引用本文: 王美儀, 彭敏芝, 邵詠賢, 林云婷, 蔡燕娜, 徐佳南, 劉麗. 11 例楓糖尿癥患兒臨床特征、代謝標志物及遺傳異質性分析. 華西醫學, 2025, 40(1): 23-28. doi: 10.7507/1002-0179.202403293 復制
版權信息: ?四川大學華西醫院華西期刊社《華西醫學》版權所有,未經授權不得轉載、改編
楓糖尿癥(maple syrup urine disease, MSUD)是一種罕見的常染色體隱性遺傳病,由 BCKDHA、BCKDHB、DBT、DLD 或 BCAT2 基因變異,導致支鏈 α-酮酸脫氫酶活性下降引起[1-2]。該病在我國的發病率約為 1/
1 對象與方法
1.1 研究對象
回顧收集 2011 年 1 月—2016 年 10 月廣州醫科大學附屬婦女兒童醫療中心遺傳與內分泌科收治的 11 例 MSUD 患兒的臨床資料。依據患兒的臨床表現和對藥物的反應,將患兒分為經典型組(n=6)和中間型/硫胺素有效型組(n=5)。本研究獲得廣州醫科大學附屬婦女兒童醫療中心倫理委員會批準,資料采集獲得患兒監護人的知情同意。
1.2 方法
1.2.1 臨床表型研究
收集患兒發病年齡、臨床診斷分型、首發臨床癥狀、影像學檢測結果、預后情況等基本臨床資料。
1.2.2 體液生化檢測結果
比較兩組患兒首診時實驗室常規生化檢查[包括丙氨酸轉氨酶(alanine aminotransferase, ALT)、天冬氨酸轉氨酶(aspartate aminotransferase, AST)、總膽汁酸、γ-谷氨酰轉移酶(gamma glutamyltransferase, γ-GT)、球蛋白、乳酸等]及血漿支鏈氨基酸(包括亮氨酸、異亮氨酸、纈氨酸)分析結果。
1.2.3 測序分析 BCKDHA、BCKDHB 和 DBT 基因
所有患兒均接受 3 個與 MUSD 相關的基因測序。乙二胺四乙酸抗凝管采集 2 mL 靜脈血,用 DNA 提取試劑盒(德國 Qiagen 公司)提取基因組 DNA。BCKDHA(NG_013004)、BCKDHB(NG_009775)和 DBT(NG_011852)基因的標準核酸序列從美國國家生物技術信息中心的 GenBank 數據庫下載,采用 Primer Premier 5 設計聚合酶鏈反應擴增引物。擴增產物采用 Sanger 測序法測序。
1.2.4 新變異的致病性預測和蛋白質結構分析
在人類基因突變數據庫上搜索檢測到的變異。對于尚未報道的新變異,使用 PolyPhen2 和 Mutation Taster 預測致病性。從 Uniprot 數據庫下載 3 個基因的標準氨基酸序列,用 Bioedit 對新變異進行多序列比對和保守性分析。在 PDB 數據庫下載蛋白的三維結構,采用 PyMOL 軟件分析變異引起的蛋白結構變化。
1.3 統計學方法
使用 SPSS 25.0 軟件進行統計。計量資料采用 Shapiro-Wilk 法進行正態性檢驗,服從正態分布的用均數±標準差表示,否則用中位數(下四分位數,上四分位數)表示,組間比較均用 Mann-Whitney U 檢驗。計數資料采用例數表示,組間比較采用 Fisher 確切概率法。分析兩連續變量的線性關系時,服從雙變量正態分布的采用 Pearson 相關系數,否則采用 Spearman 相關系數。雙側檢驗水準 α=0.05。
2 結果
2.1 主要臨床特征
11 例患兒來自 11 個無血緣關系的家庭,父母均體健,非近親結婚。患兒中男 8 例,女 3 例;多在新生兒期起病,最晚 3 歲發病;主要表現為反應差、喂養困難和運動發育遲緩(表1)。部分患兒抽搐后短時間進展至昏迷、呼吸困難或呼吸暫停。6 例患兒在起病 15~20 d 內夭折。5 例患兒表現為肌張力低,1 例肌張力高伴四肢間歇性僵直;3 例新生兒原始反射(包括吸吮反射、握持反射、踏步反射等)和/或雙膝反射弱或未引出。另有 2 例男性患兒存在雙側聽覺傳導異常,其中 1 例伴有壞死性小腸結腸炎。患兒頭顱 MRI 檢查主要提示雙側基底節異常信號,其余受累部位各患兒間有差異,主要包括雙側小腦齒狀核、雙側大腦腳、雙側腦室、背側丘腦等,受累部位特征性表現為 T1 加權像低信號,T2 加權像、T2 加權-液體衰減反轉恢復及磁共振彌散加權成像高信號。
2.2 MSUD 患兒生化代謝物與臨床表型的關系
經典型組男 5 例,女 1 例;年齡 1~12 d。中間型/硫胺素有效型組男 3 例,女 2 例;年齡 8 個月~3 歲。兩組性別分布差異無統計學意義(P=0.545);中間型/硫胺素有效型組年齡更大,差異有統計學意義(Z=–2.751,P=0.006)。
與中間型/硫胺素有效型組相比,經典型組患兒血清 γ-GT 水平升高(P=0.004),且高出正常值上限 2.7 倍;球蛋白(P=0.018)和乳酸(P=0.030)偏低;兩組 ALT、AST、總膽汁酸等肝膽功能相關指標組間差異無統計學意義(P>0.05);經典型組患兒亮氨酸水平升高(P=0.004),且高出正常值上限 19 倍;兩組異亮氨酸及纈氨酸組間差異無統計學意義(P>0.05)。見表2。
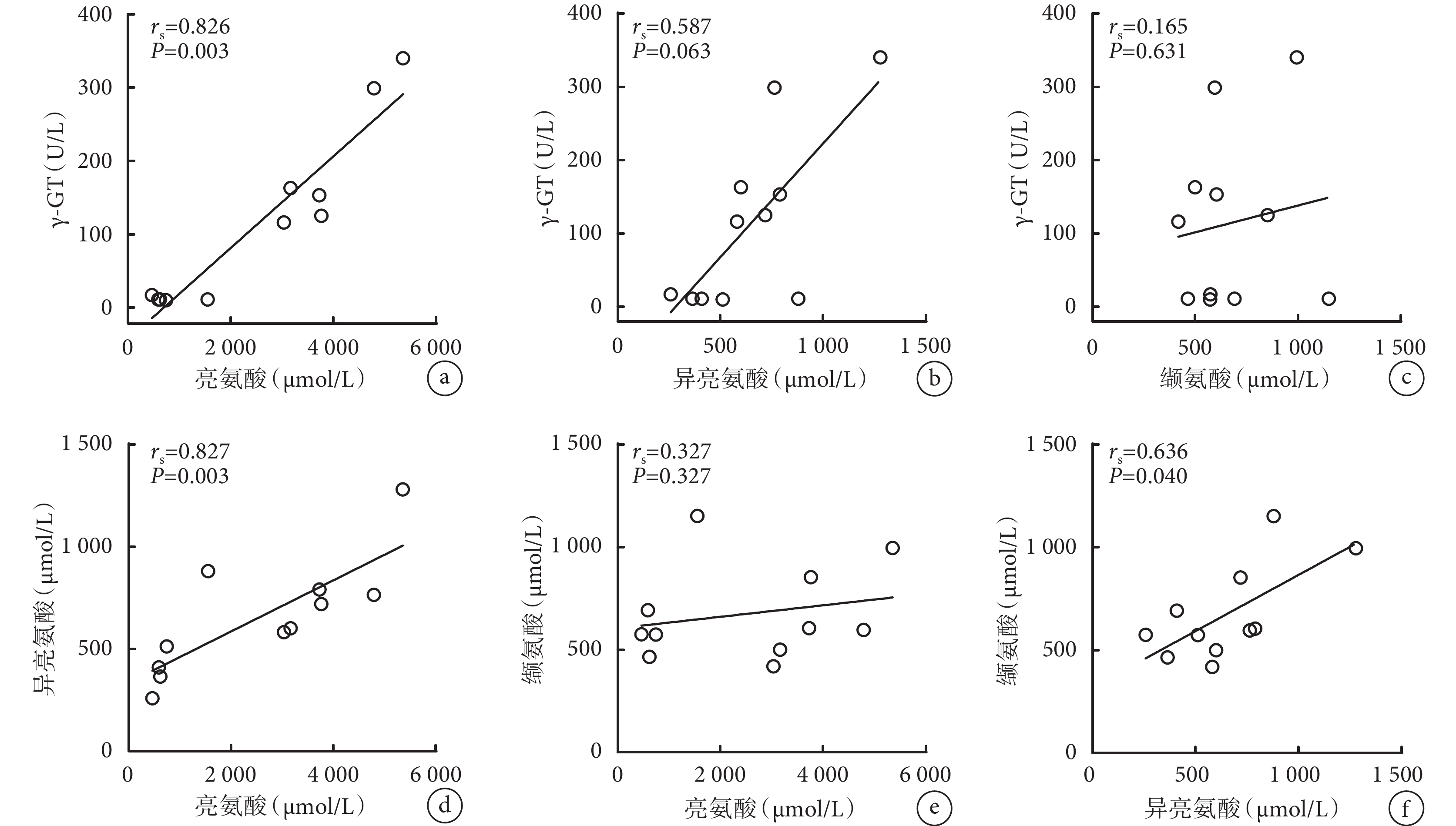
相關性分析發現,亮氨酸與 γ-GT(rs=0.826,P=0.003)、亮氨酸與異亮氨酸(rs=0.827,P=0.003)以及異亮氨酸與纈氨酸(rs=0.636,P=0.040)存在明顯正相關性(圖1)。
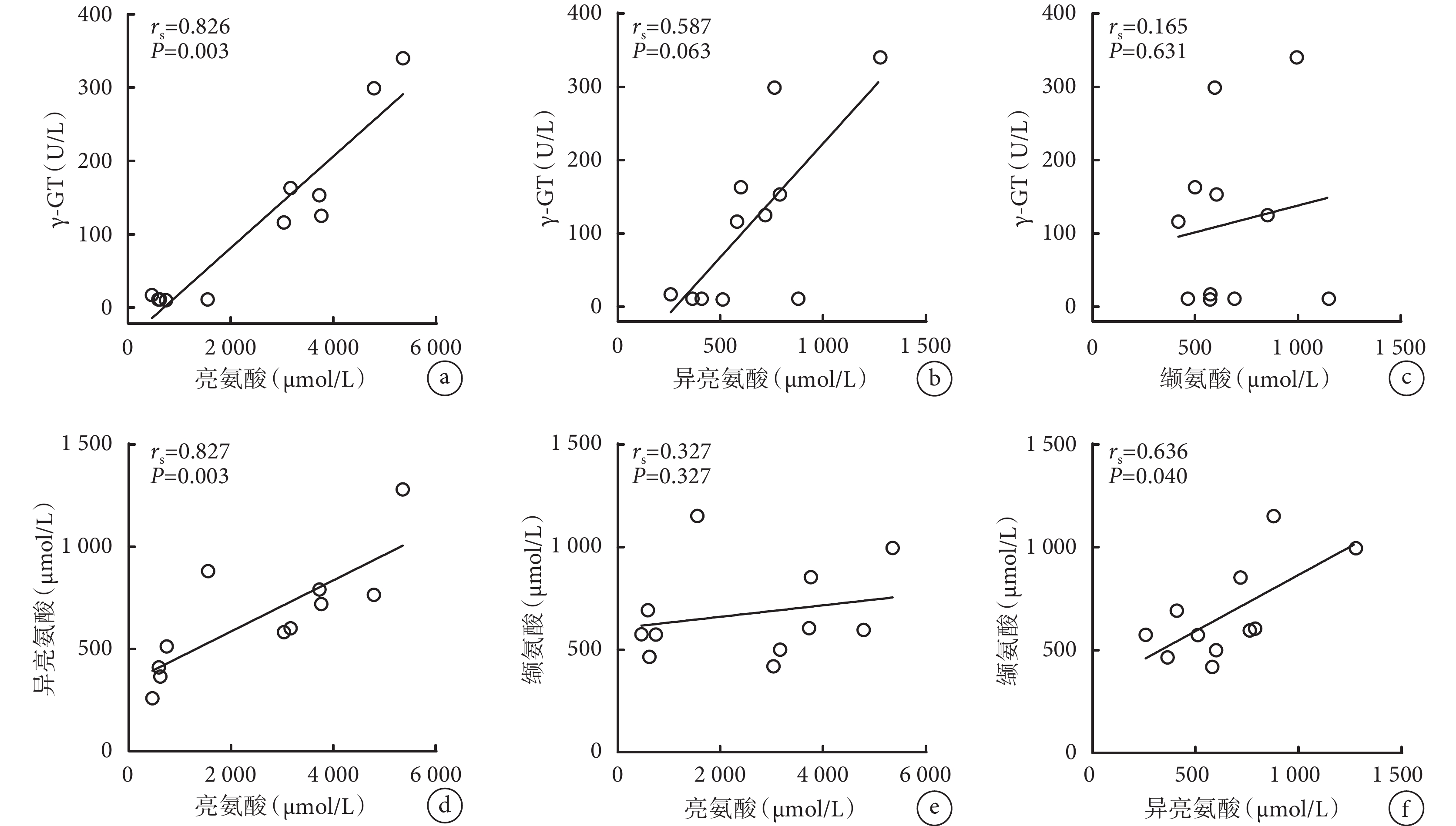 圖1
γ-GT、亮氨酸、異亮氨酸和纈氨酸的相關性
圖1
γ-GT、亮氨酸、異亮氨酸和纈氨酸的相關性
γ-GT:γ-谷氨酰轉移酶
2.3 MSUD 相關基因變異分布特征
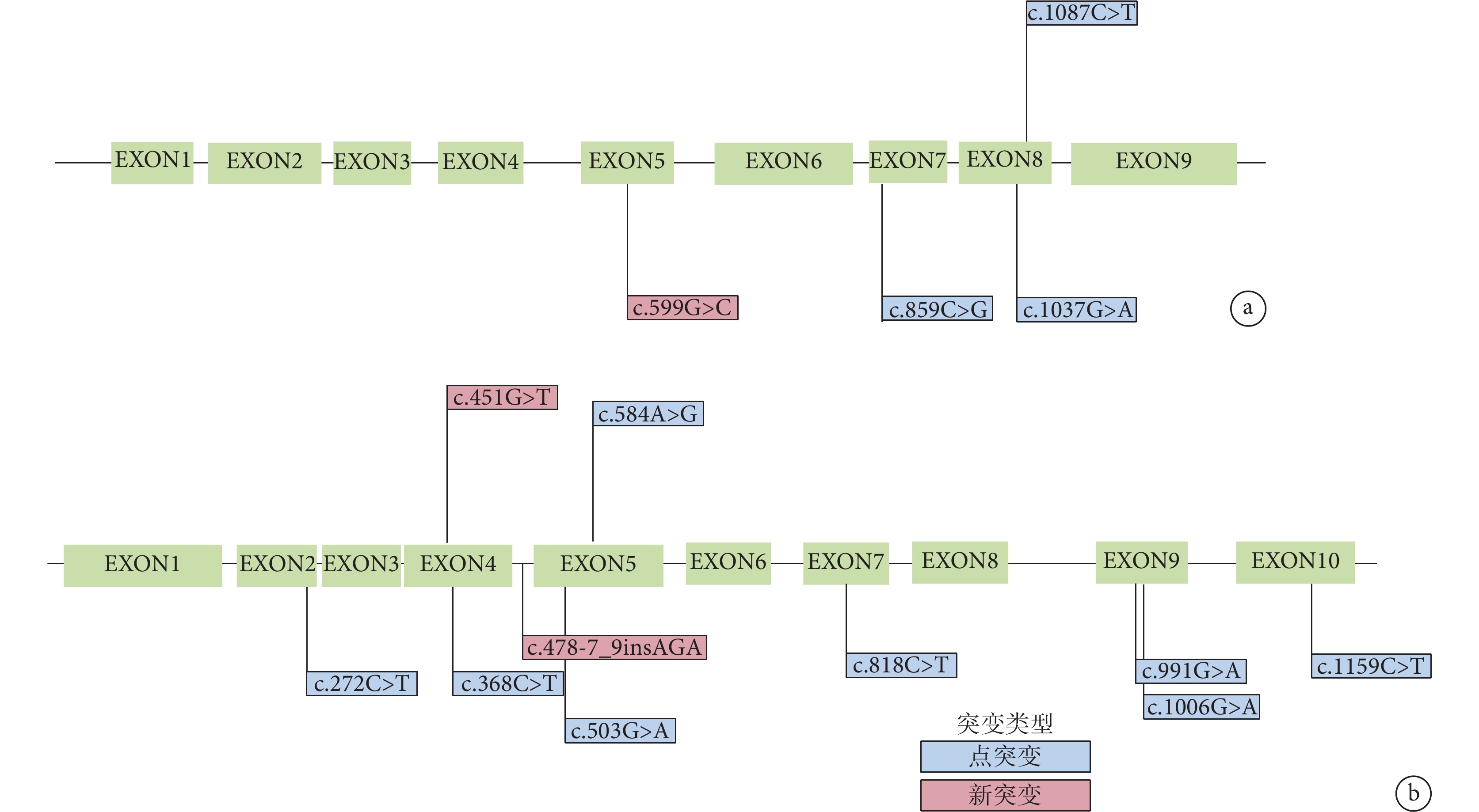
11 例患兒均進行 MSUD 相關的基因分析。結果顯示 11 例患兒分別攜帶有 BCKDHA(n=4)和 BCKDHB(n=7)基因變異。共檢測出 15 個變異點,其中 11 個為已知致病變異;4 個為新變異,其中 3 個經 polyphen-2 和 MutationTaster 預測可能致病,另 1 個 BCKDHB 基因插入變異在病例 1 中發現,MutationTaster 預測為可能無害的多態性變異。病例 5 存在 BCKDHB 基因 c.451G>T 已知純合致病變異和 DBT 基因 c.168_170delAAC 雜合變異,該變異 MutationTaster 預測為可能致病(表3、圖2)。
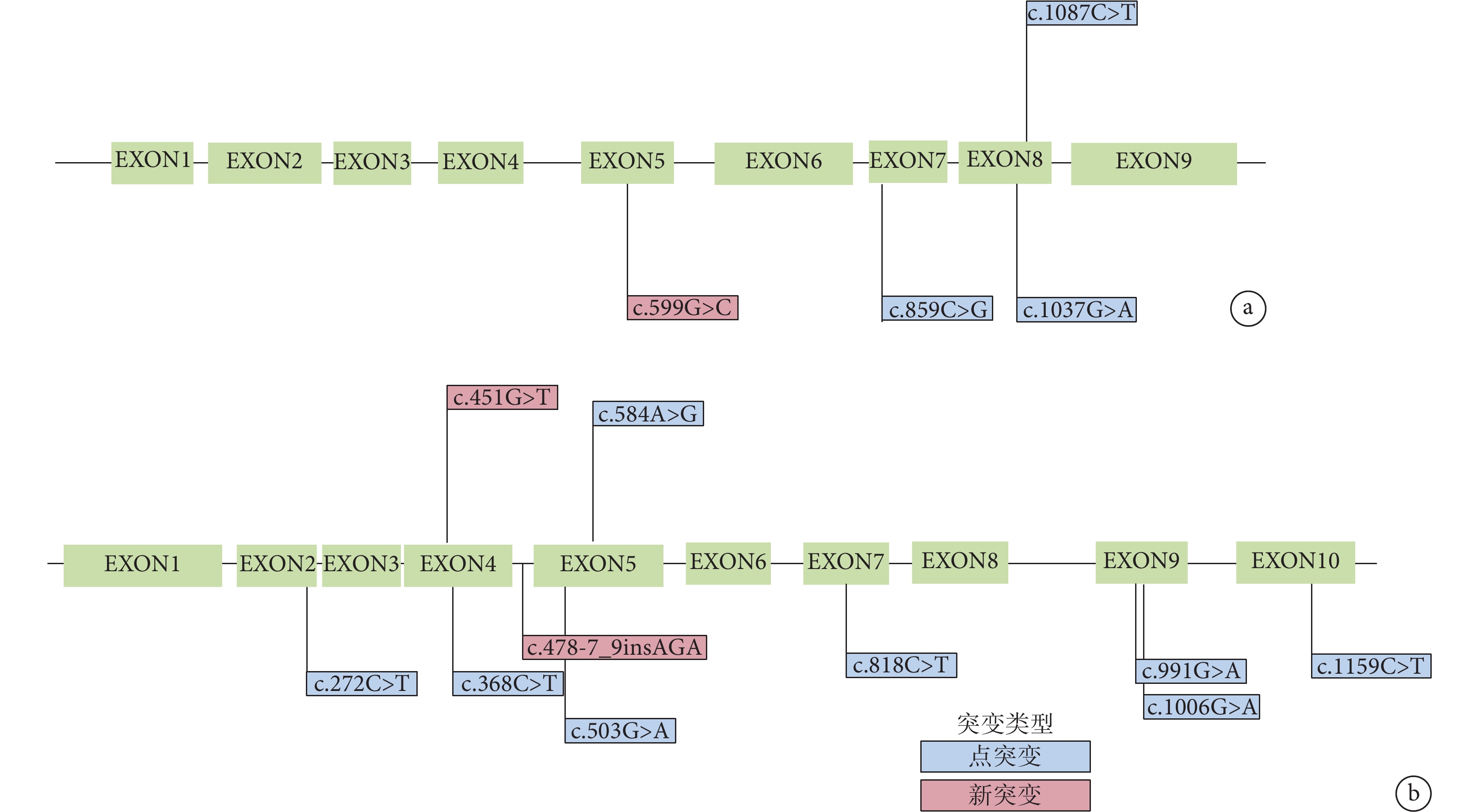 圖2
BCKDHA 和 BCKHDB 基因變異分布
圖2
BCKDHA 和 BCKHDB 基因變異分布
a.
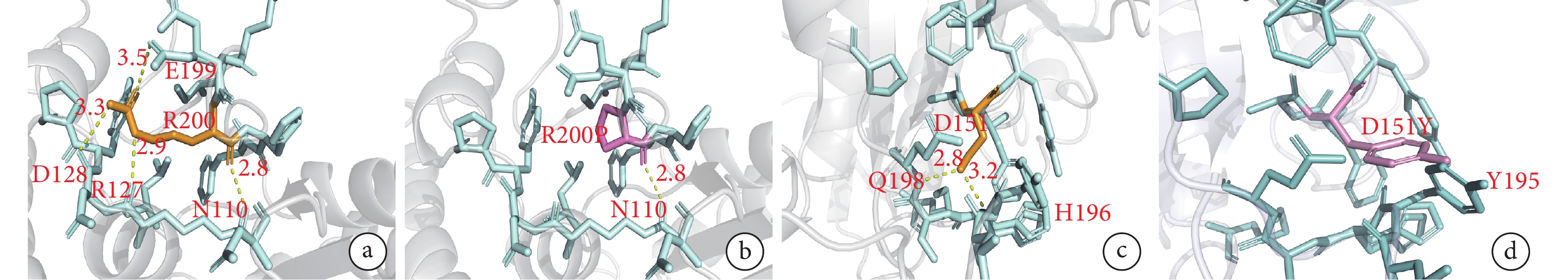
BCKDHA 基因 c.599G>C 雜合變異和 BCKDHB 基因 c.451G>T 雜合變異這兩個新變異在 7 個物種中高度保守(附件圖 1),polyphen-2 和 MutationTaster 預測有較強致病性。如圖3a、3b 所示,BCKDHA 基因第 200 位精氨酸位于蛋白二級結構的一個 β 轉角上,與其他氨基酸形成 4 個氫鍵。變異為脯氨酸后,脯氨酸僅與其他氨基酸形成 1 個氫鍵,影響了 BCKDHA 基因編碼的 α-酮酸脫氫酶 E1α 亞基穩定性,進而影響蛋白質的結構與功能。在 BCKDHB 基因中,151 位的天冬氨酸位于二級結構的 α-螺旋上,與 198 位谷氨酰胺和 196 位組氨酸分別形成氫鍵。變異為酪氨酸后,兩個氫鍵消失,同時 151 位酪氨酸上苯環的羥基與 195 位酪氨酸的苯環形成 p-pi 共軛,p-pi 共軛的形成會增加苯環上的電子云密度并激活苯環。苯環激活后,對氨基酸穩定性產生影響,蛋白質容易降解(圖3c、3d)。
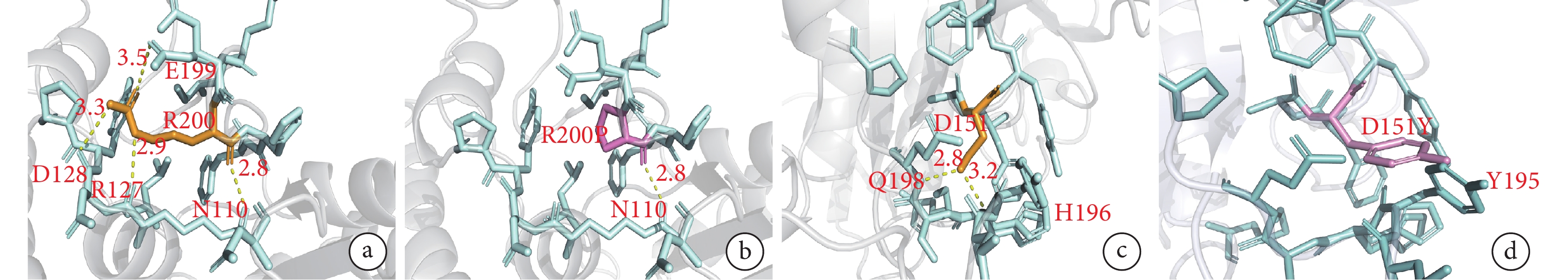 圖3
BCKDHA 基因和 BCKDHB 基因兩個新變異的三維晶體結構分析
圖3
BCKDHA 基因和 BCKDHB 基因兩個新變異的三維晶體結構分析
a、b.
3 討論
本研究回顧并分析 2011 年—2016 年就診于廣州醫科大學附屬婦女兒童醫療中心遺傳與內分泌科的 11 例 MSUD 患兒的臨床資料,比較分析不同表型患兒生化代謝標志物差異,探討基因型和表型之間的關系。本研究發現,除支鏈氨基酸外,血清 γ-GT 水平與 MSUD 患兒的臨床表型密切相關;BCKDHB 基因變異的 MSUD 患兒臨床癥狀嚴重,預后差。因此 MSUD 的疾病預后與支鏈氨基酸、γ-GT 和基因型三者均密切相關。
本研究發現經典型患兒血清 γ-GT 水平特征性增高,該結果與已發表文章相符[6-7]。γ-GT 水平與亮氨酸有顯著正相關關系。血清 γ-GT 水平升高在膽汁淤積[8]和肝功能異常[9]患兒中最為常見。然而 MSUD 患兒 ALT、AST 和總膽汁酸水平等肝膽功能相關指標均在正常范圍。已有研究發現血清 γ-GT 升高可損傷細胞膜,導致有毒過渡金屬釋放,引起氧化應激和 DNA 損傷[10],另外支鏈氨基酸和支鏈 α-酮酸單獨作用或協同作用可增加自由基的產生,引起 DNA 損傷[11]。γ-GT 主要參與 γ 谷氨酰循環中谷胱甘肽的分解代謝,已有研究發現 MSUD 小鼠模型大腦谷胱甘肽減少[12],另外 MSUD 患者大腦中的甲硫氨酸和谷氨酰胺降低,這兩種物質對谷胱甘肽的合成至關重要[13-14]。因此 MSUD 患者血清 γ-GT 升高可能與谷胱甘肽及其相關代謝物的合成與分解相關,具體機制需深入研究。γ-GT 與支鏈氨基酸的關系在 MSUD 的發病機制中的作用,也有待進一步研究。
當前尚無報道明確指出 MSUD 基因型與表型之間的關系。本研究發現 6 例經典型 MSUD 患兒均存在 BCKDHB 基因變異,且這些患兒的變異位點廣泛分布在 BCKDHB 基因各個外顯子區域,無明顯分布規律。已有研究也發現在亞洲,BCKDHB 基因變異的患兒為經典型 MSUD 的占比較大[15-16]。而以白種人為主的歐洲、地中海和埃及,多數 BCKDHB 基因變異患者臨床表型較輕,預后相對好[17-18]。通過比較本研究結果與已發表文章結果[14],我們發現同一個 BCKDHB 基因變異在不同人群的臨床表型輕重是有差異的。這可能是由于遺傳背景差異和環境因素的影響導致同一基因變異在不同人群中的表現度不同[19]。在本研究中,病例 1 僅發現 1 個 BCKDHB 基因的致病變異,但其臨床表現、影像學檢查和實驗室檢查與 MSUD 的診斷高度一致。不排除患兒還存在其他與 MSUD 相關的基因變異。另外,盡管該患兒的 BCKDHB 基因另一個位于內含子區域的插入變異尚無報道,且預測為可能無害變異,但是不排除今后還有類似的 MSUD 病例也存在該變異,有更多的證據支持該插入變異為致病變異。無義變異導致終止密碼子提前出現可能會嚴重影響蛋白質活性[17, 20],導致患兒表現為經典型 MSUD。本研究中的病例 3 攜帶無義變異 p.Arg387X,為經典型 MSUD,其他已報道的 MSUD 病例亦如此[21]。
綜上所述,本研究分析了 11 例 MSUD 患兒臨床特征、體液中生化代謝標志物濃度變化,并探討了臨床表型和基因變異的相關性,結果表明 MSUD 患兒的疾病發展及預后與血 γ-GT 水平、血漿支鏈氨基酸濃度和基因型密切相關。建議在 MSUD 的診治中,將 γ-GT 作為 MSUD 實驗室檢查的重要生化指標之一。
利益沖突:所有作者聲明不存在利益沖突。
請掃描本文首頁二維碼后在“補充材料”中下載查看附件。
楓糖尿癥(maple syrup urine disease, MSUD)是一種罕見的常染色體隱性遺傳病,由 BCKDHA、BCKDHB、DBT、DLD 或 BCAT2 基因變異,導致支鏈 α-酮酸脫氫酶活性下降引起[1-2]。該病在我國的發病率約為 1/
1 對象與方法
1.1 研究對象
回顧收集 2011 年 1 月—2016 年 10 月廣州醫科大學附屬婦女兒童醫療中心遺傳與內分泌科收治的 11 例 MSUD 患兒的臨床資料。依據患兒的臨床表現和對藥物的反應,將患兒分為經典型組(n=6)和中間型/硫胺素有效型組(n=5)。本研究獲得廣州醫科大學附屬婦女兒童醫療中心倫理委員會批準,資料采集獲得患兒監護人的知情同意。
1.2 方法
1.2.1 臨床表型研究
收集患兒發病年齡、臨床診斷分型、首發臨床癥狀、影像學檢測結果、預后情況等基本臨床資料。
1.2.2 體液生化檢測結果
比較兩組患兒首診時實驗室常規生化檢查[包括丙氨酸轉氨酶(alanine aminotransferase, ALT)、天冬氨酸轉氨酶(aspartate aminotransferase, AST)、總膽汁酸、γ-谷氨酰轉移酶(gamma glutamyltransferase, γ-GT)、球蛋白、乳酸等]及血漿支鏈氨基酸(包括亮氨酸、異亮氨酸、纈氨酸)分析結果。
1.2.3 測序分析 BCKDHA、BCKDHB 和 DBT 基因
所有患兒均接受 3 個與 MUSD 相關的基因測序。乙二胺四乙酸抗凝管采集 2 mL 靜脈血,用 DNA 提取試劑盒(德國 Qiagen 公司)提取基因組 DNA。BCKDHA(NG_013004)、BCKDHB(NG_009775)和 DBT(NG_011852)基因的標準核酸序列從美國國家生物技術信息中心的 GenBank 數據庫下載,采用 Primer Premier 5 設計聚合酶鏈反應擴增引物。擴增產物采用 Sanger 測序法測序。
1.2.4 新變異的致病性預測和蛋白質結構分析
在人類基因突變數據庫上搜索檢測到的變異。對于尚未報道的新變異,使用 PolyPhen2 和 Mutation Taster 預測致病性。從 Uniprot 數據庫下載 3 個基因的標準氨基酸序列,用 Bioedit 對新變異進行多序列比對和保守性分析。在 PDB 數據庫下載蛋白的三維結構,采用 PyMOL 軟件分析變異引起的蛋白結構變化。
1.3 統計學方法
使用 SPSS 25.0 軟件進行統計。計量資料采用 Shapiro-Wilk 法進行正態性檢驗,服從正態分布的用均數±標準差表示,否則用中位數(下四分位數,上四分位數)表示,組間比較均用 Mann-Whitney U 檢驗。計數資料采用例數表示,組間比較采用 Fisher 確切概率法。分析兩連續變量的線性關系時,服從雙變量正態分布的采用 Pearson 相關系數,否則采用 Spearman 相關系數。雙側檢驗水準 α=0.05。
2 結果
2.1 主要臨床特征
11 例患兒來自 11 個無血緣關系的家庭,父母均體健,非近親結婚。患兒中男 8 例,女 3 例;多在新生兒期起病,最晚 3 歲發病;主要表現為反應差、喂養困難和運動發育遲緩(表1)。部分患兒抽搐后短時間進展至昏迷、呼吸困難或呼吸暫停。6 例患兒在起病 15~20 d 內夭折。5 例患兒表現為肌張力低,1 例肌張力高伴四肢間歇性僵直;3 例新生兒原始反射(包括吸吮反射、握持反射、踏步反射等)和/或雙膝反射弱或未引出。另有 2 例男性患兒存在雙側聽覺傳導異常,其中 1 例伴有壞死性小腸結腸炎。患兒頭顱 MRI 檢查主要提示雙側基底節異常信號,其余受累部位各患兒間有差異,主要包括雙側小腦齒狀核、雙側大腦腳、雙側腦室、背側丘腦等,受累部位特征性表現為 T1 加權像低信號,T2 加權像、T2 加權-液體衰減反轉恢復及磁共振彌散加權成像高信號。
2.2 MSUD 患兒生化代謝物與臨床表型的關系
經典型組男 5 例,女 1 例;年齡 1~12 d。中間型/硫胺素有效型組男 3 例,女 2 例;年齡 8 個月~3 歲。兩組性別分布差異無統計學意義(P=0.545);中間型/硫胺素有效型組年齡更大,差異有統計學意義(Z=–2.751,P=0.006)。
與中間型/硫胺素有效型組相比,經典型組患兒血清 γ-GT 水平升高(P=0.004),且高出正常值上限 2.7 倍;球蛋白(P=0.018)和乳酸(P=0.030)偏低;兩組 ALT、AST、總膽汁酸等肝膽功能相關指標組間差異無統計學意義(P>0.05);經典型組患兒亮氨酸水平升高(P=0.004),且高出正常值上限 19 倍;兩組異亮氨酸及纈氨酸組間差異無統計學意義(P>0.05)。見表2。
相關性分析發現,亮氨酸與 γ-GT(rs=0.826,P=0.003)、亮氨酸與異亮氨酸(rs=0.827,P=0.003)以及異亮氨酸與纈氨酸(rs=0.636,P=0.040)存在明顯正相關性(圖1)。
圖1
γ-GT、亮氨酸、異亮氨酸和纈氨酸的相關性
γ-GT:γ-谷氨酰轉移酶
2.3 MSUD 相關基因變異分布特征
11 例患兒均進行 MSUD 相關的基因分析。結果顯示 11 例患兒分別攜帶有 BCKDHA(n=4)和 BCKDHB(n=7)基因變異。共檢測出 15 個變異點,其中 11 個為已知致病變異;4 個為新變異,其中 3 個經 polyphen-2 和 MutationTaster 預測可能致病,另 1 個 BCKDHB 基因插入變異在病例 1 中發現,MutationTaster 預測為可能無害的多態性變異。病例 5 存在 BCKDHB 基因 c.451G>T 已知純合致病變異和 DBT 基因 c.168_170delAAC 雜合變異,該變異 MutationTaster 預測為可能致病(表3、圖2)。
圖2
BCKDHA 和 BCKHDB 基因變異分布
a.
BCKDHA 基因 c.599G>C 雜合變異和 BCKDHB 基因 c.451G>T 雜合變異這兩個新變異在 7 個物種中高度保守(附件圖 1),polyphen-2 和 MutationTaster 預測有較強致病性。如圖3a、3b 所示,BCKDHA 基因第 200 位精氨酸位于蛋白二級結構的一個 β 轉角上,與其他氨基酸形成 4 個氫鍵。變異為脯氨酸后,脯氨酸僅與其他氨基酸形成 1 個氫鍵,影響了 BCKDHA 基因編碼的 α-酮酸脫氫酶 E1α 亞基穩定性,進而影響蛋白質的結構與功能。在 BCKDHB 基因中,151 位的天冬氨酸位于二級結構的 α-螺旋上,與 198 位谷氨酰胺和 196 位組氨酸分別形成氫鍵。變異為酪氨酸后,兩個氫鍵消失,同時 151 位酪氨酸上苯環的羥基與 195 位酪氨酸的苯環形成 p-pi 共軛,p-pi 共軛的形成會增加苯環上的電子云密度并激活苯環。苯環激活后,對氨基酸穩定性產生影響,蛋白質容易降解(圖3c、3d)。
圖3
BCKDHA 基因和 BCKDHB 基因兩個新變異的三維晶體結構分析
a、b.
3 討論
本研究回顧并分析 2011 年—2016 年就診于廣州醫科大學附屬婦女兒童醫療中心遺傳與內分泌科的 11 例 MSUD 患兒的臨床資料,比較分析不同表型患兒生化代謝標志物差異,探討基因型和表型之間的關系。本研究發現,除支鏈氨基酸外,血清 γ-GT 水平與 MSUD 患兒的臨床表型密切相關;BCKDHB 基因變異的 MSUD 患兒臨床癥狀嚴重,預后差。因此 MSUD 的疾病預后與支鏈氨基酸、γ-GT 和基因型三者均密切相關。
本研究發現經典型患兒血清 γ-GT 水平特征性增高,該結果與已發表文章相符[6-7]。γ-GT 水平與亮氨酸有顯著正相關關系。血清 γ-GT 水平升高在膽汁淤積[8]和肝功能異常[9]患兒中最為常見。然而 MSUD 患兒 ALT、AST 和總膽汁酸水平等肝膽功能相關指標均在正常范圍。已有研究發現血清 γ-GT 升高可損傷細胞膜,導致有毒過渡金屬釋放,引起氧化應激和 DNA 損傷[10],另外支鏈氨基酸和支鏈 α-酮酸單獨作用或協同作用可增加自由基的產生,引起 DNA 損傷[11]。γ-GT 主要參與 γ 谷氨酰循環中谷胱甘肽的分解代謝,已有研究發現 MSUD 小鼠模型大腦谷胱甘肽減少[12],另外 MSUD 患者大腦中的甲硫氨酸和谷氨酰胺降低,這兩種物質對谷胱甘肽的合成至關重要[13-14]。因此 MSUD 患者血清 γ-GT 升高可能與谷胱甘肽及其相關代謝物的合成與分解相關,具體機制需深入研究。γ-GT 與支鏈氨基酸的關系在 MSUD 的發病機制中的作用,也有待進一步研究。
當前尚無報道明確指出 MSUD 基因型與表型之間的關系。本研究發現 6 例經典型 MSUD 患兒均存在 BCKDHB 基因變異,且這些患兒的變異位點廣泛分布在 BCKDHB 基因各個外顯子區域,無明顯分布規律。已有研究也發現在亞洲,BCKDHB 基因變異的患兒為經典型 MSUD 的占比較大[15-16]。而以白種人為主的歐洲、地中海和埃及,多數 BCKDHB 基因變異患者臨床表型較輕,預后相對好[17-18]。通過比較本研究結果與已發表文章結果[14],我們發現同一個 BCKDHB 基因變異在不同人群的臨床表型輕重是有差異的。這可能是由于遺傳背景差異和環境因素的影響導致同一基因變異在不同人群中的表現度不同[19]。在本研究中,病例 1 僅發現 1 個 BCKDHB 基因的致病變異,但其臨床表現、影像學檢查和實驗室檢查與 MSUD 的診斷高度一致。不排除患兒還存在其他與 MSUD 相關的基因變異。另外,盡管該患兒的 BCKDHB 基因另一個位于內含子區域的插入變異尚無報道,且預測為可能無害變異,但是不排除今后還有類似的 MSUD 病例也存在該變異,有更多的證據支持該插入變異為致病變異。無義變異導致終止密碼子提前出現可能會嚴重影響蛋白質活性[17, 20],導致患兒表現為經典型 MSUD。本研究中的病例 3 攜帶無義變異 p.Arg387X,為經典型 MSUD,其他已報道的 MSUD 病例亦如此[21]。
綜上所述,本研究分析了 11 例 MSUD 患兒臨床特征、體液中生化代謝標志物濃度變化,并探討了臨床表型和基因變異的相關性,結果表明 MSUD 患兒的疾病發展及預后與血 γ-GT 水平、血漿支鏈氨基酸濃度和基因型密切相關。建議在 MSUD 的診治中,將 γ-GT 作為 MSUD 實驗室檢查的重要生化指標之一。
利益沖突:所有作者聲明不存在利益沖突。
請掃描本文首頁二維碼后在“補充材料”中下載查看附件。