孟德爾隨機化是工具變量分析的一種類型,由于其自身特殊的優勢及基因組學的迅速發展,近年來受到醫學研究領域的青睞。了解孟德爾隨機化的原理、方法及局限性對于正確應用孟德爾隨機化方法及解讀結果至關重要。本文旨在介紹孟德爾隨機化的基本概念、原理、方法和局限性,以期為研究者進行孟德爾隨機化研究提供指導。
引用本文: 于天琦, 徐文濤, 蘇雅娜, 李靜. 孟德爾隨機化研究基本原理、方法和局限性. 中國循證醫學雜志, 2021, 21(10): 1227-1234. doi: 10.7507/1672-2531.202107008 復制
版權信息: ?四川大學華西醫院華西期刊社《中國循證醫學雜志》版權所有,未經授權不得轉載、改編
傳統的觀察性研究中,潛在混雜和反向因果關系會影響其因果推斷能力[1, 2]。孟德爾隨機化(Mendelian randomization,MR)是工具變量(instrumental variable,IV)分析的一種類型,它使用遺傳變異作為 IV 來檢測和量化因果關系[3]。由于能克服潛在混雜和反向因果關系的影響,近年來 MR 在觀察性研究中的應用越來越廣泛[4, 5]。早期的 MR 研究通常在小樣本人群中進行,且僅使用了少量的遺傳變異[6],這使得 MR 研究的效力較低。然而,隨著生物學界發現了大量與特定性狀緊密相關的遺傳變異,加上許多大樣本全基因組關聯研究(genome-wide association study,GWAS[5])公開發布了數十萬個暴露和疾病與遺傳變異關系的匯總數據[7],這一領域發生了一場革命。這些匯總數據使得研究者能估計大樣本數據中的遺傳關聯,從而促進了 MR 研究發展。近年來,該領域在方法學上也迅速更新,新方法克服了傳統 MR 方法的一些特定限制[8-11],但其同樣存在局限性。只有正確了解 MR 背后的原理、局限性及不同方法的適用條件才能針對不同的研究問題和特定的數據正確應用 MR。本文介紹 MR 的基本概念和原理,并解讀其在使用中的一些問題和局限性,介紹 MR 研究迄今為止常用的幾種方法,以期為研究者進行孟德爾隨機化研究提供指導。
1 MR 基本原理和假設
MR 是 IV 分析的一種特殊類型,它遵循 IV 分析的基本原理和相關假設[12]。
1.1 IV 與 MR 基本原理
IV 分析在經濟學中的應用已有許多年,它是許多計量經濟學統計分析方法的基礎[13]。但在醫學研究中,它最早用于處理隨機臨床試驗中的依從性問題[14]。因此,本文將通過闡述 IV 如何處理隨機試驗的依從性問題,并比較多因素回歸、傾向性評分(propensity score,PS)與 IV 分析在因果推斷中的異同點,以幫助讀者理解其原理和本質。
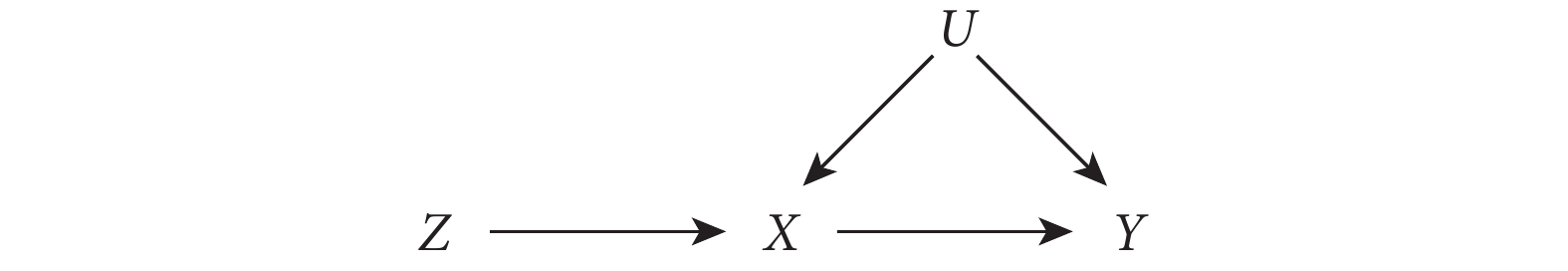
首先,假設一個隨機雙盲對照試驗,在此試驗中,研究者隨機將患者分配到干預組(表示為 Z=1)和對照組(表示為 Z=0)。由于存在不依從的情況,患者實際是否接受干預可能與分配情況不符,用 X 表示患者實際是否接受干預的情況(X=1 接受干預,X=0 未接受干預);用 Y 表示結局;U 代表所有混雜因素(包括可測和不可測的混雜因素)。圖 1 中的有向無環圖(directed acyclic graph,DAG)展示了以上變量間的關系。
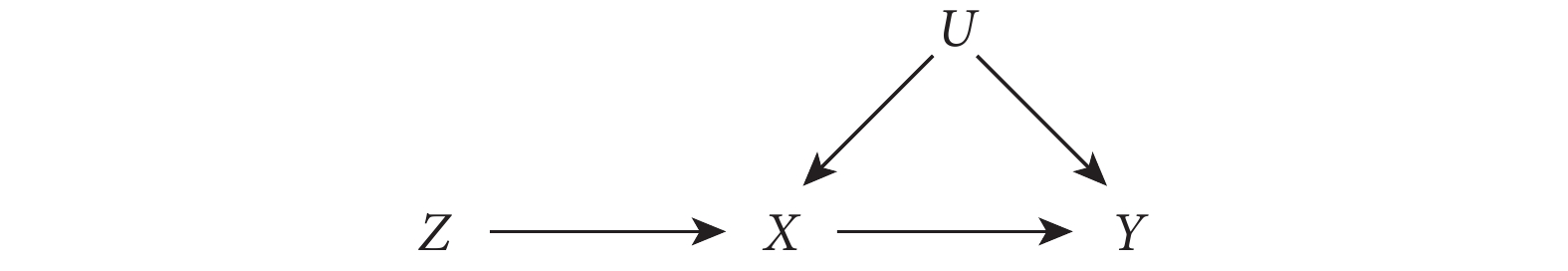 圖1
IV 分析各變量關系的有向無環圖
圖1
IV 分析各變量關系的有向無環圖
在此試驗中,若直接估計  與 Y 之間的因果效應會因不依從問題而受到混雜因素的影響,這是因為患者最終接受的干預狀態并不是隨機的。但我們可利用隨機分配狀態變量
與 Y 之間的因果效應會因不依從問題而受到混雜因素的影響,這是因為患者最終接受的干預狀態并不是隨機的。但我們可利用隨機分配狀態變量  作為有效的 IV,來得到因果效應的估計。通過
作為有效的 IV,來得到因果效應的估計。通過  來估計
來估計  與 Y 之間的平均因果效應是無偏的,因為它不會受到任何可測和不可測的混雜因素的影響。此時,X 對 Y 效應的 IV 估計可表示為:
與 Y 之間的平均因果效應是無偏的,因為它不會受到任何可測和不可測的混雜因素的影響。此時,X 對 Y 效應的 IV 估計可表示為:
 |
其中,分子代表的是 Z 對 Y 的效應,分母代表的是 Z 對 X 的效應。由圖 1 可知, 與 Y 之間及
與 Y 之間及  與 X 之間無混雜因素影響,因此分子、分母都是無偏估計。用上式來計算 X 對 Y 的因果效應,相當于只使用了結果 Y 中由 Z 引起的部分變化,從而避免了 Y 與 X 之間混雜因素的影響。
與 X 之間無混雜因素影響,因此分子、分母都是無偏估計。用上式來計算 X 對 Y 的因果效應,相當于只使用了結果 Y 中由 Z 引起的部分變化,從而避免了 Y 與 X 之間混雜因素的影響。
在觀察性研究中,研究對象的分配不受研究者控制,各組之間的混雜因素可能存在重要差異,因此結果的差異可能是由于暴露本身、可測量的和不可測量的混雜因素在組間的差異,或兩者共同造成的。多因素回歸分析常用于校正由可測量的混雜因素引起的偏倚。然而,研究者很難通過回歸模型發現暴露組之間的混雜因素沒有充分重疊的情況,從而使得結果的可信性降低[15]。另外,許多回歸模型的準確性依賴于足夠的結局事件數。例如,為獲得準確和穩定結果,Logistic 回歸模型要求結局事件數/協變量數大于 8~10 個[16]。由 Rubin 和 Rosenbaum[17]提出的 PS 分析能夠克服以上難題。PS 被定義為給定一組可測量的協變量,個體接受暴露或治療的條件概率,在評估觀察性研究中的因果效應時,可用來調整選擇偏倚[17]。與傳統回歸模型相比,分析中考慮的潛在混雜因素的數量是不受限制的。因此,PS 方法理論上可通過對所有可能的可測量混雜因素創建偽隨機化來增加組間的可比性。然而,與多因素回歸類似,PS 分析也無法校正由于不可測量或未知混雜因素造成的偏倚。IV 分析與前兩種分析方法最大的區別在于它并非將研究對象按照是否接受暴露或治療進行比較,而是將研究對象根據 IV 的情況進行分組比較[15]。由于 IV 在研究對象中的分布是隨機的,理論上,如果實施得當,它能同時校正可測量與不可測量混雜因素產生的偏倚。
1.2 基因作為 IV
雖然 IV 分析能夠克服觀察性研究中最棘手的不可觀測混雜的問題,但一個有效的 IV 所需滿足的假設非常嚴格,要找到一個可用的 IV 十分困難[18]。隨著近十年來基因組學迅速發展,研究者們逐漸意識到以基因作為 IV 能在很大程度上克服以上難題[19]。
基因作為 IV 的優勢要從孟德爾第二定律[20]說起:“每一對具有區別的性狀或行為與父母之間的其他差異無關,等位基因從父母隨機分離后遺傳給后代。”此定律表明一個性狀的遺傳獨立于其他性狀的遺傳且呈現隨機性,因此后代基因型不太可能與人群中的環境混雜因素相關[21]。其次,基因型分布在時間上先于后天暴露,基因型與疾病之間的聯系不會受到反向因果關系的影響。再次,與暴露相關的基因型通常從出生到成年都與之相關,因此,在因果推斷中可避免因誤差而造成的衰減(回歸稀釋偏倚)[19]。此外,這一定律在人群中是普遍存在的,避免了隨機對照試驗(randomized controlled trials,RCT)中的代表性問題。最后,在 GWAS 和高通量基因組技術的時代,人群遺傳數據通常可方便地從大型公開數據庫中獲得[1]。
1.3 IV 分析與 MR 基本假設
MR 與 IV 的基本假設在本質上是一致的,但因 MR 是將遺傳變異作為 IV,因此它在說法上具有一些特殊性。
假設一:Z 與感興趣的暴露 X 相關。
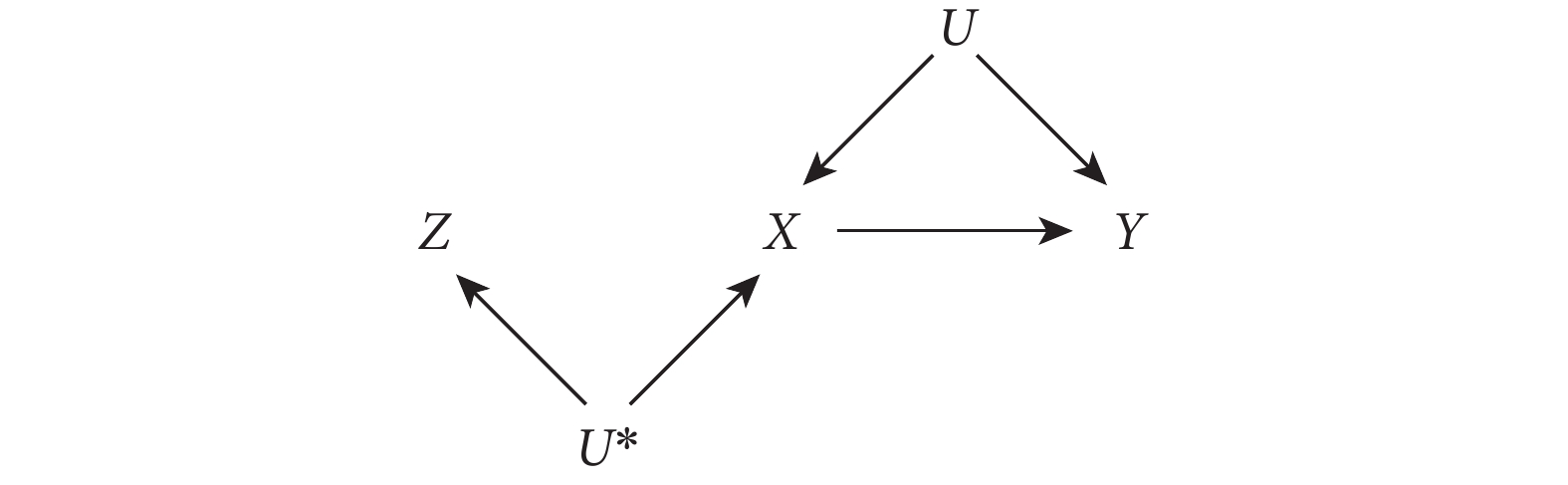
此假設在 MR 中的表達為遺傳變異與(非遺傳的)感興趣的暴露相關。這一假設中,Z(遺傳變異)與暴露 X 的相關關系并不要求一定是因果關系,如圖 2 所示的 Z 也滿足這一假設。需要注意的是,在實際研究中采用 IV 分析時,需要 Z 與 X 有較強相關關系。當這種相關關系較弱時,Z 就被稱為“弱工具”。這種弱工具會給 MR 研究造成很大局限性。
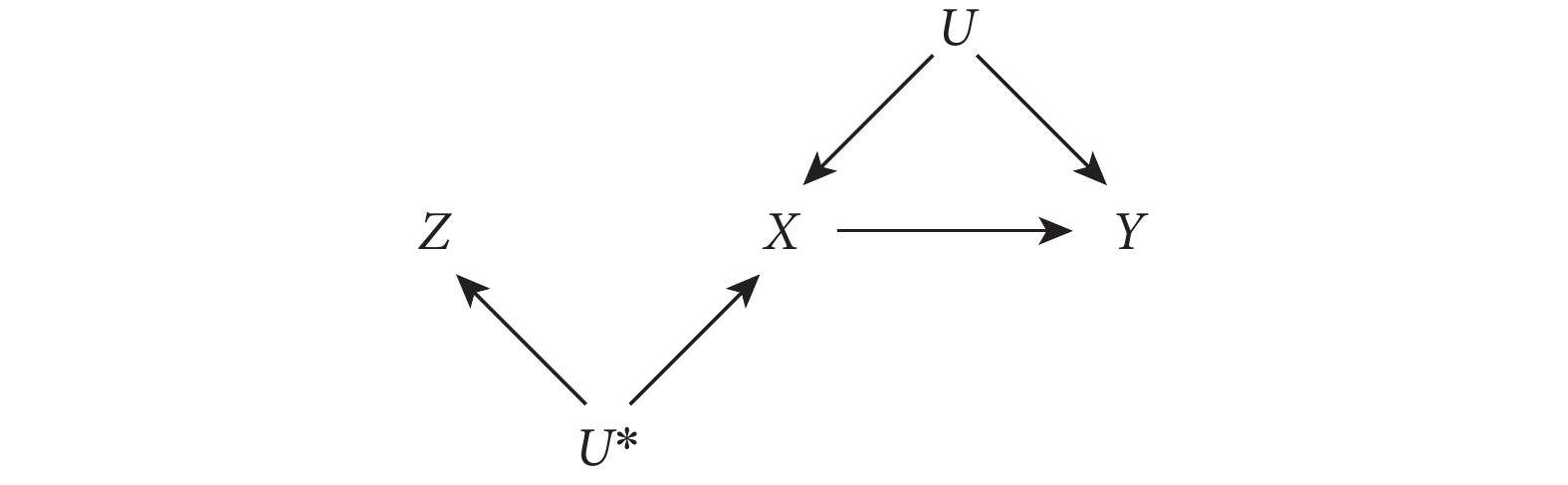 圖2
有效 IV 與暴露無因果關系的有向無環圖
圖2
有效 IV 與暴露無因果關系的有向無環圖
假設二:Z 獨立于暴露 X 與結局 Y 之間的混雜因素 U。
MR 中基因型不應與暴露-結局關系間的混雜因素相關。雖然這一假設通常很難直接證明,但有時候可通過比較暴露-結果關系的變異和已知混雜因素之間的聯系證偽[22]。
假設三:Z 對結果 Y 無直接影響,它只通過暴露 X 來影響結果。
這一假設又被叫做排除限制標準,在 MR 中又叫無多效性假設。過去在研究中很難證明這一假設成立,但近年發展出的一些方法能在違反此假設的情況下,檢測其存在,并對暴露與結局的因果效應進行無偏估計[8]。
2 MR 的潛在問題與局限性
基因特殊性質使其很可能作為一個有效的 IV。然而,在一些情況下,MR(IV)的假設及其生物學合理性可能不成立。孟德爾第二定律并非對所有遺傳變異適用,因為并不是決定所有性狀的基因都是獨立(隨機)分離的。這一現象被叫做連鎖不平衡(linkage disequilibrium,LD)。另外,基因作為 IV 無法避免弱工具、人群分層及發育代償等問題造成的偏倚。了解這些問題帶來的局限性和潛在偏倚來源對于合理進行 MR 研究至關重要。
2.1 LD
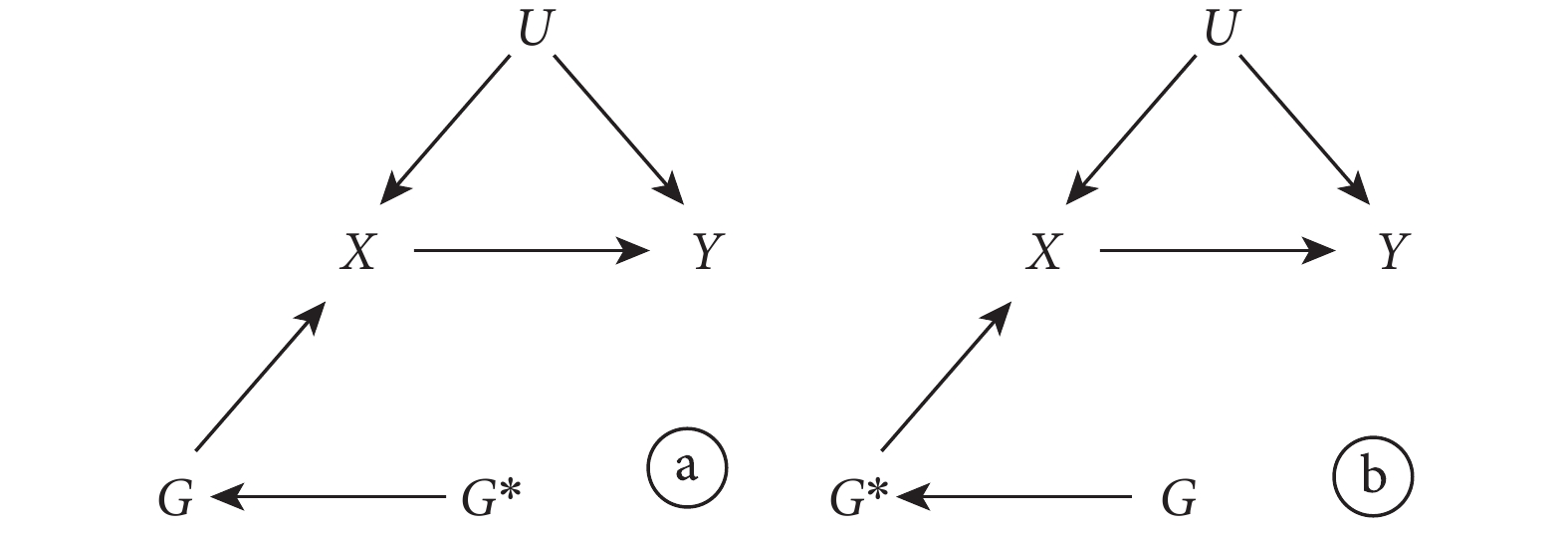
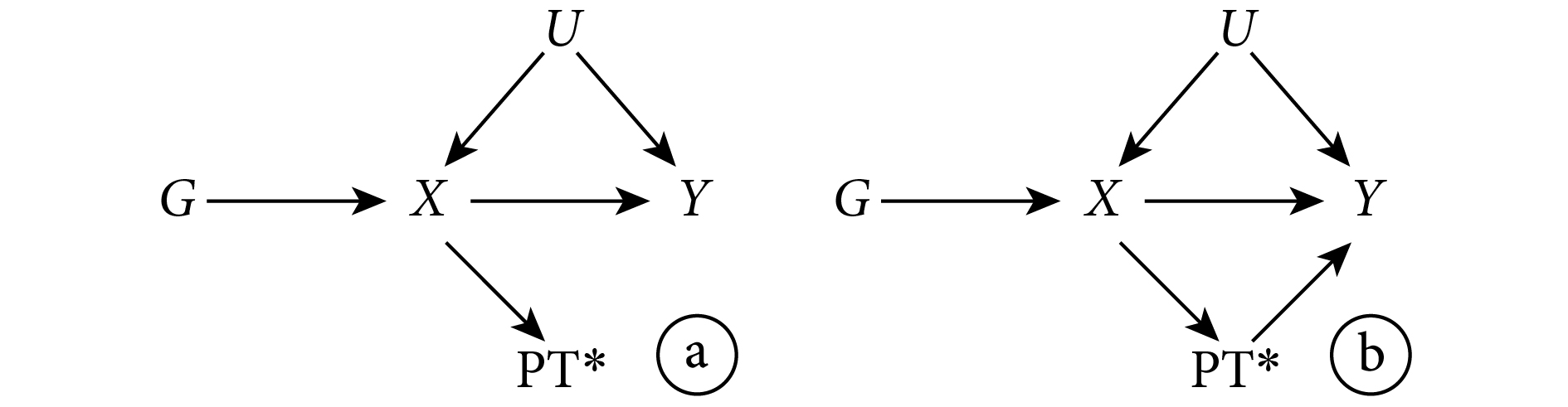
LD 是用來描述非同源染色體上由于遺傳位點彼此接近而產生的相關性[3]。根據這一定義,如果用來作為 IV 的遺傳變異具有連鎖不平衡性,那么它與其他遺傳變異之間就存在關聯。此時,該遺傳變異作為 IV 是否造成偏倚取決于與它存在關聯的變量與結果的關系。圖 3 表示的是 LD 不會造成偏倚的情況。此時,雖然存在連鎖不平衡性,但作為 IV 的遺傳變異 G 對結果 Y 影響的路徑均需通過暴露 X( 和
和  ),依然滿足 IV 的所有假設。相反,圖 4 表示了 LD 會造成偏倚的情況。此時,G 對結果 Y 的影響部分不通過暴露 X(
),依然滿足 IV 的所有假設。相反,圖 4 表示了 LD 會造成偏倚的情況。此時,G 對結果 Y 的影響部分不通過暴露 X( ),IV 的第三條假設遭到違反,從而導致偏倚。對于此問題可能的解決方法是在不同 LD 結構的人群中進行 MR 研究。此外,解決多效性問題的方法也可被用于解決此問題(見下文)。
),IV 的第三條假設遭到違反,從而導致偏倚。對于此問題可能的解決方法是在不同 LD 結構的人群中進行 MR 研究。此外,解決多效性問題的方法也可被用于解決此問題(見下文)。
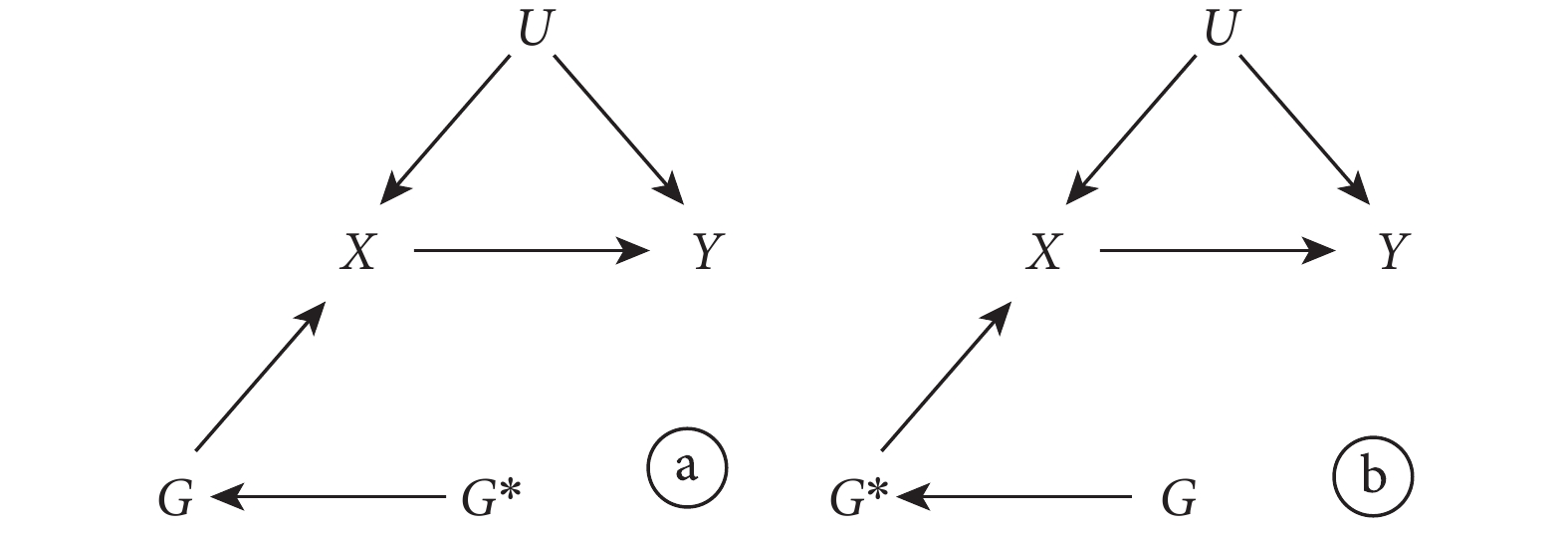 圖3
選用的遺傳變異存在連鎖不平衡的孟德爾隨機化研究示意圖,其中 IV 分析的假設未被違反
圖3
選用的遺傳變異存在連鎖不平衡的孟德爾隨機化研究示意圖,其中 IV 分析的假設未被違反
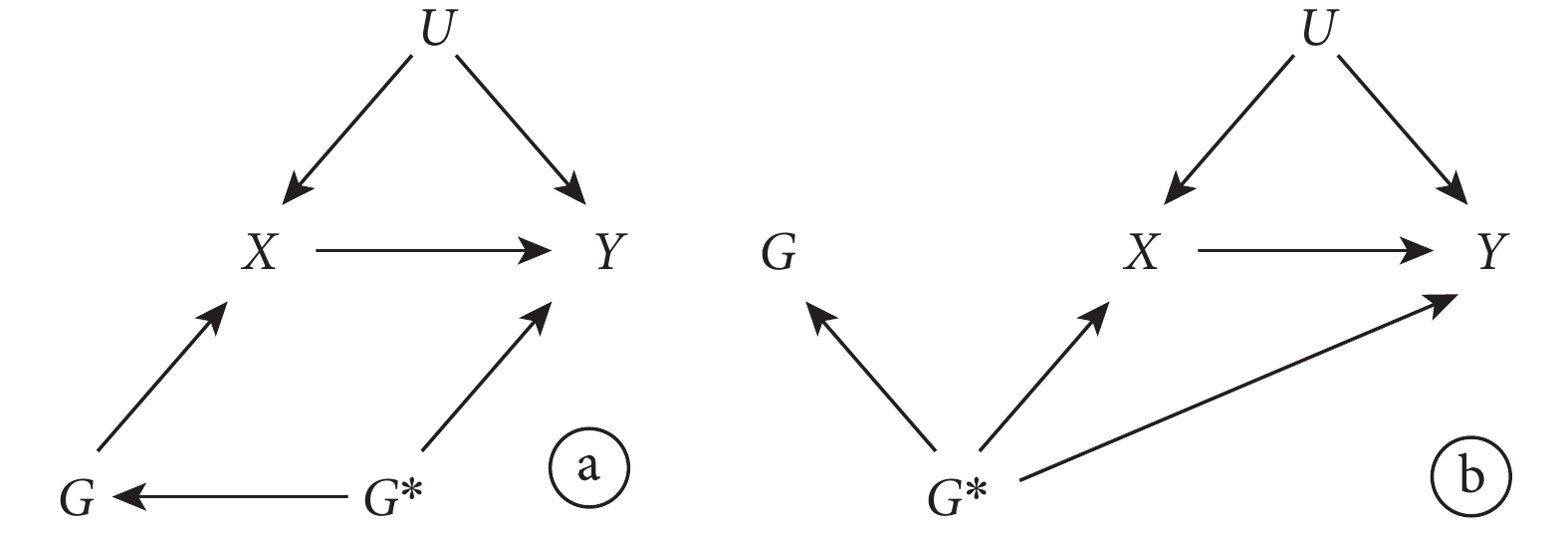 圖4
選用的遺傳變異存在連鎖不平衡的孟德爾隨機化研究示意圖,其中 IV 分析的假設違反
圖4
選用的遺傳變異存在連鎖不平衡的孟德爾隨機化研究示意圖,其中 IV 分析的假設違反
2.2 多效性
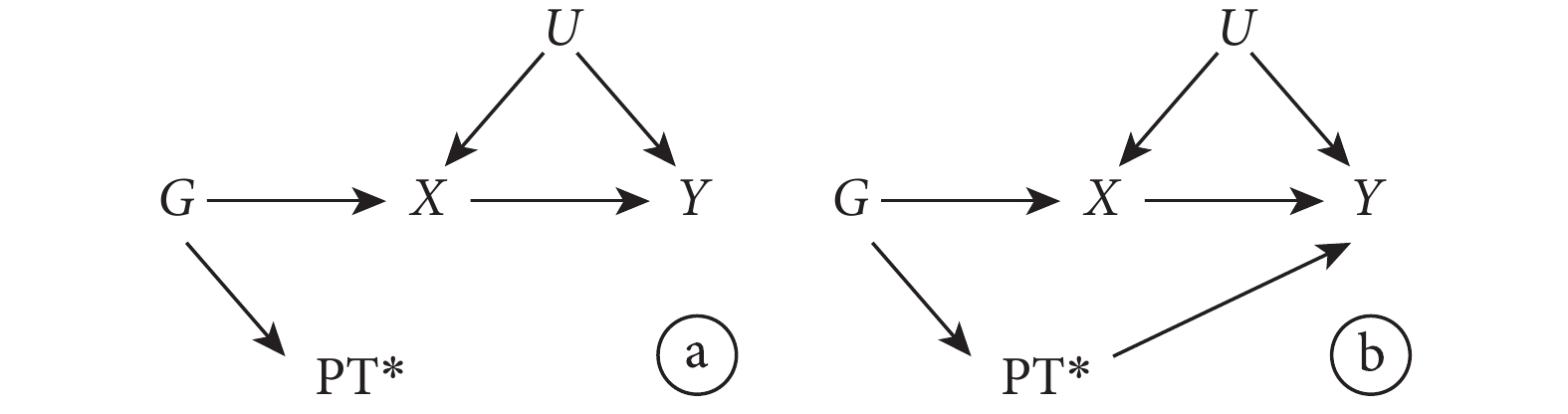
多效性是指一個基因位點影響多種表型的現象[23]。多效性是否導致偏倚取決于多效性的類型。在 MR 的背景下,多效性發生有兩種機制[1]:① 一個位點影響一個表型(暴露),通過此表型(暴露)又影響其他表型;② 單個位點直接影響多個表型。前者被稱為“Ⅱ型多效性”[24];后者被稱為“Ⅰ型多效性”[24]。
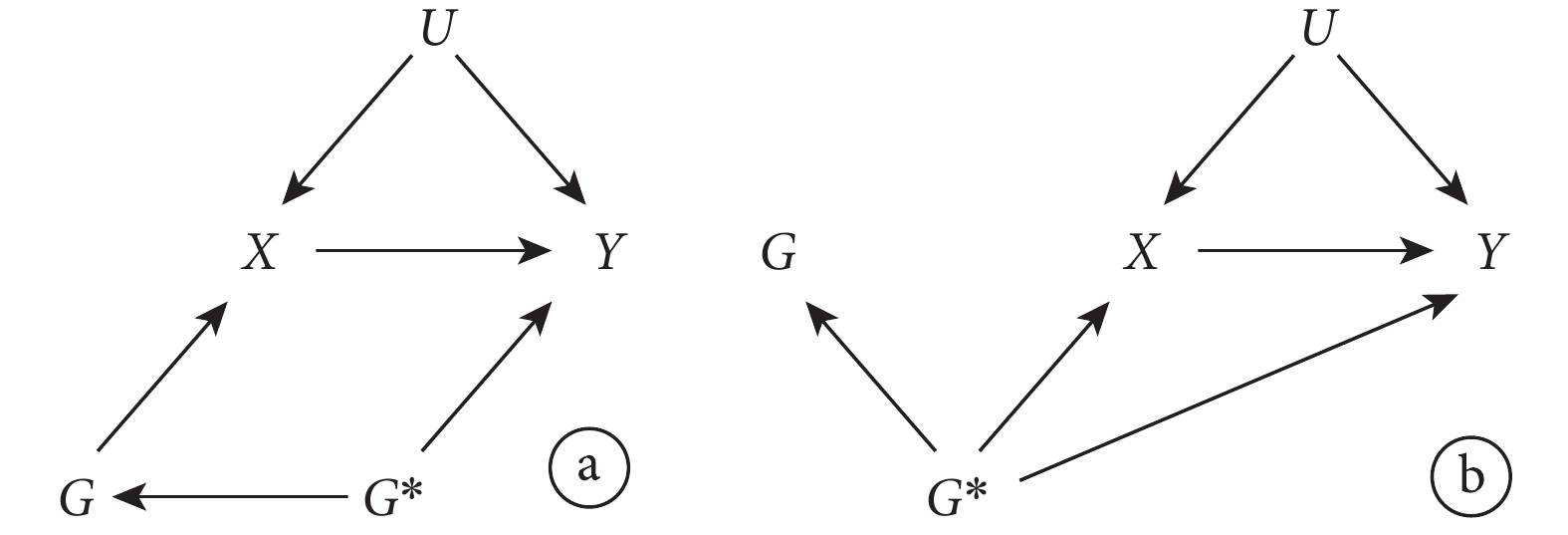
Ⅱ型多效性不但不會帶來偏倚,它還是 MR 方法的精髓所在。在該方法中,如圖 5 所示,通過使用與該表型(暴露)X 相關的遺傳變異 G 來估計的因果效應包括了下游的間接因果效應( )。此時無論其他的基因表型
)。此時無論其他的基因表型  是否與結果有關聯,IV 假設都不會被違反。因 G 對 Y 影響的兩條路徑都需經過 X(
是否與結果有關聯,IV 假設都不會被違反。因 G 對 Y 影響的兩條路徑都需經過 X( 和
和  )。
)。
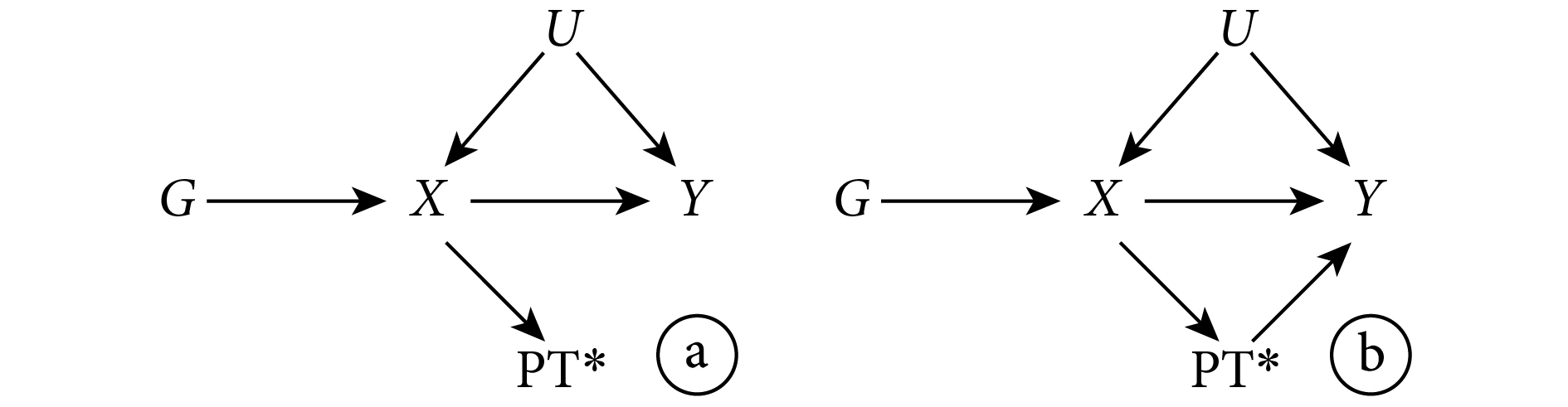 圖5
選用的遺傳變異存在基因多效性的孟德爾隨機化研究示意圖,其中 IV 分析的假設未被違反
圖5
選用的遺傳變異存在基因多效性的孟德爾隨機化研究示意圖,其中 IV 分析的假設未被違反
然而,Ⅰ型多效性卻很可能影響 MR 研究結果的可信性。如圖 6 所示,當  與結果 Y 無關聯時,IV 假設成立。但當
與結果 Y 無關聯時,IV 假設成立。但當  與結果 Y 有關聯時,G 對 Y 的影響有一條路徑不通過 X(
與結果 Y 有關聯時,G 對 Y 的影響有一條路徑不通過 X( ),IV 假設不成立。此時可使用一些統計學方法如 MR-Egger 回歸,中位數估計等,進行敏感性分析以檢測和校正多效性產生的偏倚。
),IV 假設不成立。此時可使用一些統計學方法如 MR-Egger 回歸,中位數估計等,進行敏感性分析以檢測和校正多效性產生的偏倚。
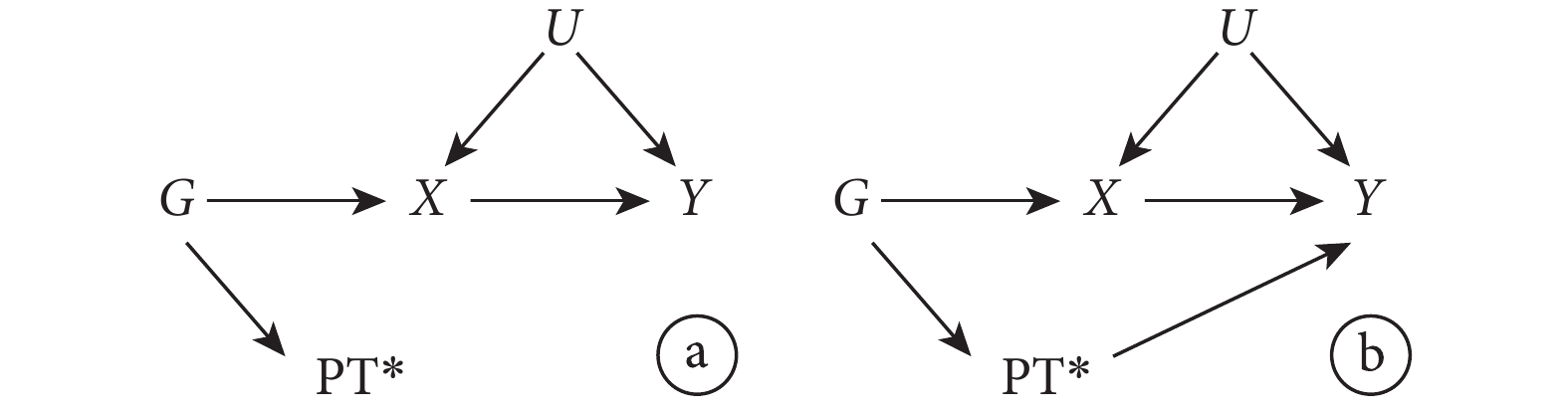 圖6
選用的遺傳變異存在基因多效性的孟德爾隨機化研究示意圖,其中 IV 分析的假設違反
圖6
選用的遺傳變異存在基因多效性的孟德爾隨機化研究示意圖,其中 IV 分析的假設違反
2.3 人群分層
人群分層是指存在不同疾病發生率(或不同性狀分布)和不同等位基因頻率的群體[25]。當基因(IV)-暴露因素相關性與基因-結果相關性在不同的研究人群中獲得時,整個研究人群中基因型和疾病之間就很可能存在虛假的(混雜的)關聯。解決這一問題的主要方法有:對種族同質群體進行分析,并使用祖先信息標記或對全基因組數據的主要成分進行校正[1];或將研究限制在家庭背景下,例如在兄弟姐妹之間進行分析[26]。
2.4 發育代償
發育代償是指個體試圖對干擾發育的環境或基因啟動代償性反應以對抗作為工具的遺傳變異所導致的影響[27, 28]。這種反應可通過改變暴露-結果的效應使得 MR 研究的結果不準確。但在發育期之后(發育代償期后)首次出現的遺傳變異不會受到發育代償的影響[11]。此外,當用母體基因型來估計子宮內暴露對后代結果的影響時,發育失代償也不會帶來偏倚[3]。目前,針對這一問題,還沒有發展出通用的校正和估計方法。只能利用特定的生物學知識,了解遺傳變異對表型產生影響的時期以判斷是否需要警惕發育代償可能產生的影響。
2.5 弱工具
當一個作為 IV 的遺傳變異對暴露影響很小時,該遺傳變異就是一個“弱工具”。如果 IV 非常弱,它就只能提供有關暴露很少的信息。原則上,這會導致對因果效應的不精確的估計、第一類錯誤概率增加(對零假設的過度拒絕),及放大違背其他核心假設所帶來的偏倚[29]。單樣本設定中,由于暴露和結果之間的混雜因素,分子和分母的回歸系數之間存在相關性,弱工具偏倚會導致高估暴露-結局關聯[30];兩樣本設定中,弱工具可能帶來回歸稀釋偏倚,從而低估暴露-結局關聯[31]。利用兩階段最小二乘法(two-stage least square,2SLS)中第一階段回歸的 Cragg-Donald F 統計量可檢驗 IV 強度和樣本大小的結合是否容易受到弱工具偏倚的影響。F 統計量大于 10 通常表示 IV 方法的有效性較好[31]。在 GWAS 背景下,還通常采用 P<5×10-8對 IV 進行篩選[32]。然而,P 值為 5×10-8對應的 F 值約為 29。這一閾值較為嚴格,容易導致從 GWAS 中獲得的平均估計效應比真實的遺傳效應小。這一現象也被稱為“贏家詛咒”[32]。對 MR-Egger 來說,F 統計量不足以作為儀器強度的指標,它會低估 MR-Egger 中的回歸稀釋偏倚。近年來,Bowden[31]等提出  統計量可用來定量估計這種回歸稀釋偏倚。目前已發展出許多控制弱 IV 偏倚的方法,如采用基因風險評分[33],有限信息最大似然(limited information maximum likelihood,LIML)估計[34]及持續更新估計(continuously updating estimator,CUE)[34]。
統計量可用來定量估計這種回歸稀釋偏倚。目前已發展出許多控制弱 IV 偏倚的方法,如采用基因風險評分[33],有限信息最大似然(limited information maximum likelihood,LIML)估計[34]及持續更新估計(continuously updating estimator,CUE)[34]。
2.6 樣本重疊
在兩樣本 MR 研究中,不同的數據集往往不是完全不同的,一些人群數據可能同時存在于兩個主流數據集中。因此,表面上看起來為兩樣本設定的 MR 研究,樣本重疊也可能導致弱工具造成的偏倚更類似于單樣本設定[30]。對于 IVW 方法,隨著樣本重疊比例的增加,弱工具偏倚的方向呈現出從雙樣本設置(低估暴露-結果效應)到單樣本設置(高估暴露-結果效應)線性變化[35]。對于 MR-Egger 方法是否遵循這一規律,還有待進一步研究。
3 MR 基本設定和方法
迄今為止,MR 發展出了許多的設定和衍生方法。本文僅介紹最常用的兩種設定:單樣本 MR 和兩樣本 MR 及幾種常用方法:2SLS、逆方差加權(Inverse-variance weighted,IVW)、MR-Egger 回歸、中值估計及基于個體數據(individual personal data,IPD)的校正方法。本文介紹的所有方法都能夠用于包含多個 IV 的數據中。
3.1 單樣本與兩樣本 MR
遺傳變異與暴露及遺傳變異與結果的關系均在同一樣本中獲得的 MR 研究叫做單樣本 MR。10 年前,MR 的研究幾乎都是在單樣本設定下進行的。雖然在單樣本中,IV 分析的估計值是漸近無偏的,但在有限樣本中卻可能存在很大偏倚[36, 37]。在單樣本中,分子和分母的回歸系數之間的相關性造成了弱工具偏倚,它會高估暴露和結果之間的關聯,其偏倚大小取決于 IV 和暴露之間的關聯強度[9]。
在實踐中,研究者或許并不能從同一樣本中獲得完整的遺傳變異與暴露及遺傳變異與結果關系的數據。如果分別從兩個不重疊的數據集中獲得這兩類數據,這樣的研究就稱為兩樣本 MR[38]。使用兩樣本設定相對于單樣本設定有許多優勢。首先,在兩樣本設定中,弱工具偏倚是偏向于零假設的,零假設方向的偏倚相對于相關性方向的偏倚更容易接受,因為這一方向的偏倚比較保守,不會增加假陽性結果的可能性。其次,在一些情況下,同一組個體暴露和結局的數據難以同時測量,兩樣本 MR 大大增加了 MR 研究的應用范圍。在所有 MR 研究中,使用兩樣本設計的比例從 2011 年的接近 0% 上升至 2016 年的 40% 左右[39]。
3.2 2SLS
最簡單的 MR 估計方法是 2SLS[3]。此方法有兩個基本步驟。首先,以 Z 為自變量,以感興趣的暴露因素 X 為因變量進行最小二乘回歸;第二步,以第一步回歸的預測值為自變量,以結果 Y 為因變量進行最小二乘回歸。對于多個 IV,2SLS 估計可被認為是每一個 IV 估計值的加權平均值,其中權重由第一階段回歸中 IV 的相對強度決定[12]。假設有 j 個可用的 IV,每個個體對應的 IV 的值用  表示。第一階段的回歸模型可表示為:
表示。第一階段的回歸模型可表示為:
 |
根據此模型可得到  的預測值:
的預測值:
 。將此預測值帶入第二階段回歸模型:
。將此預測值帶入第二階段回歸模型:
 |
其中, 和
和  是誤差項。通過此方法得出的
是誤差項。通過此方法得出的  是第 j 個 IV 所對應的暴露-結果效應的估計值。
是第 j 個 IV 所對應的暴露-結果效應的估計值。
然而,2SLS 可能出現弱工具偏倚。此外,在無法獲得個體層面的數據、只能獲得關于遺傳變異與暴露因素和結果之間關系的匯總數據時,不能采用 2SLS 方法[7]。
3.3 逆方差加權(inverse-variance weighted,IVW)
逆方差加權法是 MR 匯總數據的標準方法[40]。它不需要個體層面的數據,可直接利用匯總數據計算因果效應值。在使用多個遺傳變異作為 IV 的數據中,對于第 j 個 IV,如果 IV 的相關假設滿足,暴露對結果的因果效應的估計值  為第 j 個遺傳變異與結果相關性的估計值
為第 j 個遺傳變異與結果相關性的估計值  和其與暴露的相關性估計值
和其與暴露的相關性估計值  的比值[3],即:
的比值[3],即:
 |
如果遺傳變異不相關(非連鎖不平衡),那么每個遺傳變異所對應的估計值可總和成一個整體的加權估計,即:
 |
 是第 j 個 IV 的基因-結果關聯估計值的方差。如果遺傳變量之間不相關,IVW 的估計值與用于個體水平數據 2SLS 方法的估計值相等[41]。然而,與所有 IV 方法一樣,IVW 方法也容易受到弱工具偏倚的影響。有模擬研究結果表明,IVW 法的弱工具偏倚水平與 2SLS 法相同,其大小可通過 F 統計量進行量化[42]。
是第 j 個 IV 的基因-結果關聯估計值的方差。如果遺傳變量之間不相關,IVW 的估計值與用于個體水平數據 2SLS 方法的估計值相等[41]。然而,與所有 IV 方法一樣,IVW 方法也容易受到弱工具偏倚的影響。有模擬研究結果表明,IVW 法的弱工具偏倚水平與 2SLS 法相同,其大小可通過 F 統計量進行量化[42]。
3.4 MR-Egger 回歸
MR-Egger 回歸是近年提出的檢測和調整 MR 分析中多效性的方法。此方法中,給定一組遺傳變異,首先估計每個遺傳變異與結果相關性  及遺傳變異與暴露的相關性
及遺傳變異與暴露的相關性  ,然后擬合線性函數[8]:
,然后擬合線性函數[8]:
 |
暴露對結果的因果效應的估計值  可通過下式計算[8]:
可通過下式計算[8]:
 |
MR-Egger 回歸的截距估計值  是各遺傳變異多效性效應估計值的平均值。MR-Egger 方法放寬了 IVW 方法中遺傳變異之間不存在多效性的要求。它假設工具-暴露和工具-結果的關聯是獨立的。這被稱為 InSIDE 假設[43](instrument strength independent of direct effect,InSIDE),與嚴格的排除限制標準相比,這一假設相對較弱。但是,IVW 和 MR-Egger 回歸方法理論上都需假設基因變異-暴露關聯是無誤差的測量(no measurement error,NOME)[31]。MR-Egger 放寬多效性假設的代價是其違反 NOME 假設后,帶來的偏倚比 IVW 估計更大,且特別容易受到弱工具偏倚的影響[31]。另外,只有在基因多效性具有方向性的時候(即多效性具有非零平均值),MR-Egger 回歸才能檢測出多效性[8]。因為只有在這種情況下,
是各遺傳變異多效性效應估計值的平均值。MR-Egger 方法放寬了 IVW 方法中遺傳變異之間不存在多效性的要求。它假設工具-暴露和工具-結果的關聯是獨立的。這被稱為 InSIDE 假設[43](instrument strength independent of direct effect,InSIDE),與嚴格的排除限制標準相比,這一假設相對較弱。但是,IVW 和 MR-Egger 回歸方法理論上都需假設基因變異-暴露關聯是無誤差的測量(no measurement error,NOME)[31]。MR-Egger 放寬多效性假設的代價是其違反 NOME 假設后,帶來的偏倚比 IVW 估計更大,且特別容易受到弱工具偏倚的影響[31]。另外,只有在基因多效性具有方向性的時候(即多效性具有非零平均值),MR-Egger 回歸才能檢測出多效性[8]。因為只有在這種情況下, 才會是非零的值。例如,當所有的遺傳變異都表現出多效性但其方向不同,但在平均水平下它們相互抵消(這一情況被稱為平衡多效性[8]),MR-Egger 回歸就無法檢測出多效性。
才會是非零的值。例如,當所有的遺傳變異都表現出多效性但其方向不同,但在平均水平下它們相互抵消(這一情況被稱為平衡多效性[8]),MR-Egger 回歸就無法檢測出多效性。
3.5 中值估計
中值估計包括簡單的中位數估計、加權中值估計和懲罰加權中值估計。簡單的中位數估計方法非常容易理解。設  表示第 j 個遺傳變異所對應的暴露-結果效應的估計值(從最小到最大排列)。如果遺傳變異的總數為奇數(J=2k+1),簡單中值估計就取其中間值
表示第 j 個遺傳變異所對應的暴露-結果效應的估計值(從最小到最大排列)。如果遺傳變異的總數為奇數(J=2k+1),簡單中值估計就取其中間值  。如果是偶數(J=2k),其估計值就取
。如果是偶數(J=2k),其估計值就取  。我們可將簡單的中值估計理解為具有相同權重的加權中值估計。但在不同遺傳變異所對應的估計精度差異很大時,該方法具有低效性[9]。
。我們可將簡單的中值估計理解為具有相同權重的加權中值估計。但在不同遺傳變異所對應的估計精度差異很大時,該方法具有低效性[9]。
加權中值估計考慮了估計精度差異大的問題。在此方法中,設  為第 j 個遺傳變異估計值的權重,設
為第 j 個遺傳變異估計值的權重,設  為 j 個估計值(從最小到最大排列)權重的總和。如果進行了標準化,則
為 j 個估計值(從最小到最大排列)權重的總和。如果進行了標準化,則  等于 1。加權中值估計是取
等于 1。加權中值估計是取  等于 50% 的估計值,其中
等于 50% 的估計值,其中  。與 IVW 方法類似,該方法的權重
。與 IVW 方法類似,該方法的權重  一般使用每個遺傳變異的方差逆權重[44]:
一般使用每個遺傳變異的方差逆權重[44]:
 |
值得注意的是,簡單的中位數估計要求至少 50% 的遺傳變異為有效的 IV,而加權中值估計僅要求至少 50% 由遺傳變異貢獻的權重是有效的。
雖然無效 IV 的存在不影響中值估計的漸進無偏性,但在有限樣本中,它還是可能導致偏倚。當無效 IV 的估計在真實的因果效應兩邊出現不平衡時(如一個研究中有多個無效 IV,而這些 IV 的估計值全部大于或小于真實的估計值),就可能出現偏倚。在這種情況下,可采用懲罰加權中值估計進行校正,以降低具有異質性的估計值的遺傳變異的權重。采用此方法時,應首先用 Cochran’s Q 值量化估計值間的異質性[9]:
 |
其中, 為 IVW 方法得出的估計值[45]。在所有遺傳變異均為有效 IV 及所有變量均可識別出相同的因果關系的零假設下,
為 IVW 方法得出的估計值[45]。在所有遺傳變異均為有效 IV 及所有變量均可識別出相同的因果關系的零假設下, 服從自由度為 1 的卡方分布。通過這一分布,找到與每個遺傳變異
服從自由度為 1 的卡方分布。通過這一分布,找到與每個遺傳變異  值對應的 P 值(用
值對應的 P 值(用  表示)。然后將權重乘以 P 值再乘以 20(如果 P 值大于 0.05,則乘以 1)進行懲罰。最終得到懲罰后的權重(
表示)。然后將權重乘以 P 值再乘以 20(如果 P 值大于 0.05,則乘以 1)進行懲罰。最終得到懲罰后的權重( )[9]:
)[9]:
 |
3.6 基于 IPD 的校正方法
MR-Egger 回歸與中值估計均是針對匯總數據的對于無效 IV 的校正方法。針對 IPD,目前常見的方法有:TSHT(two-stage hard thresholding)[46]、限制 IV 法(constrained instrumental variable,CIV)[47]和 sisVIVE(some invalid some valid IV estimator)[48]。
TSHT 是一種基于眾數的估計方法。它是通過對不同候選工具的估計進行兩兩比較來實現的,當兩種基因變異的估算值相似時,它們就會“投票”給對方。最后,基于這些選票中最多的一組基因變異得到總的估計[46]。當無效的遺傳變異估計的都是不同的因果效應時,即使少于 50% 的遺傳變異是有效工具,真正的因果效應也能夠被識別[49]。然而,由于這些“投票”是由一個固定的閾值決定的,當比較度量接近閾值時,TSHT 的估計對數據的微小變化將非常敏感[49]。CIV 方法通過將潛在多效表型的相關性縮小到零來消除多效效應[47]。同時,CIV 的懲罰算法對有效基因型的選擇較為嚴格,并考慮了稀疏性問題。當基因型數量大于樣本數量時,CIV 能減少由于使用多個基因型而導致的過擬合問題[47]。然而,當研究者只能獲取部分多效表型而不是全部多效表型時,CIV 就無法消除多效表型的影響[47]。sisVIVE 的中心思想是通過 LASSO 型懲罰對遺傳變異進行稀疏選擇[48]。在存在多效性及 IV 對結局有直接因果效應時,sisVIVE 的估計也較為穩健[48]。
迄今為止,MR 除單樣本和兩樣本設定之外,還發展出了許多其他設計形式,如雙向 MR[50]、兩步 MR[51]和析因 MR[52]等。同樣,除了上述的四種計算方法外,近年來還發展出了更多新方法,如其他 Mode-based 估計[53]、穩健多效性 MR[54]和貝葉斯模型平均[55]等。
4 小結
MR 是一種結合了靈活和穩健性的統計方法。MR 在觀察性研究中使用遺傳變異作為 IV 來檢測和量化因果關系。隨著數據生成成本的持續降低,它的應用范圍將會持續擴大。使用遺傳變異作為 IV 可能避免觀察性研究(混雜、反向因果關系、回歸稀釋偏倚)和 RCT(代表性和可行性問題)在進行因果推斷時的一些限制。但這種方法本身也同樣有許多局限性(LD、多效性、發育補償等)。另外,目前許多 MR 研究存在報告不規范問題。Burgess 等[56]于 2019 年發布了 MR 應用和規范報告的指南。在國內,CSCO 生物統計學專家委員會 RWS 方法學組也于近期發表了《孟德爾隨機化模型及其規范化應用的統計學共識》[57]。研究者應遵循上述指南和共識規范應用和報告 MR 研究。
總之,本文介紹了 MR 的基本原理和 3 個核心假設,討論了 MR 的 6 個主要局限性,介紹了常見的多種估計方法,希望能幫助研究者加深對 MR 研究的認識,有助于提高 MR 研究的質量。
傳統的觀察性研究中,潛在混雜和反向因果關系會影響其因果推斷能力[1, 2]。孟德爾隨機化(Mendelian randomization,MR)是工具變量(instrumental variable,IV)分析的一種類型,它使用遺傳變異作為 IV 來檢測和量化因果關系[3]。由于能克服潛在混雜和反向因果關系的影響,近年來 MR 在觀察性研究中的應用越來越廣泛[4, 5]。早期的 MR 研究通常在小樣本人群中進行,且僅使用了少量的遺傳變異[6],這使得 MR 研究的效力較低。然而,隨著生物學界發現了大量與特定性狀緊密相關的遺傳變異,加上許多大樣本全基因組關聯研究(genome-wide association study,GWAS[5])公開發布了數十萬個暴露和疾病與遺傳變異關系的匯總數據[7],這一領域發生了一場革命。這些匯總數據使得研究者能估計大樣本數據中的遺傳關聯,從而促進了 MR 研究發展。近年來,該領域在方法學上也迅速更新,新方法克服了傳統 MR 方法的一些特定限制[8-11],但其同樣存在局限性。只有正確了解 MR 背后的原理、局限性及不同方法的適用條件才能針對不同的研究問題和特定的數據正確應用 MR。本文介紹 MR 的基本概念和原理,并解讀其在使用中的一些問題和局限性,介紹 MR 研究迄今為止常用的幾種方法,以期為研究者進行孟德爾隨機化研究提供指導。
1 MR 基本原理和假設
MR 是 IV 分析的一種特殊類型,它遵循 IV 分析的基本原理和相關假設[12]。
1.1 IV 與 MR 基本原理
IV 分析在經濟學中的應用已有許多年,它是許多計量經濟學統計分析方法的基礎[13]。但在醫學研究中,它最早用于處理隨機臨床試驗中的依從性問題[14]。因此,本文將通過闡述 IV 如何處理隨機試驗的依從性問題,并比較多因素回歸、傾向性評分(propensity score,PS)與 IV 分析在因果推斷中的異同點,以幫助讀者理解其原理和本質。
首先,假設一個隨機雙盲對照試驗,在此試驗中,研究者隨機將患者分配到干預組(表示為 Z=1)和對照組(表示為 Z=0)。由于存在不依從的情況,患者實際是否接受干預可能與分配情況不符,用 X 表示患者實際是否接受干預的情況(X=1 接受干預,X=0 未接受干預);用 Y 表示結局;U 代表所有混雜因素(包括可測和不可測的混雜因素)。圖 1 中的有向無環圖(directed acyclic graph,DAG)展示了以上變量間的關系。
圖1
IV 分析各變量關系的有向無環圖
在此試驗中,若直接估計 與 Y 之間的因果效應會因不依從問題而受到混雜因素的影響,這是因為患者最終接受的干預狀態并不是隨機的。但我們可利用隨機分配狀態變量 作為有效的 IV,來得到因果效應的估計。通過 來估計 與 Y 之間的平均因果效應是無偏的,因為它不會受到任何可測和不可測的混雜因素的影響。此時,X 對 Y 效應的 IV 估計可表示為:
|
其中,分子代表的是 Z 對 Y 的效應,分母代表的是 Z 對 X 的效應。由圖 1 可知, 與 Y 之間及 與 X 之間無混雜因素影響,因此分子、分母都是無偏估計。用上式來計算 X 對 Y 的因果效應,相當于只使用了結果 Y 中由 Z 引起的部分變化,從而避免了 Y 與 X 之間混雜因素的影響。
在觀察性研究中,研究對象的分配不受研究者控制,各組之間的混雜因素可能存在重要差異,因此結果的差異可能是由于暴露本身、可測量的和不可測量的混雜因素在組間的差異,或兩者共同造成的。多因素回歸分析常用于校正由可測量的混雜因素引起的偏倚。然而,研究者很難通過回歸模型發現暴露組之間的混雜因素沒有充分重疊的情況,從而使得結果的可信性降低[15]。另外,許多回歸模型的準確性依賴于足夠的結局事件數。例如,為獲得準確和穩定結果,Logistic 回歸模型要求結局事件數/協變量數大于 8~10 個[16]。由 Rubin 和 Rosenbaum[17]提出的 PS 分析能夠克服以上難題。PS 被定義為給定一組可測量的協變量,個體接受暴露或治療的條件概率,在評估觀察性研究中的因果效應時,可用來調整選擇偏倚[17]。與傳統回歸模型相比,分析中考慮的潛在混雜因素的數量是不受限制的。因此,PS 方法理論上可通過對所有可能的可測量混雜因素創建偽隨機化來增加組間的可比性。然而,與多因素回歸類似,PS 分析也無法校正由于不可測量或未知混雜因素造成的偏倚。IV 分析與前兩種分析方法最大的區別在于它并非將研究對象按照是否接受暴露或治療進行比較,而是將研究對象根據 IV 的情況進行分組比較[15]。由于 IV 在研究對象中的分布是隨機的,理論上,如果實施得當,它能同時校正可測量與不可測量混雜因素產生的偏倚。
1.2 基因作為 IV
雖然 IV 分析能夠克服觀察性研究中最棘手的不可觀測混雜的問題,但一個有效的 IV 所需滿足的假設非常嚴格,要找到一個可用的 IV 十分困難[18]。隨著近十年來基因組學迅速發展,研究者們逐漸意識到以基因作為 IV 能在很大程度上克服以上難題[19]。
基因作為 IV 的優勢要從孟德爾第二定律[20]說起:“每一對具有區別的性狀或行為與父母之間的其他差異無關,等位基因從父母隨機分離后遺傳給后代。”此定律表明一個性狀的遺傳獨立于其他性狀的遺傳且呈現隨機性,因此后代基因型不太可能與人群中的環境混雜因素相關[21]。其次,基因型分布在時間上先于后天暴露,基因型與疾病之間的聯系不會受到反向因果關系的影響。再次,與暴露相關的基因型通常從出生到成年都與之相關,因此,在因果推斷中可避免因誤差而造成的衰減(回歸稀釋偏倚)[19]。此外,這一定律在人群中是普遍存在的,避免了隨機對照試驗(randomized controlled trials,RCT)中的代表性問題。最后,在 GWAS 和高通量基因組技術的時代,人群遺傳數據通常可方便地從大型公開數據庫中獲得[1]。
1.3 IV 分析與 MR 基本假設
MR 與 IV 的基本假設在本質上是一致的,但因 MR 是將遺傳變異作為 IV,因此它在說法上具有一些特殊性。
假設一:Z 與感興趣的暴露 X 相關。
此假設在 MR 中的表達為遺傳變異與(非遺傳的)感興趣的暴露相關。這一假設中,Z(遺傳變異)與暴露 X 的相關關系并不要求一定是因果關系,如圖 2 所示的 Z 也滿足這一假設。需要注意的是,在實際研究中采用 IV 分析時,需要 Z 與 X 有較強相關關系。當這種相關關系較弱時,Z 就被稱為“弱工具”。這種弱工具會給 MR 研究造成很大局限性。
圖2
有效 IV 與暴露無因果關系的有向無環圖
假設二:Z 獨立于暴露 X 與結局 Y 之間的混雜因素 U。
MR 中基因型不應與暴露-結局關系間的混雜因素相關。雖然這一假設通常很難直接證明,但有時候可通過比較暴露-結果關系的變異和已知混雜因素之間的聯系證偽[22]。
假設三:Z 對結果 Y 無直接影響,它只通過暴露 X 來影響結果。
這一假設又被叫做排除限制標準,在 MR 中又叫無多效性假設。過去在研究中很難證明這一假設成立,但近年發展出的一些方法能在違反此假設的情況下,檢測其存在,并對暴露與結局的因果效應進行無偏估計[8]。
2 MR 的潛在問題與局限性
基因特殊性質使其很可能作為一個有效的 IV。然而,在一些情況下,MR(IV)的假設及其生物學合理性可能不成立。孟德爾第二定律并非對所有遺傳變異適用,因為并不是決定所有性狀的基因都是獨立(隨機)分離的。這一現象被叫做連鎖不平衡(linkage disequilibrium,LD)。另外,基因作為 IV 無法避免弱工具、人群分層及發育代償等問題造成的偏倚。了解這些問題帶來的局限性和潛在偏倚來源對于合理進行 MR 研究至關重要。
2.1 LD
LD 是用來描述非同源染色體上由于遺傳位點彼此接近而產生的相關性[3]。根據這一定義,如果用來作為 IV 的遺傳變異具有連鎖不平衡性,那么它與其他遺傳變異之間就存在關聯。此時,該遺傳變異作為 IV 是否造成偏倚取決于與它存在關聯的變量與結果的關系。圖 3 表示的是 LD 不會造成偏倚的情況。此時,雖然存在連鎖不平衡性,但作為 IV 的遺傳變異 G 對結果 Y 影響的路徑均需通過暴露 X( 和 ),依然滿足 IV 的所有假設。相反,圖 4 表示了 LD 會造成偏倚的情況。此時,G 對結果 Y 的影響部分不通過暴露 X(),IV 的第三條假設遭到違反,從而導致偏倚。對于此問題可能的解決方法是在不同 LD 結構的人群中進行 MR 研究。此外,解決多效性問題的方法也可被用于解決此問題(見下文)。
圖3
選用的遺傳變異存在連鎖不平衡的孟德爾隨機化研究示意圖,其中 IV 分析的假設未被違反
圖4
選用的遺傳變異存在連鎖不平衡的孟德爾隨機化研究示意圖,其中 IV 分析的假設違反
2.2 多效性
多效性是指一個基因位點影響多種表型的現象[23]。多效性是否導致偏倚取決于多效性的類型。在 MR 的背景下,多效性發生有兩種機制[1]:① 一個位點影響一個表型(暴露),通過此表型(暴露)又影響其他表型;② 單個位點直接影響多個表型。前者被稱為“Ⅱ型多效性”[24];后者被稱為“Ⅰ型多效性”[24]。
Ⅱ型多效性不但不會帶來偏倚,它還是 MR 方法的精髓所在。在該方法中,如圖 5 所示,通過使用與該表型(暴露)X 相關的遺傳變異 G 來估計的因果效應包括了下游的間接因果效應()。此時無論其他的基因表型 是否與結果有關聯,IV 假設都不會被違反。因 G 對 Y 影響的兩條路徑都需經過 X( 和 )。
圖5
選用的遺傳變異存在基因多效性的孟德爾隨機化研究示意圖,其中 IV 分析的假設未被違反
然而,Ⅰ型多效性卻很可能影響 MR 研究結果的可信性。如圖 6 所示,當 與結果 Y 無關聯時,IV 假設成立。但當 與結果 Y 有關聯時,G 對 Y 的影響有一條路徑不通過 X(),IV 假設不成立。此時可使用一些統計學方法如 MR-Egger 回歸,中位數估計等,進行敏感性分析以檢測和校正多效性產生的偏倚。
圖6
選用的遺傳變異存在基因多效性的孟德爾隨機化研究示意圖,其中 IV 分析的假設違反
2.3 人群分層
人群分層是指存在不同疾病發生率(或不同性狀分布)和不同等位基因頻率的群體[25]。當基因(IV)-暴露因素相關性與基因-結果相關性在不同的研究人群中獲得時,整個研究人群中基因型和疾病之間就很可能存在虛假的(混雜的)關聯。解決這一問題的主要方法有:對種族同質群體進行分析,并使用祖先信息標記或對全基因組數據的主要成分進行校正[1];或將研究限制在家庭背景下,例如在兄弟姐妹之間進行分析[26]。
2.4 發育代償
發育代償是指個體試圖對干擾發育的環境或基因啟動代償性反應以對抗作為工具的遺傳變異所導致的影響[27, 28]。這種反應可通過改變暴露-結果的效應使得 MR 研究的結果不準確。但在發育期之后(發育代償期后)首次出現的遺傳變異不會受到發育代償的影響[11]。此外,當用母體基因型來估計子宮內暴露對后代結果的影響時,發育失代償也不會帶來偏倚[3]。目前,針對這一問題,還沒有發展出通用的校正和估計方法。只能利用特定的生物學知識,了解遺傳變異對表型產生影響的時期以判斷是否需要警惕發育代償可能產生的影響。
2.5 弱工具
當一個作為 IV 的遺傳變異對暴露影響很小時,該遺傳變異就是一個“弱工具”。如果 IV 非常弱,它就只能提供有關暴露很少的信息。原則上,這會導致對因果效應的不精確的估計、第一類錯誤概率增加(對零假設的過度拒絕),及放大違背其他核心假設所帶來的偏倚[29]。單樣本設定中,由于暴露和結果之間的混雜因素,分子和分母的回歸系數之間存在相關性,弱工具偏倚會導致高估暴露-結局關聯[30];兩樣本設定中,弱工具可能帶來回歸稀釋偏倚,從而低估暴露-結局關聯[31]。利用兩階段最小二乘法(two-stage least square,2SLS)中第一階段回歸的 Cragg-Donald F 統計量可檢驗 IV 強度和樣本大小的結合是否容易受到弱工具偏倚的影響。F 統計量大于 10 通常表示 IV 方法的有效性較好[31]。在 GWAS 背景下,還通常采用 P<5×10-8對 IV 進行篩選[32]。然而,P 值為 5×10-8對應的 F 值約為 29。這一閾值較為嚴格,容易導致從 GWAS 中獲得的平均估計效應比真實的遺傳效應小。這一現象也被稱為“贏家詛咒”[32]。對 MR-Egger 來說,F 統計量不足以作為儀器強度的指標,它會低估 MR-Egger 中的回歸稀釋偏倚。近年來,Bowden[31]等提出 統計量可用來定量估計這種回歸稀釋偏倚。目前已發展出許多控制弱 IV 偏倚的方法,如采用基因風險評分[33],有限信息最大似然(limited information maximum likelihood,LIML)估計[34]及持續更新估計(continuously updating estimator,CUE)[34]。
2.6 樣本重疊
在兩樣本 MR 研究中,不同的數據集往往不是完全不同的,一些人群數據可能同時存在于兩個主流數據集中。因此,表面上看起來為兩樣本設定的 MR 研究,樣本重疊也可能導致弱工具造成的偏倚更類似于單樣本設定[30]。對于 IVW 方法,隨著樣本重疊比例的增加,弱工具偏倚的方向呈現出從雙樣本設置(低估暴露-結果效應)到單樣本設置(高估暴露-結果效應)線性變化[35]。對于 MR-Egger 方法是否遵循這一規律,還有待進一步研究。
3 MR 基本設定和方法
迄今為止,MR 發展出了許多的設定和衍生方法。本文僅介紹最常用的兩種設定:單樣本 MR 和兩樣本 MR 及幾種常用方法:2SLS、逆方差加權(Inverse-variance weighted,IVW)、MR-Egger 回歸、中值估計及基于個體數據(individual personal data,IPD)的校正方法。本文介紹的所有方法都能夠用于包含多個 IV 的數據中。
3.1 單樣本與兩樣本 MR
遺傳變異與暴露及遺傳變異與結果的關系均在同一樣本中獲得的 MR 研究叫做單樣本 MR。10 年前,MR 的研究幾乎都是在單樣本設定下進行的。雖然在單樣本中,IV 分析的估計值是漸近無偏的,但在有限樣本中卻可能存在很大偏倚[36, 37]。在單樣本中,分子和分母的回歸系數之間的相關性造成了弱工具偏倚,它會高估暴露和結果之間的關聯,其偏倚大小取決于 IV 和暴露之間的關聯強度[9]。
在實踐中,研究者或許并不能從同一樣本中獲得完整的遺傳變異與暴露及遺傳變異與結果關系的數據。如果分別從兩個不重疊的數據集中獲得這兩類數據,這樣的研究就稱為兩樣本 MR[38]。使用兩樣本設定相對于單樣本設定有許多優勢。首先,在兩樣本設定中,弱工具偏倚是偏向于零假設的,零假設方向的偏倚相對于相關性方向的偏倚更容易接受,因為這一方向的偏倚比較保守,不會增加假陽性結果的可能性。其次,在一些情況下,同一組個體暴露和結局的數據難以同時測量,兩樣本 MR 大大增加了 MR 研究的應用范圍。在所有 MR 研究中,使用兩樣本設計的比例從 2011 年的接近 0% 上升至 2016 年的 40% 左右[39]。
3.2 2SLS
最簡單的 MR 估計方法是 2SLS[3]。此方法有兩個基本步驟。首先,以 Z 為自變量,以感興趣的暴露因素 X 為因變量進行最小二乘回歸;第二步,以第一步回歸的預測值為自變量,以結果 Y 為因變量進行最小二乘回歸。對于多個 IV,2SLS 估計可被認為是每一個 IV 估計值的加權平均值,其中權重由第一階段回歸中 IV 的相對強度決定[12]。假設有 j 個可用的 IV,每個個體對應的 IV 的值用 表示。第一階段的回歸模型可表示為:
|
根據此模型可得到 的預測值:。將此預測值帶入第二階段回歸模型:
|
其中, 和 是誤差項。通過此方法得出的 是第 j 個 IV 所對應的暴露-結果效應的估計值。
然而,2SLS 可能出現弱工具偏倚。此外,在無法獲得個體層面的數據、只能獲得關于遺傳變異與暴露因素和結果之間關系的匯總數據時,不能采用 2SLS 方法[7]。
3.3 逆方差加權(inverse-variance weighted,IVW)
逆方差加權法是 MR 匯總數據的標準方法[40]。它不需要個體層面的數據,可直接利用匯總數據計算因果效應值。在使用多個遺傳變異作為 IV 的數據中,對于第 j 個 IV,如果 IV 的相關假設滿足,暴露對結果的因果效應的估計值 為第 j 個遺傳變異與結果相關性的估計值 和其與暴露的相關性估計值 的比值[3],即:
|
如果遺傳變異不相關(非連鎖不平衡),那么每個遺傳變異所對應的估計值可總和成一個整體的加權估計,即:
|
是第 j 個 IV 的基因-結果關聯估計值的方差。如果遺傳變量之間不相關,IVW 的估計值與用于個體水平數據 2SLS 方法的估計值相等[41]。然而,與所有 IV 方法一樣,IVW 方法也容易受到弱工具偏倚的影響。有模擬研究結果表明,IVW 法的弱工具偏倚水平與 2SLS 法相同,其大小可通過 F 統計量進行量化[42]。
3.4 MR-Egger 回歸
MR-Egger 回歸是近年提出的檢測和調整 MR 分析中多效性的方法。此方法中,給定一組遺傳變異,首先估計每個遺傳變異與結果相關性 及遺傳變異與暴露的相關性 ,然后擬合線性函數[8]:
|
暴露對結果的因果效應的估計值 可通過下式計算[8]:
|
MR-Egger 回歸的截距估計值 是各遺傳變異多效性效應估計值的平均值。MR-Egger 方法放寬了 IVW 方法中遺傳變異之間不存在多效性的要求。它假設工具-暴露和工具-結果的關聯是獨立的。這被稱為 InSIDE 假設[43](instrument strength independent of direct effect,InSIDE),與嚴格的排除限制標準相比,這一假設相對較弱。但是,IVW 和 MR-Egger 回歸方法理論上都需假設基因變異-暴露關聯是無誤差的測量(no measurement error,NOME)[31]。MR-Egger 放寬多效性假設的代價是其違反 NOME 假設后,帶來的偏倚比 IVW 估計更大,且特別容易受到弱工具偏倚的影響[31]。另外,只有在基因多效性具有方向性的時候(即多效性具有非零平均值),MR-Egger 回歸才能檢測出多效性[8]。因為只有在這種情況下, 才會是非零的值。例如,當所有的遺傳變異都表現出多效性但其方向不同,但在平均水平下它們相互抵消(這一情況被稱為平衡多效性[8]),MR-Egger 回歸就無法檢測出多效性。
3.5 中值估計
中值估計包括簡單的中位數估計、加權中值估計和懲罰加權中值估計。簡單的中位數估計方法非常容易理解。設 表示第 j 個遺傳變異所對應的暴露-結果效應的估計值(從最小到最大排列)。如果遺傳變異的總數為奇數(J=2k+1),簡單中值估計就取其中間值 。如果是偶數(J=2k),其估計值就取 。我們可將簡單的中值估計理解為具有相同權重的加權中值估計。但在不同遺傳變異所對應的估計精度差異很大時,該方法具有低效性[9]。
加權中值估計考慮了估計精度差異大的問題。在此方法中,設 為第 j 個遺傳變異估計值的權重,設 為 j 個估計值(從最小到最大排列)權重的總和。如果進行了標準化,則 等于 1。加權中值估計是取 等于 50% 的估計值,其中 。與 IVW 方法類似,該方法的權重 一般使用每個遺傳變異的方差逆權重[44]:
|
值得注意的是,簡單的中位數估計要求至少 50% 的遺傳變異為有效的 IV,而加權中值估計僅要求至少 50% 由遺傳變異貢獻的權重是有效的。
雖然無效 IV 的存在不影響中值估計的漸進無偏性,但在有限樣本中,它還是可能導致偏倚。當無效 IV 的估計在真實的因果效應兩邊出現不平衡時(如一個研究中有多個無效 IV,而這些 IV 的估計值全部大于或小于真實的估計值),就可能出現偏倚。在這種情況下,可采用懲罰加權中值估計進行校正,以降低具有異質性的估計值的遺傳變異的權重。采用此方法時,應首先用 Cochran’s Q 值量化估計值間的異質性[9]:
|
其中, 為 IVW 方法得出的估計值[45]。在所有遺傳變異均為有效 IV 及所有變量均可識別出相同的因果關系的零假設下, 服從自由度為 1 的卡方分布。通過這一分布,找到與每個遺傳變異 值對應的 P 值(用 表示)。然后將權重乘以 P 值再乘以 20(如果 P 值大于 0.05,則乘以 1)進行懲罰。最終得到懲罰后的權重()[9]:
|
3.6 基于 IPD 的校正方法
MR-Egger 回歸與中值估計均是針對匯總數據的對于無效 IV 的校正方法。針對 IPD,目前常見的方法有:TSHT(two-stage hard thresholding)[46]、限制 IV 法(constrained instrumental variable,CIV)[47]和 sisVIVE(some invalid some valid IV estimator)[48]。
TSHT 是一種基于眾數的估計方法。它是通過對不同候選工具的估計進行兩兩比較來實現的,當兩種基因變異的估算值相似時,它們就會“投票”給對方。最后,基于這些選票中最多的一組基因變異得到總的估計[46]。當無效的遺傳變異估計的都是不同的因果效應時,即使少于 50% 的遺傳變異是有效工具,真正的因果效應也能夠被識別[49]。然而,由于這些“投票”是由一個固定的閾值決定的,當比較度量接近閾值時,TSHT 的估計對數據的微小變化將非常敏感[49]。CIV 方法通過將潛在多效表型的相關性縮小到零來消除多效效應[47]。同時,CIV 的懲罰算法對有效基因型的選擇較為嚴格,并考慮了稀疏性問題。當基因型數量大于樣本數量時,CIV 能減少由于使用多個基因型而導致的過擬合問題[47]。然而,當研究者只能獲取部分多效表型而不是全部多效表型時,CIV 就無法消除多效表型的影響[47]。sisVIVE 的中心思想是通過 LASSO 型懲罰對遺傳變異進行稀疏選擇[48]。在存在多效性及 IV 對結局有直接因果效應時,sisVIVE 的估計也較為穩健[48]。
迄今為止,MR 除單樣本和兩樣本設定之外,還發展出了許多其他設計形式,如雙向 MR[50]、兩步 MR[51]和析因 MR[52]等。同樣,除了上述的四種計算方法外,近年來還發展出了更多新方法,如其他 Mode-based 估計[53]、穩健多效性 MR[54]和貝葉斯模型平均[55]等。
4 小結
MR 是一種結合了靈活和穩健性的統計方法。MR 在觀察性研究中使用遺傳變異作為 IV 來檢測和量化因果關系。隨著數據生成成本的持續降低,它的應用范圍將會持續擴大。使用遺傳變異作為 IV 可能避免觀察性研究(混雜、反向因果關系、回歸稀釋偏倚)和 RCT(代表性和可行性問題)在進行因果推斷時的一些限制。但這種方法本身也同樣有許多局限性(LD、多效性、發育補償等)。另外,目前許多 MR 研究存在報告不規范問題。Burgess 等[56]于 2019 年發布了 MR 應用和規范報告的指南。在國內,CSCO 生物統計學專家委員會 RWS 方法學組也于近期發表了《孟德爾隨機化模型及其規范化應用的統計學共識》[57]。研究者應遵循上述指南和共識規范應用和報告 MR 研究。
總之,本文介紹了 MR 的基本原理和 3 個核心假設,討論了 MR 的 6 個主要局限性,介紹了常見的多種估計方法,希望能幫助研究者加深對 MR 研究的認識,有助于提高 MR 研究的質量。