引用本文: 崔嚴奇, 楊鯨蓉, 倪琳, 連鐸煌, 葉仕新, 廖毅, 張錦燦, 曾志勇. 基于機器學習算法構建浸潤性肺腺癌預后預測模型. 中國胸心血管外科臨床雜志, 2025, 32(1): 80-86. doi: 10.7507/1007-4848.202303059 復制
版權信息: ?四川大學華西醫院華西期刊社《中國胸心血管外科臨床雜志》版權所有,未經授權不得轉載、改編
在全球范圍內癌癥相關死亡中,肺癌仍是其主要原因[1]。肺癌主要包括兩種組織學類型:非小細胞肺癌(non-small cell lung cancer,NSCLC)和小細胞肺癌,NSCLC約占所有肺癌的80%。其中,NSCLC的主要組織學亞型是肺腺癌(lung adenocarcinoma,LUAD),約占肺癌發病率的40%。根據2011年國際肺癌研究協會(International Association for the Study of Lung Cancer,IASLC)/美國胸科學會(American Thoracic Society,ATS)/歐洲呼吸學會(European Respiratory Society,ERS)和2015年世界衛生組織(World Health Organization,WHO)肺腫瘤分類的組織學分型,浸潤性肺腺癌(invasive lung adenocarcinoma,IAC)主要分為5種病理亞型:貼壁型、腺泡型、乳頭型、微乳頭型以及實體型,并基于其最主要的生長亞型進行級別劃分:低級別以貼壁為主;中級別以腺泡或乳頭為主;高級別以實體或微乳頭為主;級別越高,侵襲性越高,患者預后越差[2-3]。目前,肺癌的治療主要依賴于組織類型和臨床分期,但由于IAC的高度異質性,即使是同樣級別組織類型和臨床分期的IAC患者預后也不同。近年來,高通量技術的進步使醫學研究人員能夠從基因層面發現癌癥中的遺傳改變,并被廣泛用于篩選潛在的腫瘤生物標志物[4-5]。同時,機器學習算法已被引入且應用于遺傳和基因組數據,并從生物醫學數據集中,通過構建預測模型來預測臨床結果。因此基于高通量測序數據來檢測IAC不同級別病理亞型的基因表達情況,以及結合生物信息學方法系統分析相關差異表達基因及其調控機制,然后運用機器學習算法構建臨床風險預后預測模型,對LUAD患者進行個體化、精準化治療具有重要意義。
在本研究中,我們篩選出與LUAD患者預后相關的特征基因,然后構建基于基因特征的風險預測模型,預測患者的生存狀況,為LUAD患者的個體化診療提供參考。
1 資料與方法
1.1 獲取數據
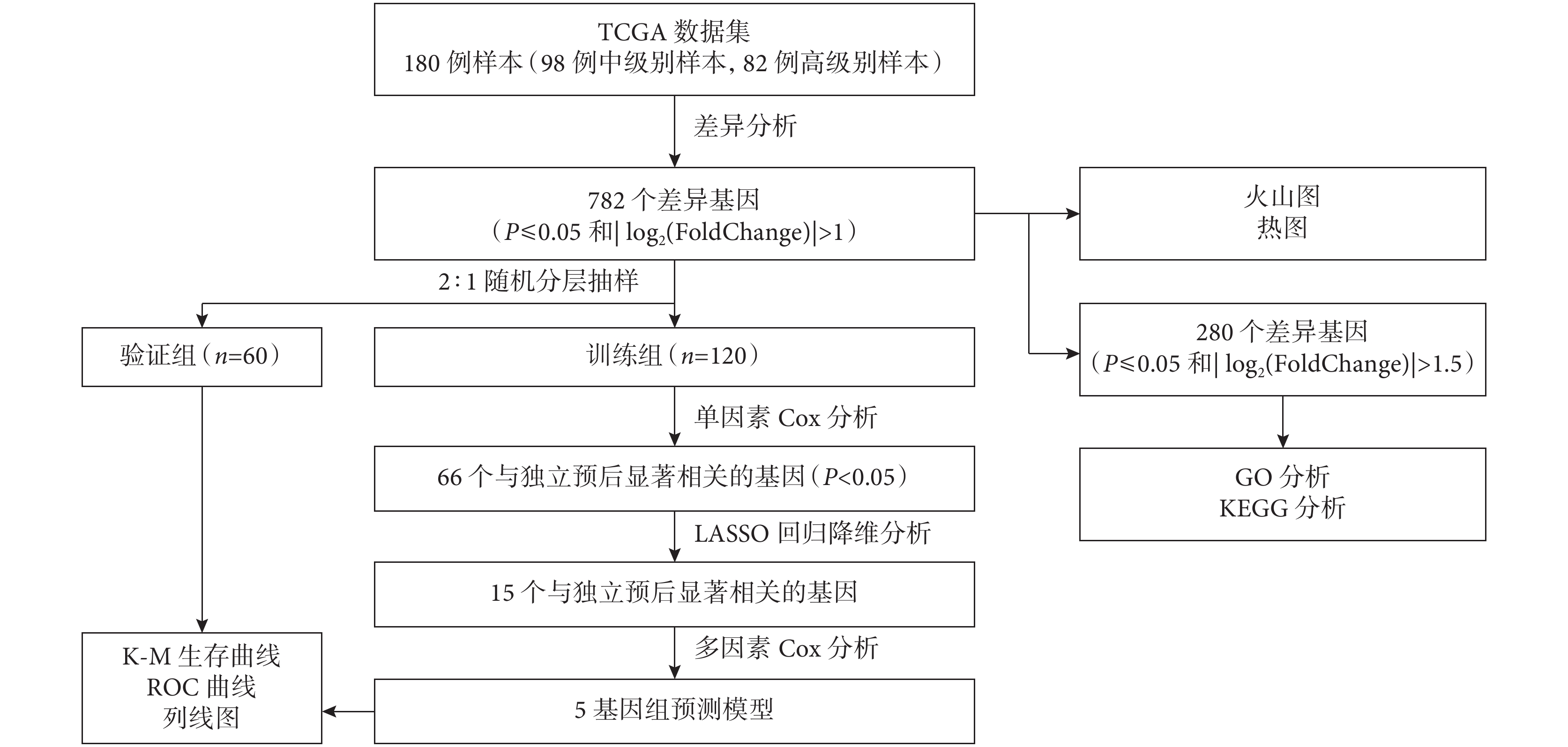
于2023年1月3日從UCSC Xena網站(https://xenabrowser.net/)下載了全部的LUAD RNA測序(RNA-Seq)數據和臨床病理數據,包括性別、年齡、腫瘤TNM分期、病理亞型、生存時間及生存狀態等。納入具有明確IAC病理亞型的樣本180例(中級別n=98,高級別n=82),用以篩選差異表達基因(differentially expressed genes,DEGs)和分析基因功能。按照2∶1隨機分層抽樣的方法分為訓練組(n=120)和驗證組(n=60)。通過訓練組樣本構建風險預測模型,驗證組樣本評價模型的準確性。研究流程圖見圖1。
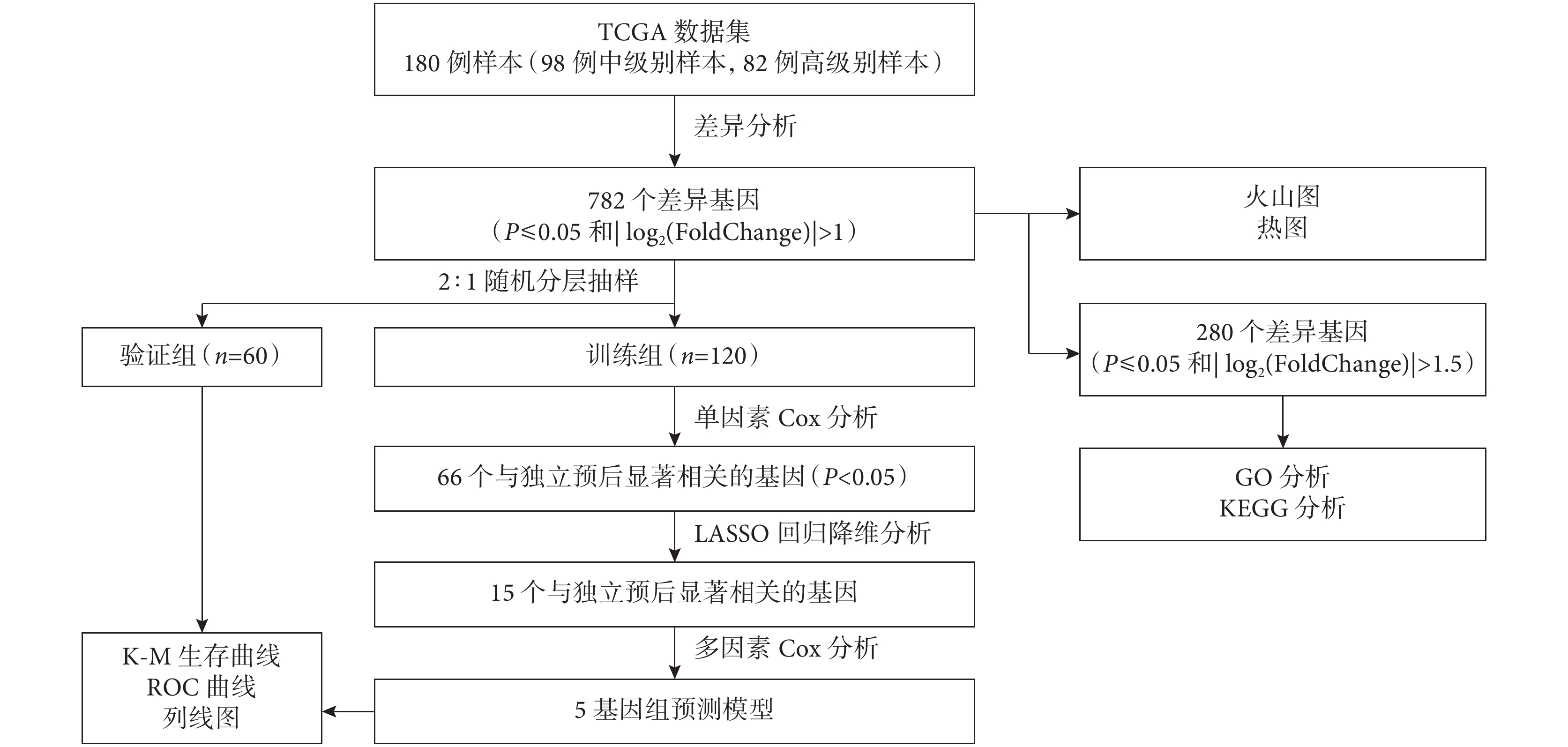 圖1
研究流程圖
圖1
研究流程圖
GO:基因本體;KEGG:京都基因與基因組百科全書;LASSO:最小絕對值收縮和選擇算子;ROC:受試者工作特征
1.2 篩選差異表達基因
使用R-4.2.1軟件中的“DESeq2”包篩選中級別與高級別病理亞型間的DEGs,根據P≤0.05和| log2(FoldChange)| >1的閾值對DEGs進行篩選。使用“pheatmap”和“ggplot2”包繪制火山圖和 熱圖。
1.3 基因功能和基因集富集分析
使用R-4.2.1軟件的“enrichplot”“org.Hs.eg.db”和“clusterProfiler”等包對DEGs進行基因本體(Gene Ontology,GO)功能富集分析、京都基因與基因組百科全書(Kyoto Encyclopedia of Genes and Genomes,KEGG)分析和基因集富集分析(Gene Set Enrichment Analysis,GSEA)。P≤0.05為差異有統計學意義,使用“ggplot2”包對結果進行可視化。
1.4 構建風險預測模型
將訓練組中120例樣本的DEGs表達量和IAC樣本生存信息合并,剔除沒有總生存時間(overall survival,OS)及生存狀態的患者后,剩余115例IAC樣本納入Cox分析。把通過P≤0.05和| log2(FoldChange)| >1篩選出來的DEGs納入單因素Cox分析,通過R-4.2.1軟件的“survival”包得到具有獨立預后相關的基因。用“glmnet”包把獨立預后相關的DEGs行最小絕對值收縮和選擇算子(least absolute shrinkage and selection operator,LASSO)回歸分析,得到的結果納入多因素Cox分析,最終構建臨床風險預測評分模型。
風險評分(RiskScore)預測模型計算公式:RiskScore=Exp1×C1+ Exp2×C2+…+ Expn×Cn(Exp:基因表達量;C:LASSO Cox回歸分析得到的回歸系數;n:DEGs在多因素Cox分析中篩選出的基因總數)。
根據上面的公式計算每例樣本的RiskScore,以RiskScore中位數為截斷值(cut-off)將IAC患者分為低風險組和高風險組,P≤0.05為差異有統計學意義。
1.5 評價預測風險模型和繪制列線圖
利用R-4.2.1軟件的“pheatmap”包繪制低風險組和高風險組樣本的臨床病理因素和候選基因表達量,同時應用“survival”“survminer”等包繪制Kaplan-Meier生存曲線來比較兩組的OS,最后通過“timeROC”包繪制時間依賴性的受試者工作特征(receiver operating characteristic,ROC)曲線,計算出訓練組樣本OS在1年、3年、5年的曲線下面積(area under the curve,AUC)值,來評價預測風險模型的準確性。將兩組的臨床病理因素和RiskScore納入Cox回歸分析,加載“rms”等包把具有獨立危險因素的變量繪制成列線圖,上部分為評分系統,下部分為預測系統。患者的1年、2年、3年、5年、10年生存率均可通過評分系統預測,并且利用校準曲線和一致性指數(C指數)來顯示生存預測的準確性。
1.6 驗證預測風險模型
用驗證組的樣本數據來驗證預測模型,通過上述的公式計算出驗證組各個樣本的RiskScore,以RiskScore中位數為cut-off值分為低風險組和高風險組,應用Kaplan-Meier生存曲線比較兩組的OS。繪制1年、3年、5年的ROC曲線,計算AUC值評估模型預測預后的能力。
2 結果
2.1 篩選差異基因和功能富集分析
以P≤0.05和| log2(FoldChange)| >1為閾值,篩選出782個DEGs,包括327個上調基因和455個下調基因,可視化火山圖、熱圖結果見附件圖1。
為了探索不同級別病理亞型相關的生物學功能和途徑,進一步縮小篩選標準,以P≤0.05和| log2(FoldChange)| >1.5得到280個DEGs,對上述280個DEGs進行GO及KEGG分析(附件圖2a~d)。GO分析顯示,DEGs主要富集在免疫反應、物質代謝及酶活性調節等相關的生物學功能;KEGG分析主要富集在細胞色素P450調節物質代謝、神經活性配體-受體相互作用通路等相關生物學功能。把P≤0.05的DEGs納入GSEA分析(附件圖2e~f),其分析結果主要富集于細胞色素P450相關的物質代謝、抗原的呈遞與自然殺傷細胞介導的免疫反應等相關生物學過程,進一步驗證了中級別與高級別病理亞型DEGs與免疫反應和物質代謝之間的密切關系。以上功能富集分析的結果解釋了IAC不同級別病理亞型之間基因層面的關系,也為以后尋找與預后相關生物學標志物的潛在靶點提供了不同的研究思路。
2.2 Cox分析、LASSO回歸分析及風險模型構建
把782個DEGs納入單因素Cox分析,得到66個與獨立預后顯著相關的基因(P<0.05),隨后進行LASSO回歸的降維分析(附件圖3),虛線標注處為logλ最小值,篩選出15個基因。進一步經多因素Cox分析,最終得到最佳構建風險模型的5個基因(MELTF,C=
根據5個基因的基因表達量和風險系數計算每個樣本的RiskScore:RiskScore=(MELTF表達量×0.185 1)+(MAGEA1表達量×
2.3 檢驗預測風險模型性能
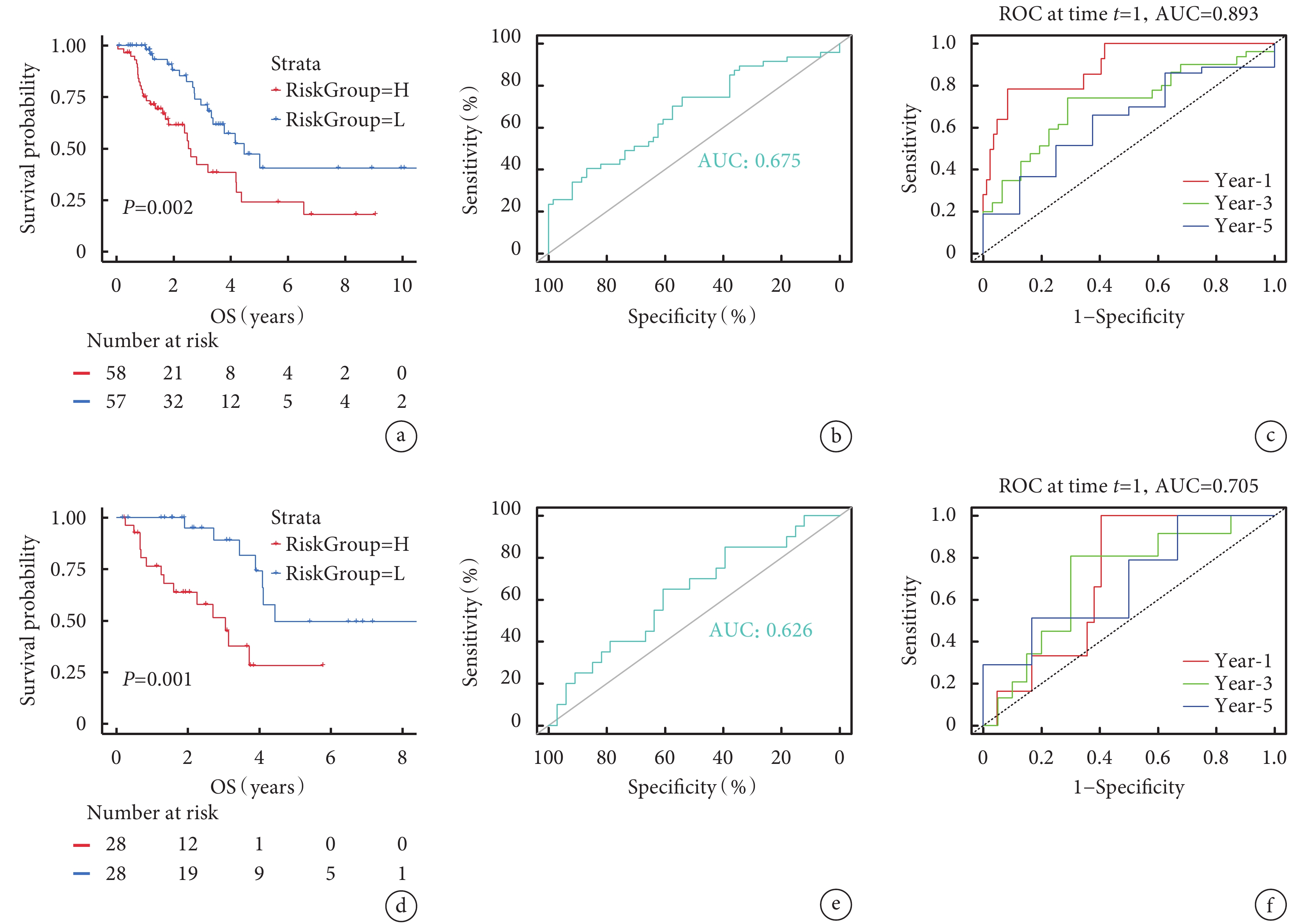
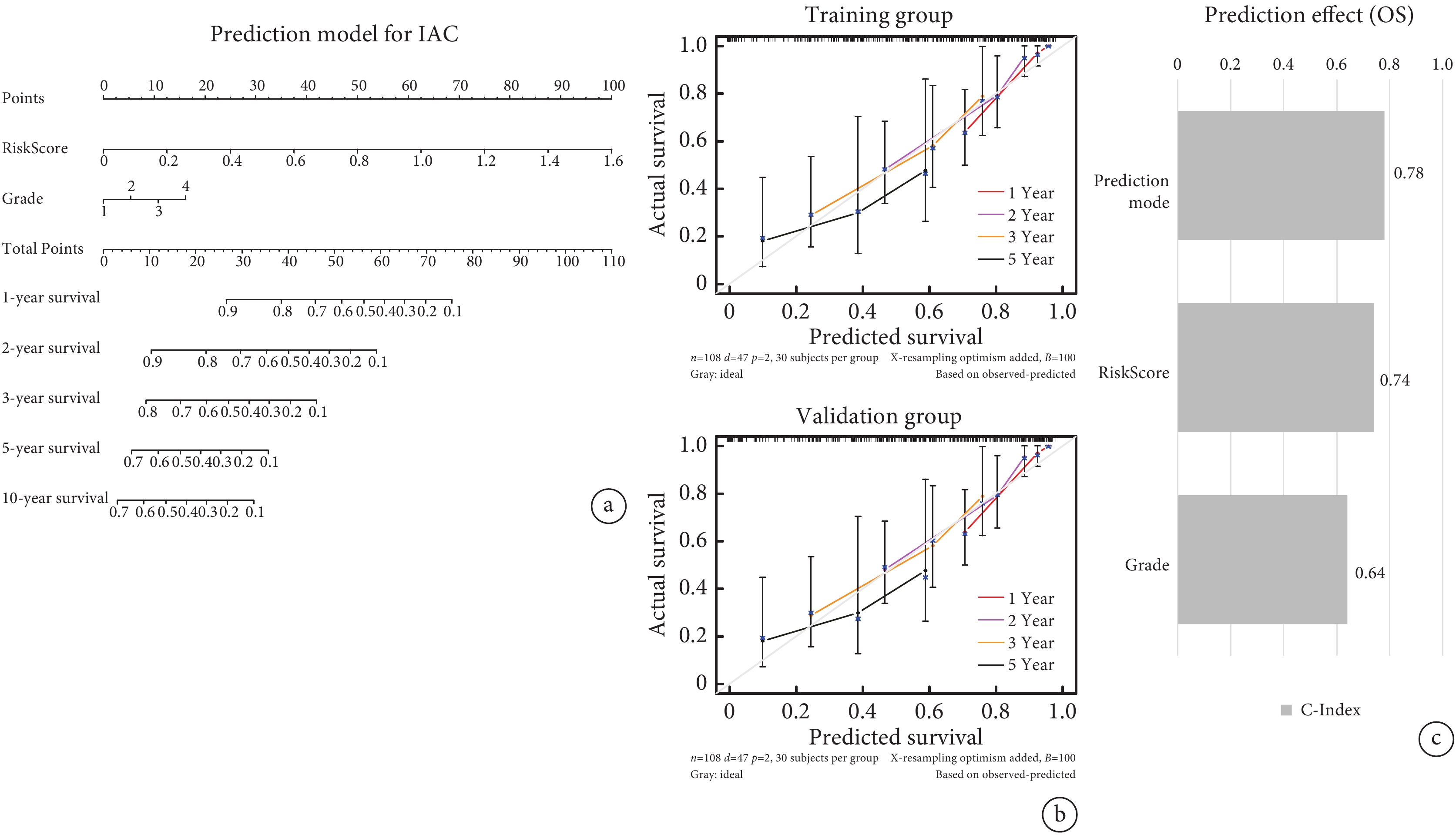
以訓練組IAC患者的RiskScore中位數為cut-off值分為低風險組(n=57)和高風險組(n=58)。不同危險組患者表現出不同的相關臨床病理特征(附件圖4a)。繪制Kaplan-Meier生存曲線(藍色代表低風險組,紅色代表高風險組),結果顯示兩組OS差異具有統計學意義(P=0.002),表明高風險組比低風險組更易具有不良預后(圖2a)。RiskScore的ROC曲線AUC值為0.675,時間依賴性ROC曲線1年、3年、5年AUC值為0.893、0.713、0.632(圖2b~c)。表明該預測風險模型具有良好的敏感性和特異性。單因素和多因素Cox分析顯示,RiskScore可作為IAC患者的獨立預后預測因子,將低風險組和高風險組的臨床病理因素及RiskScore納入Cox回歸分析,識別出具有獨立危險因素的變量,據此繪制列線圖,該個體化預測模型可估計IAC患者(1年、2年、3年、5年和10年)生存率(圖3a)。校準曲線中的列線圖和實際觀測結果在訓練組和驗證組中顯示出準確的重疊,表明具有良好的預測精度(圖3b)。并且該列線圖模型的C指數為0.78,高于其他單一因素的預測模型(圖3c)。
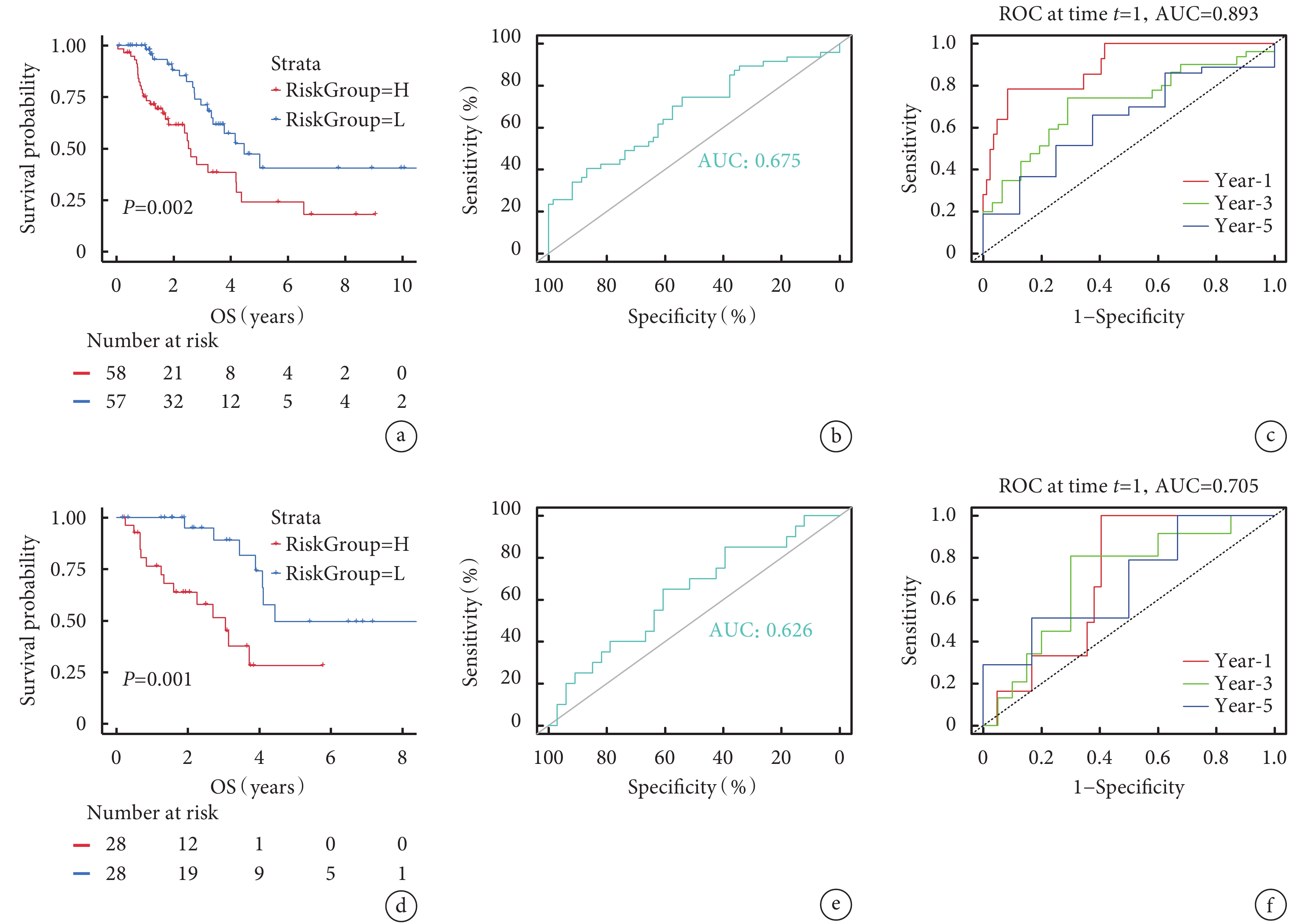 圖2
兩組Kaplan-Meier生存分析和ROC曲線
圖2
兩組Kaplan-Meier生存分析和ROC曲線
a:訓練組Kaplan-Meier生存曲線(
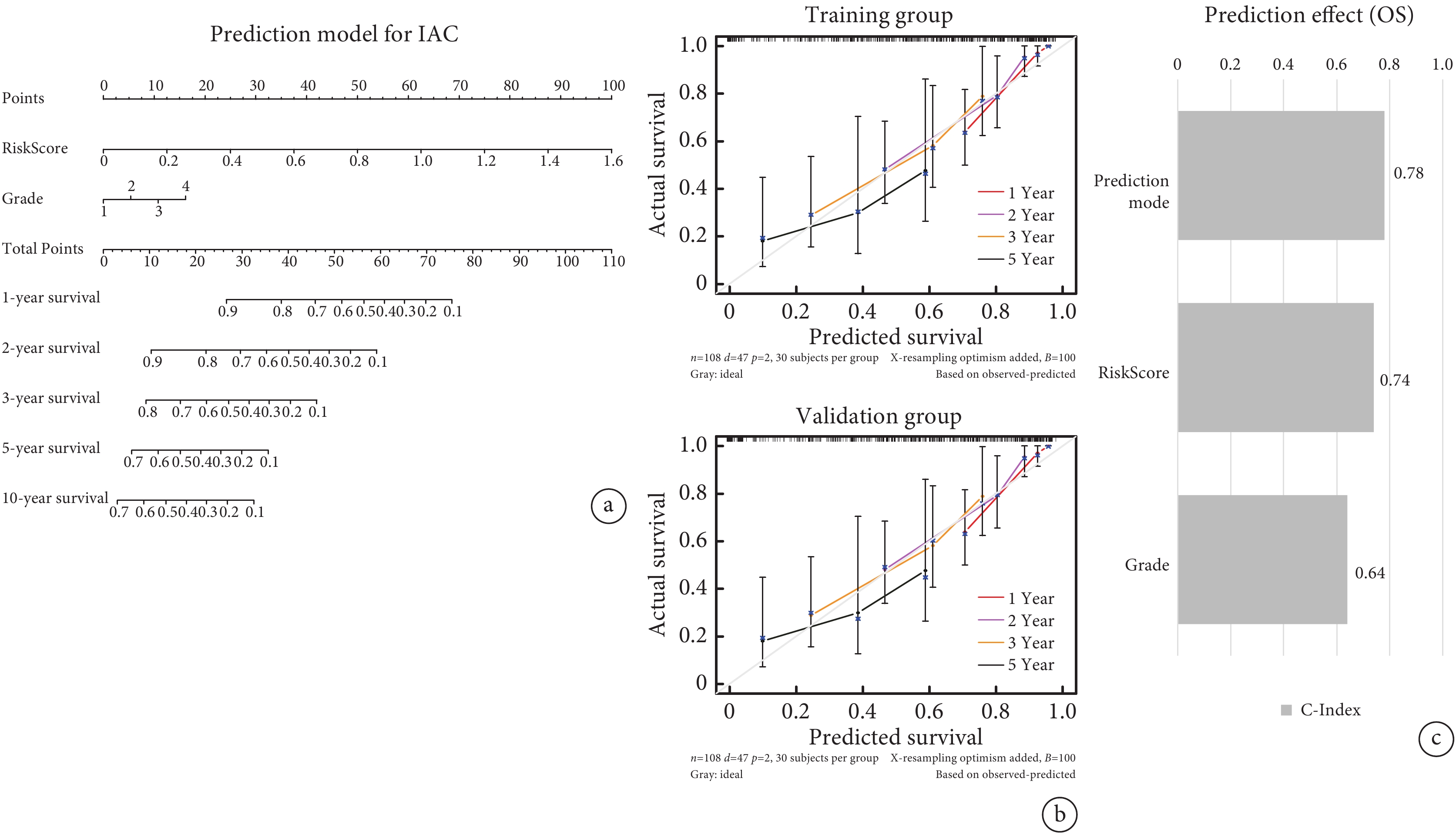 圖3
個體化預測模型
圖3
個體化預測模型
a:預測患者1年、2年、3年、5年和10年生存率的列線圖;b:校準圖顯示訓練組和驗證組1年、2年、3年和5年預測OS與實際OS之間生存概率的比較;c:采用C指數評價個體化預測模型的預測效果;IAC:浸潤性腺癌;OS:總生存時間
2.4 驗證組數據集驗證模型
將驗證組中60例樣本的DEGs表達量和臨床信息合并,經剔除無OS和生存狀態的患者后,計算剩余56例患者的RiskScore,以中位數為cut-off值分為低風險組(n=28)和高風險組(n=28),將驗證組相關臨床病理特征可視化(附件圖4b)。然后繪制Kaplan-Meier生存曲線、ROC曲線及時間依賴性ROC曲線,二者OS差異有統計學意義(P=0.001),時間依賴性ROC曲線1年、3年、5年AUC值分別為0.705、0.700、0.684(圖2d~f)。上述結果說明該模型在驗證組中仍具有較好的預測性能。
3 討論
盡管免疫治療和分子靶向治療已經取得了很大進展,但由于缺乏準確的早期診斷和預后生物學標志物,肺癌患者的OS仍然較差,5年生存率僅約15%[5]。不同肺癌亞型具有不同的臨床特征和預后,確定特異的預后生物學標志物和新的治療靶點仍然至關重要。
在本研究中,我們篩選出IAC患者中級別和高級別病理亞型間DEGs,包括327個上調基因和455個下調基因,GO、KEGG和GSEA富集分析表明,這些基因主要富集在細胞色素P450相關物質代謝、自然殺傷細胞介導的免疫反應、抗原的呈遞和酶活性調節等生物學過程。細胞色素P450(CYP)是存在于動物、植物和微生物中的一個參與物質代謝的酶超家族,其密切參與了肺癌、肝癌、乳腺癌等癌癥的相關生物學功能[6]。自然殺傷細胞是免疫系統中不可或缺的腫瘤殺傷效應細胞,通過依賴的細胞毒性和產生細胞因子進行免疫調節,相關證據表明自然殺傷細胞可以有效防止肺癌的發生和發展[7]。自然殺傷細胞介導的免疫反應在腫瘤形成的免疫監測中具有至關重要的作用[8]。通過對中級別和高級別兩組病理亞型DEGs的分析,解釋了兩組之間生物侵襲性和預后等因素基因層面的差異,可能為更深層次研究提供了一定的科研思路。
上述5個特征基因已被報道與多種癌癥的發生、發展密切相關。MELTF又稱MTf(Melanotransferrin)或MTF1(metal regulatory transcription factor 1),是鐵結合轉鐵蛋白的同源物,可作為LUAD和胃癌的預后標志物[9-10]。研究[10-11]表明MTf能促進癌細胞侵襲、遷移、增殖和上皮間充質轉化(EMT)進展,是一種具有很大潛力的靶點。MAGEA1是屬于X染色體簇的癌癥/睪丸抗原,在多種腫瘤中表達,如肺癌,其可能是免疫治療的理想抗原靶點[12]。研究[13]表明,隨著腫瘤進展,MAGEA1表達水平增加,說明MAGEA1檢測有可能作為肺癌早期診斷的輔助檢查方法。成纖維細胞生長因子(FGFs)家族由22個成員組成,分為7個亞家族,調節著一系列生物學功能和多種發育過程[14]。作為FGFs家族內分泌亞群的一員,FGF19在維持蛋白質和葡萄糖代謝方面都起著關鍵作用[15]。FGF19具有與4種類型酪氨酸激酶FGF受體(FGFR 1~4)結合的潛力,但親和力相當低,需要β-klotho (KLB)作為輔受體,一旦FGF19結合到FGFRs和KLB,它就會啟動二聚作用和自身磷酸化,最終激活細胞質信號轉導通路[15-17]。 相關研究[15]表明FGF19在NSCLC中表達明顯上調,且FGF19過表達與不良預后顯著相關,可能作為預測NSCLC患者不良預后的潛在生物學標志物。DKK4作為DKK家族中的一員,是Wnt/β-catenin信號通路的抑制劑[18]。研究[19]發現DKK4在腎細胞癌、結直腸癌、胃癌、NSCLC和上皮性卵巢癌中表達上調,在肝細胞癌中表達下調,DKK4也參與腫瘤生長、侵襲、遷移和化療耐藥等生物功能。目前已經有很多關于DKK4的研究,然而,DKK4在癌癥或非癌癥疾病中的作用機制和分子機制仍不明確,許多前沿領域有待探索,所以本文為進行更深層次的研究提供了可能的方向。研究[20]發現C14ORF105在耐紫杉醇的卵巢癌細胞中過表達,提示其可能參與了紫杉醇耐藥。到目前為止,C14ORF105在肺癌中的作用尚未被廣泛研究。綜上所述,MELTF、MAGEA1、FGF19、DKK4和C14ORF105均參與了腫瘤的發生或進展。
總的來說,我們的研究通過Cox分析和LASSO回歸分析構建了一個準確有效的5基因組預測模型。基于該5基因組的RiskScore可用于確定LUAD患者的OS。將RiskScore和臨床參數相結合的列線圖可用于預測LUAD患者1年、2年、3年、5年和10年的生存率,所以,它將有助于LUAD患者的預后和隨訪監測,為LUAD患者的個體化診療提供參考。但需要指出的是,我們的研究還存在一定的局限性。首先,我們的研究數據主要來自TCGA數據集,有必要在大型獨立臨床隊列中評估其預測效能。其次,我們納入的分組僅包括了中級別和高級別病理亞型,排除了樣本量較少的低級別病理亞型,并且需要進一步外部數據集的驗證。最后,這5個基因在LUAD發病中的生物學機制有待進一步通過功能研究來闡明。
利益沖突:無。
作者貢獻:崔嚴奇負責病例篩選、數據整理與論文設計、初稿撰寫等;曾志勇、廖毅、倪琳、連鐸煌、楊鯨蓉、葉仕新和張錦燦負責論文審閱與修改。
本文附件圖1~4見本刊網站電子版(https://www. tcsurg.org/article/10.7507/1007-4848.202303059)。
在全球范圍內癌癥相關死亡中,肺癌仍是其主要原因[1]。肺癌主要包括兩種組織學類型:非小細胞肺癌(non-small cell lung cancer,NSCLC)和小細胞肺癌,NSCLC約占所有肺癌的80%。其中,NSCLC的主要組織學亞型是肺腺癌(lung adenocarcinoma,LUAD),約占肺癌發病率的40%。根據2011年國際肺癌研究協會(International Association for the Study of Lung Cancer,IASLC)/美國胸科學會(American Thoracic Society,ATS)/歐洲呼吸學會(European Respiratory Society,ERS)和2015年世界衛生組織(World Health Organization,WHO)肺腫瘤分類的組織學分型,浸潤性肺腺癌(invasive lung adenocarcinoma,IAC)主要分為5種病理亞型:貼壁型、腺泡型、乳頭型、微乳頭型以及實體型,并基于其最主要的生長亞型進行級別劃分:低級別以貼壁為主;中級別以腺泡或乳頭為主;高級別以實體或微乳頭為主;級別越高,侵襲性越高,患者預后越差[2-3]。目前,肺癌的治療主要依賴于組織類型和臨床分期,但由于IAC的高度異質性,即使是同樣級別組織類型和臨床分期的IAC患者預后也不同。近年來,高通量技術的進步使醫學研究人員能夠從基因層面發現癌癥中的遺傳改變,并被廣泛用于篩選潛在的腫瘤生物標志物[4-5]。同時,機器學習算法已被引入且應用于遺傳和基因組數據,并從生物醫學數據集中,通過構建預測模型來預測臨床結果。因此基于高通量測序數據來檢測IAC不同級別病理亞型的基因表達情況,以及結合生物信息學方法系統分析相關差異表達基因及其調控機制,然后運用機器學習算法構建臨床風險預后預測模型,對LUAD患者進行個體化、精準化治療具有重要意義。
在本研究中,我們篩選出與LUAD患者預后相關的特征基因,然后構建基于基因特征的風險預測模型,預測患者的生存狀況,為LUAD患者的個體化診療提供參考。
1 資料與方法
1.1 獲取數據
于2023年1月3日從UCSC Xena網站(https://xenabrowser.net/)下載了全部的LUAD RNA測序(RNA-Seq)數據和臨床病理數據,包括性別、年齡、腫瘤TNM分期、病理亞型、生存時間及生存狀態等。納入具有明確IAC病理亞型的樣本180例(中級別n=98,高級別n=82),用以篩選差異表達基因(differentially expressed genes,DEGs)和分析基因功能。按照2∶1隨機分層抽樣的方法分為訓練組(n=120)和驗證組(n=60)。通過訓練組樣本構建風險預測模型,驗證組樣本評價模型的準確性。研究流程圖見圖1。
圖1
研究流程圖
GO:基因本體;KEGG:京都基因與基因組百科全書;LASSO:最小絕對值收縮和選擇算子;ROC:受試者工作特征
1.2 篩選差異表達基因
使用R-4.2.1軟件中的“DESeq2”包篩選中級別與高級別病理亞型間的DEGs,根據P≤0.05和| log2(FoldChange)| >1的閾值對DEGs進行篩選。使用“pheatmap”和“ggplot2”包繪制火山圖和 熱圖。
1.3 基因功能和基因集富集分析
使用R-4.2.1軟件的“enrichplot”“org.Hs.eg.db”和“clusterProfiler”等包對DEGs進行基因本體(Gene Ontology,GO)功能富集分析、京都基因與基因組百科全書(Kyoto Encyclopedia of Genes and Genomes,KEGG)分析和基因集富集分析(Gene Set Enrichment Analysis,GSEA)。P≤0.05為差異有統計學意義,使用“ggplot2”包對結果進行可視化。
1.4 構建風險預測模型
將訓練組中120例樣本的DEGs表達量和IAC樣本生存信息合并,剔除沒有總生存時間(overall survival,OS)及生存狀態的患者后,剩余115例IAC樣本納入Cox分析。把通過P≤0.05和| log2(FoldChange)| >1篩選出來的DEGs納入單因素Cox分析,通過R-4.2.1軟件的“survival”包得到具有獨立預后相關的基因。用“glmnet”包把獨立預后相關的DEGs行最小絕對值收縮和選擇算子(least absolute shrinkage and selection operator,LASSO)回歸分析,得到的結果納入多因素Cox分析,最終構建臨床風險預測評分模型。
風險評分(RiskScore)預測模型計算公式:RiskScore=Exp1×C1+ Exp2×C2+…+ Expn×Cn(Exp:基因表達量;C:LASSO Cox回歸分析得到的回歸系數;n:DEGs在多因素Cox分析中篩選出的基因總數)。
根據上面的公式計算每例樣本的RiskScore,以RiskScore中位數為截斷值(cut-off)將IAC患者分為低風險組和高風險組,P≤0.05為差異有統計學意義。
1.5 評價預測風險模型和繪制列線圖
利用R-4.2.1軟件的“pheatmap”包繪制低風險組和高風險組樣本的臨床病理因素和候選基因表達量,同時應用“survival”“survminer”等包繪制Kaplan-Meier生存曲線來比較兩組的OS,最后通過“timeROC”包繪制時間依賴性的受試者工作特征(receiver operating characteristic,ROC)曲線,計算出訓練組樣本OS在1年、3年、5年的曲線下面積(area under the curve,AUC)值,來評價預測風險模型的準確性。將兩組的臨床病理因素和RiskScore納入Cox回歸分析,加載“rms”等包把具有獨立危險因素的變量繪制成列線圖,上部分為評分系統,下部分為預測系統。患者的1年、2年、3年、5年、10年生存率均可通過評分系統預測,并且利用校準曲線和一致性指數(C指數)來顯示生存預測的準確性。
1.6 驗證預測風險模型
用驗證組的樣本數據來驗證預測模型,通過上述的公式計算出驗證組各個樣本的RiskScore,以RiskScore中位數為cut-off值分為低風險組和高風險組,應用Kaplan-Meier生存曲線比較兩組的OS。繪制1年、3年、5年的ROC曲線,計算AUC值評估模型預測預后的能力。
2 結果
2.1 篩選差異基因和功能富集分析
以P≤0.05和| log2(FoldChange)| >1為閾值,篩選出782個DEGs,包括327個上調基因和455個下調基因,可視化火山圖、熱圖結果見附件圖1。
為了探索不同級別病理亞型相關的生物學功能和途徑,進一步縮小篩選標準,以P≤0.05和| log2(FoldChange)| >1.5得到280個DEGs,對上述280個DEGs進行GO及KEGG分析(附件圖2a~d)。GO分析顯示,DEGs主要富集在免疫反應、物質代謝及酶活性調節等相關的生物學功能;KEGG分析主要富集在細胞色素P450調節物質代謝、神經活性配體-受體相互作用通路等相關生物學功能。把P≤0.05的DEGs納入GSEA分析(附件圖2e~f),其分析結果主要富集于細胞色素P450相關的物質代謝、抗原的呈遞與自然殺傷細胞介導的免疫反應等相關生物學過程,進一步驗證了中級別與高級別病理亞型DEGs與免疫反應和物質代謝之間的密切關系。以上功能富集分析的結果解釋了IAC不同級別病理亞型之間基因層面的關系,也為以后尋找與預后相關生物學標志物的潛在靶點提供了不同的研究思路。
2.2 Cox分析、LASSO回歸分析及風險模型構建
把782個DEGs納入單因素Cox分析,得到66個與獨立預后顯著相關的基因(P<0.05),隨后進行LASSO回歸的降維分析(附件圖3),虛線標注處為logλ最小值,篩選出15個基因。進一步經多因素Cox分析,最終得到最佳構建風險模型的5個基因(MELTF,C=
根據5個基因的基因表達量和風險系數計算每個樣本的RiskScore:RiskScore=(MELTF表達量×0.185 1)+(MAGEA1表達量×
2.3 檢驗預測風險模型性能
以訓練組IAC患者的RiskScore中位數為cut-off值分為低風險組(n=57)和高風險組(n=58)。不同危險組患者表現出不同的相關臨床病理特征(附件圖4a)。繪制Kaplan-Meier生存曲線(藍色代表低風險組,紅色代表高風險組),結果顯示兩組OS差異具有統計學意義(P=0.002),表明高風險組比低風險組更易具有不良預后(圖2a)。RiskScore的ROC曲線AUC值為0.675,時間依賴性ROC曲線1年、3年、5年AUC值為0.893、0.713、0.632(圖2b~c)。表明該預測風險模型具有良好的敏感性和特異性。單因素和多因素Cox分析顯示,RiskScore可作為IAC患者的獨立預后預測因子,將低風險組和高風險組的臨床病理因素及RiskScore納入Cox回歸分析,識別出具有獨立危險因素的變量,據此繪制列線圖,該個體化預測模型可估計IAC患者(1年、2年、3年、5年和10年)生存率(圖3a)。校準曲線中的列線圖和實際觀測結果在訓練組和驗證組中顯示出準確的重疊,表明具有良好的預測精度(圖3b)。并且該列線圖模型的C指數為0.78,高于其他單一因素的預測模型(圖3c)。
圖2
兩組Kaplan-Meier生存分析和ROC曲線
a:訓練組Kaplan-Meier生存曲線(
圖3
個體化預測模型
a:預測患者1年、2年、3年、5年和10年生存率的列線圖;b:校準圖顯示訓練組和驗證組1年、2年、3年和5年預測OS與實際OS之間生存概率的比較;c:采用C指數評價個體化預測模型的預測效果;IAC:浸潤性腺癌;OS:總生存時間
2.4 驗證組數據集驗證模型
將驗證組中60例樣本的DEGs表達量和臨床信息合并,經剔除無OS和生存狀態的患者后,計算剩余56例患者的RiskScore,以中位數為cut-off值分為低風險組(n=28)和高風險組(n=28),將驗證組相關臨床病理特征可視化(附件圖4b)。然后繪制Kaplan-Meier生存曲線、ROC曲線及時間依賴性ROC曲線,二者OS差異有統計學意義(P=0.001),時間依賴性ROC曲線1年、3年、5年AUC值分別為0.705、0.700、0.684(圖2d~f)。上述結果說明該模型在驗證組中仍具有較好的預測性能。
3 討論
盡管免疫治療和分子靶向治療已經取得了很大進展,但由于缺乏準確的早期診斷和預后生物學標志物,肺癌患者的OS仍然較差,5年生存率僅約15%[5]。不同肺癌亞型具有不同的臨床特征和預后,確定特異的預后生物學標志物和新的治療靶點仍然至關重要。
在本研究中,我們篩選出IAC患者中級別和高級別病理亞型間DEGs,包括327個上調基因和455個下調基因,GO、KEGG和GSEA富集分析表明,這些基因主要富集在細胞色素P450相關物質代謝、自然殺傷細胞介導的免疫反應、抗原的呈遞和酶活性調節等生物學過程。細胞色素P450(CYP)是存在于動物、植物和微生物中的一個參與物質代謝的酶超家族,其密切參與了肺癌、肝癌、乳腺癌等癌癥的相關生物學功能[6]。自然殺傷細胞是免疫系統中不可或缺的腫瘤殺傷效應細胞,通過依賴的細胞毒性和產生細胞因子進行免疫調節,相關證據表明自然殺傷細胞可以有效防止肺癌的發生和發展[7]。自然殺傷細胞介導的免疫反應在腫瘤形成的免疫監測中具有至關重要的作用[8]。通過對中級別和高級別兩組病理亞型DEGs的分析,解釋了兩組之間生物侵襲性和預后等因素基因層面的差異,可能為更深層次研究提供了一定的科研思路。
上述5個特征基因已被報道與多種癌癥的發生、發展密切相關。MELTF又稱MTf(Melanotransferrin)或MTF1(metal regulatory transcription factor 1),是鐵結合轉鐵蛋白的同源物,可作為LUAD和胃癌的預后標志物[9-10]。研究[10-11]表明MTf能促進癌細胞侵襲、遷移、增殖和上皮間充質轉化(EMT)進展,是一種具有很大潛力的靶點。MAGEA1是屬于X染色體簇的癌癥/睪丸抗原,在多種腫瘤中表達,如肺癌,其可能是免疫治療的理想抗原靶點[12]。研究[13]表明,隨著腫瘤進展,MAGEA1表達水平增加,說明MAGEA1檢測有可能作為肺癌早期診斷的輔助檢查方法。成纖維細胞生長因子(FGFs)家族由22個成員組成,分為7個亞家族,調節著一系列生物學功能和多種發育過程[14]。作為FGFs家族內分泌亞群的一員,FGF19在維持蛋白質和葡萄糖代謝方面都起著關鍵作用[15]。FGF19具有與4種類型酪氨酸激酶FGF受體(FGFR 1~4)結合的潛力,但親和力相當低,需要β-klotho (KLB)作為輔受體,一旦FGF19結合到FGFRs和KLB,它就會啟動二聚作用和自身磷酸化,最終激活細胞質信號轉導通路[15-17]。 相關研究[15]表明FGF19在NSCLC中表達明顯上調,且FGF19過表達與不良預后顯著相關,可能作為預測NSCLC患者不良預后的潛在生物學標志物。DKK4作為DKK家族中的一員,是Wnt/β-catenin信號通路的抑制劑[18]。研究[19]發現DKK4在腎細胞癌、結直腸癌、胃癌、NSCLC和上皮性卵巢癌中表達上調,在肝細胞癌中表達下調,DKK4也參與腫瘤生長、侵襲、遷移和化療耐藥等生物功能。目前已經有很多關于DKK4的研究,然而,DKK4在癌癥或非癌癥疾病中的作用機制和分子機制仍不明確,許多前沿領域有待探索,所以本文為進行更深層次的研究提供了可能的方向。研究[20]發現C14ORF105在耐紫杉醇的卵巢癌細胞中過表達,提示其可能參與了紫杉醇耐藥。到目前為止,C14ORF105在肺癌中的作用尚未被廣泛研究。綜上所述,MELTF、MAGEA1、FGF19、DKK4和C14ORF105均參與了腫瘤的發生或進展。
總的來說,我們的研究通過Cox分析和LASSO回歸分析構建了一個準確有效的5基因組預測模型。基于該5基因組的RiskScore可用于確定LUAD患者的OS。將RiskScore和臨床參數相結合的列線圖可用于預測LUAD患者1年、2年、3年、5年和10年的生存率,所以,它將有助于LUAD患者的預后和隨訪監測,為LUAD患者的個體化診療提供參考。但需要指出的是,我們的研究還存在一定的局限性。首先,我們的研究數據主要來自TCGA數據集,有必要在大型獨立臨床隊列中評估其預測效能。其次,我們納入的分組僅包括了中級別和高級別病理亞型,排除了樣本量較少的低級別病理亞型,并且需要進一步外部數據集的驗證。最后,這5個基因在LUAD發病中的生物學機制有待進一步通過功能研究來闡明。
利益沖突:無。
作者貢獻:崔嚴奇負責病例篩選、數據整理與論文設計、初稿撰寫等;曾志勇、廖毅、倪琳、連鐸煌、楊鯨蓉、葉仕新和張錦燦負責論文審閱與修改。
本文附件圖1~4見本刊網站電子版(https://www. tcsurg.org/article/10.7507/1007-4848.202303059)。