因的火山圖和韋恩圖
a. GSE182193 數據集中差異表達基因的火山圖,FC:差異倍數;b. GSE182193 數據集與 GSE217811 數據集差異表達基因的韋恩圖
2.2 PPI 網絡的構建
對 GSE182193 數據集中的 110 個差異表達基因進行 PPI 網絡構建,通過 STRING 在線網絡工具,去除沒有連接的點后,獲得含 50 個點、58 條邊的 PPI 網絡,通過 Cytohubba 對連接點進行篩選選出了 10 個連接度最高的基因(CPA3、HDC、FCER1A、IL3RA、ENPP3、PTGDR2、VTN、SPP1、SERPINE1 和 S100A8),并進行差異分析,結果顯示 10 個基因中,有 8 個基因(CPA3、HDC、IL3RA、ENPP3、PTGDR2、VTN、SPP1 和 SERPINE1)在健康志愿者和抑郁癥患者中表達具有差異(P<0.05)。見圖3。
圖3
關鍵基因的篩選
a. 通過差異表達基因構建的蛋白質相互作用網絡;b. 前 10 個關鍵基因的蛋白質相互作用網絡;c. 前 10 個關鍵基因的差異分析結果,對照組為健康志愿者樣本,抑郁組為抑郁癥患者樣本,*代表
2.3 差異基因的富集分析
2.3.1 GO 富集分析
GO 富集分析結果顯示,上調的基因在生物學過程(biological process, BP)主要富集在生理節律的維持中,如節律過程、晝夜節律、機械感覺行為;細胞組分(cellular component, CC)主要富集在各種細胞器上;分子功能(molecular function, MF)主要富集在各種蛋白結合,尤其是 Wnt-蛋白質結合。下調的基因在 BP 主要集中在炎癥反應中,如正向調節炎癥反應、對真菌的反應等;同上調的 CC,下調的 CC 也主要富集在各種細胞器中;在 MF 中,其主要富集在各種受體的結合,尤其是鈣依賴蛋白結合、晚期糖基化終產物受體結合、Toll 樣受體結合。見圖4。
圖4
差異表達基因的 GO 富集分析
a. 在抑郁癥患者中上調的差異表達基因的富集分析結果;b. 在抑郁癥患者中下調的差異表達基因的富集分析結果。GO:基因本體論;BP:生物學過程;CC:細胞組分;MF:分子功能;RAGE:晚期糖基化終產物受體;TLR:Toll 樣受體;GeneRatio:基因比率
2.3.2 KEGG 富集分析
KEGG 通路富集分析結果顯示,上調的基因主要富集在多通路的代謝失調,尤其是癌癥中的轉錄失調、晝夜節律、煙酸和煙酰胺代謝、嘧啶代謝、FcεRI 信號通路。下調的基因主要富集在細胞外基質-受體相互作用、白細胞介素-17 信號通路、Toll 樣受體信號通路以及癌癥中的轉錄失調通路。見圖5。
圖5
差異表達基因的 KEGG 富集分析
a. 在抑郁癥患者中上調的差異表達基因的富集分析結果;b. 在抑郁癥患者中下調的差異表達基因的富集分析結果。KEGG:京都基因與基因組數據庫;ECM:細胞外基質;IL-17:白細胞介素-17;TLR:Toll 樣受體;GeneRatio:基因比率
2.4 單細胞測序分析
對 6 個獲驗證的差異表達基因(SALL2、AKAP12、GCSAML、CPA3、FCRL3 和 MS4A3)在不同細胞類型中的分布情況進行詳細的映射,除了 FCRL3 未在映射結果中觀察到外,其他 5 個差異表達基因主要在星形膠質細胞和神經細胞中顯著表達。見圖6。
圖6
GSE144136 數據集中重度抑郁癥患者差異表達基因的單細胞測序分析
a~e. 分別為
2.5 孟德爾隨機化分析
全部差異表達基因相關 SNP 的孟德爾隨機化分析結果表明,不同方法得到的 β 系數和標準誤差各異。MR Egger 方法的 β 系數為
考慮到較高的異質性,我們對 6 個與抑郁癥差異表達基因(SALL2、AKAP12、GCSAML、CPA3、FCRL3 和 MS4A3)相關的 SNP 進行了孟德爾隨機化分析,以探討這些基因在抑郁癥遺傳易感性中的作用。分析結果顯示,大多數基因與抑郁癥之間的關聯無統計學意義(P>0.05)。然而,FCRL3 基因的兩種分析方法(加權中位數法和逆方差加權法)表明該基因與抑郁癥之間的關聯有統計學意義(P=
2.6 藥物預測
8 個關鍵基因中,因 ENPP3 基因進行探針轉換時沒有找到其對應探針,因此只納入了 7 個基因進行 Cmap 藥物預測,通過對 Cmap 數據庫的結果進行篩選,前 10 個潛在的抑郁癥治療藥物分別是頭孢替安、駱駝蓬醇、坎那定、CP-863187、2-氨基苯磺酰胺、林可霉素、病毒唑、苯噻林、α-育亨賓、桿菌肽(表1)。
3 討論
本研究通過對抑郁癥患者和健康志愿者的基因表達進行差異分析,找到其潛在的致病基因,分別是 CPA3、HDC、FCER1A、IL3RA、ENPP3、PTGDR2、VTN、SPP1、SERPINE1 和 S100A8,其中 S100A8、FCER1A 在健康志愿者和抑郁癥患者中差異無統計學意義(P>0.05)。綜合目前的研究,Han 等[9]研究發現在肝癌和抑郁癥中 SERPINE1 基因表達具有差異,可能與細胞死亡或凋亡有關。有趣的是,本研究未發現 S100A8 基因在健康志愿者和抑郁癥患者之間存在差異表達,但現有研究顯示,S100A8 基因可能與抑郁癥患者刺激性行為有關[10]。S100A8 和 FCER1A 在本研究中未顯示出有統計學意義的差異,這可能指向抑郁癥病理機制的復雜性及個體間的異質性。此外,考慮到抑郁癥的多因素性質,未來研究中探索與這些基因相互作用的其他生物標志物,可能揭示更加細致的病理過程。
通過對抑郁癥差異表達的基因進行 GO、KEGG 富集分析,我們發現上調的基因在 GO 術語的 BP 中主要富集在生理節律的維持,CC 主要富集在各種細胞器上,MF 主要富集在各種蛋白結合,下調的基因在 GO 術語的 BP 中主要集中在炎癥反應中,CC 主要富集在各種細胞器中,MF 主要富集在各種受體的結合,由于抑郁癥與體內的生物化學反應相關,我們推測其可能與生理節律失調、炎癥反應有關。另外,KEGG 通路富集分析顯示晝夜節律紊亂明顯。另外,相關研究發現晝夜節律紊亂是抑郁癥的病理生理學基礎[11-12],這與我們的研究相一致。重要的是,在煙酸和煙酰胺代謝通路中,Ma 等[13]研究發現綠原酸可以調節該通路而發揮其抗抑郁作用。此外,Zhao 等[14]研究發現抑郁癥大學生的心率變異性與嘧啶代謝途徑有關。并且,中藥大黃素可能通過白細胞介素-17 信號通路發揮其抗抑郁的作用[15]。另外,Toll 樣受體信號通路可能與針灸發揮抗抑郁作用有關[16]。以上通路均與抑郁癥有關,因此,研究相關的靶向藥物可能對改善抑郁癥患者的癥狀提供依據。
在本研究中,FCRL3 基因在抑郁癥患者與健康對照組之間表現出差異性表達。鑒于近年來研究發現抑郁癥可能與身體炎癥狀態相關,我們猜想 FCRL3 可能通過影響炎癥反應途徑,間接參與到抑郁癥的發病機制中[17]。雖然在單細胞測序數據分析中未檢測到 FCRL3 的特定表達模式,這可能由于樣本的選擇或細胞類型的限制。然而,通過孟德爾隨機化分析,我們發現與 FCRL3 相關的特定 SNP 在抑郁癥的遺傳易感性中顯示出明顯的相關性,為 FCRL3 在抑郁癥中可能扮演的角色提供了初步證據。這一點強調了 FCRL3 作為一個潛在的雙重功能靶點,在免疫調節和精神障礙發病機制中的重要性。
有研究通過使用 Cmap 數據庫對潛在治療抑郁癥藥物進行預測,發現駱駝蓬醇中的吲哚生物堿與神經遞質血清素有關[18],它可以通過 γ-氨基丁酸受體發揮抗抑郁的作用[19]。令人詫異的是,結合當下的研究,我們發現在丙型病毒性肝炎患者中利巴韋林和聚乙二醇干擾素可誘發抑郁發作[20]。其余藥物均未發現其用于治療抑郁癥。因此,本研究可為藥物生產企業研發控制抑郁癥藥物提供參考。
綜上所述,本研究鑒定了與抑郁癥相關的差異表達基因,并通過富集分析揭示了其可能涉及的致病通路。進一步分析中,我們識別出抑郁癥的 10 個潛在關鍵基因,其中 8 個基因(CPA3、HDC、IL3RA、ENPP3、PTGDR2、VTN、SPP1 和 SERPINE1)在健康志愿者和抑郁癥患者之間的表達水平存在差異,并基于這些基因進行了藥物預測,得到了若干可能用于抑郁癥治療的候選藥物。此外,本研究結果提示晝夜節律紊亂可能是抑郁癥發病的重要機制之一。整體而言,本研究為抑郁癥的病理機制和潛在治療靶點提供了新的見解,有望改善抑郁癥患者的預后,提高生活質量,并減輕家庭和社會的負擔。
利益沖突:所有作者聲明不存在利益沖突。
請掃描本文首頁二維碼后在“補充材料”中下載查看附件。