統計分析計劃(statistical analysis plan,SAP)能夠增加臨床研究的可信度、透明度和減少統計分析過程的偏倚。SAP報告指南在開發時,主要針對后期(late phase)臨床研究,即Ⅱ期和Ⅲ期的隨機對照試驗。目前,針對早期(early phase)臨床研究,即I期臨床研究和Ⅱ期非隨機對照試驗的SAP擴展版報告指南,主要從試驗目的、設計、貝葉斯統計、數據模擬、樣本量和ICH E9(R1)的應用6個角度在原報告指南基礎上進行了擴展。擴展版報告指南能夠使早期臨床試驗的SAP規范化,提高早期臨床研究的透明性、可重復性,從而提高早期臨床研究的質量,對后期臨床研究亦起到關鍵作用。
引用本文: 李宇飛, 劉建平, 張文倩, 張穎. 統計分析計劃(SAP)擴展版報告指南的解讀及其對新藥早期臨床研究設計的啟示. 中國循證醫學雜志, 2024, 24(11): 1347-1351. doi: 10.7507/1672-2531.202405191 復制
版權信息: ?四川大學華西醫院華西期刊社《中國循證醫學雜志》版權所有,未經授權不得轉載、改編
統計分析過程是臨床研究的重要組成部分,其統計結果對最終的研究結論有著直接的影響。非透明的統計分析過程可能會出現大量的事后分析(post-hoc analysis),并由此做出偏倚風險較高的臨床結論。例如,揭盲后仍修改統計分析方法、不完整報告全部統計方法導致統計過程無法復現等。因此,統計分析方法的透明化報告在臨床研究中越來越得到研究者的重視。統計分析計劃(statistical analysis plan,SAP)可以對臨床研究的統計分析方法進行事先約束,全面地對臨床研究的統計分析過程和內容進行描述,有利于增加臨床研究的可信度、透明度,其統計結果以及結論也有更強的說服力。2017年12月,針對后期(late phase)臨床研究的SAP報告指南(下文簡稱《指南》)在JAMA發布[1]。2019年,對《指南》的解讀文章發表[2],該文章對《指南》進行了翻譯并進行解讀,說明了發表SAP的重要性。2022年1月,該指南的開發團隊針對早期(early phase)臨床研究,即I期臨床試驗和Ⅱ期的非隨機對照試驗,在BMJ發布了SAP早期臨床試驗擴展版報告指南(下文簡稱《擴展版指南》)[3]。本研究分析了在《指南》發表后SAP的發表現狀,對《擴展版指南》的發布緣起以及主要修訂內容進行介紹和解讀,并討論了《擴展版指南》對早期臨床研究的借鑒意義。
1 SAP的發表現狀及《指南》擴展動因
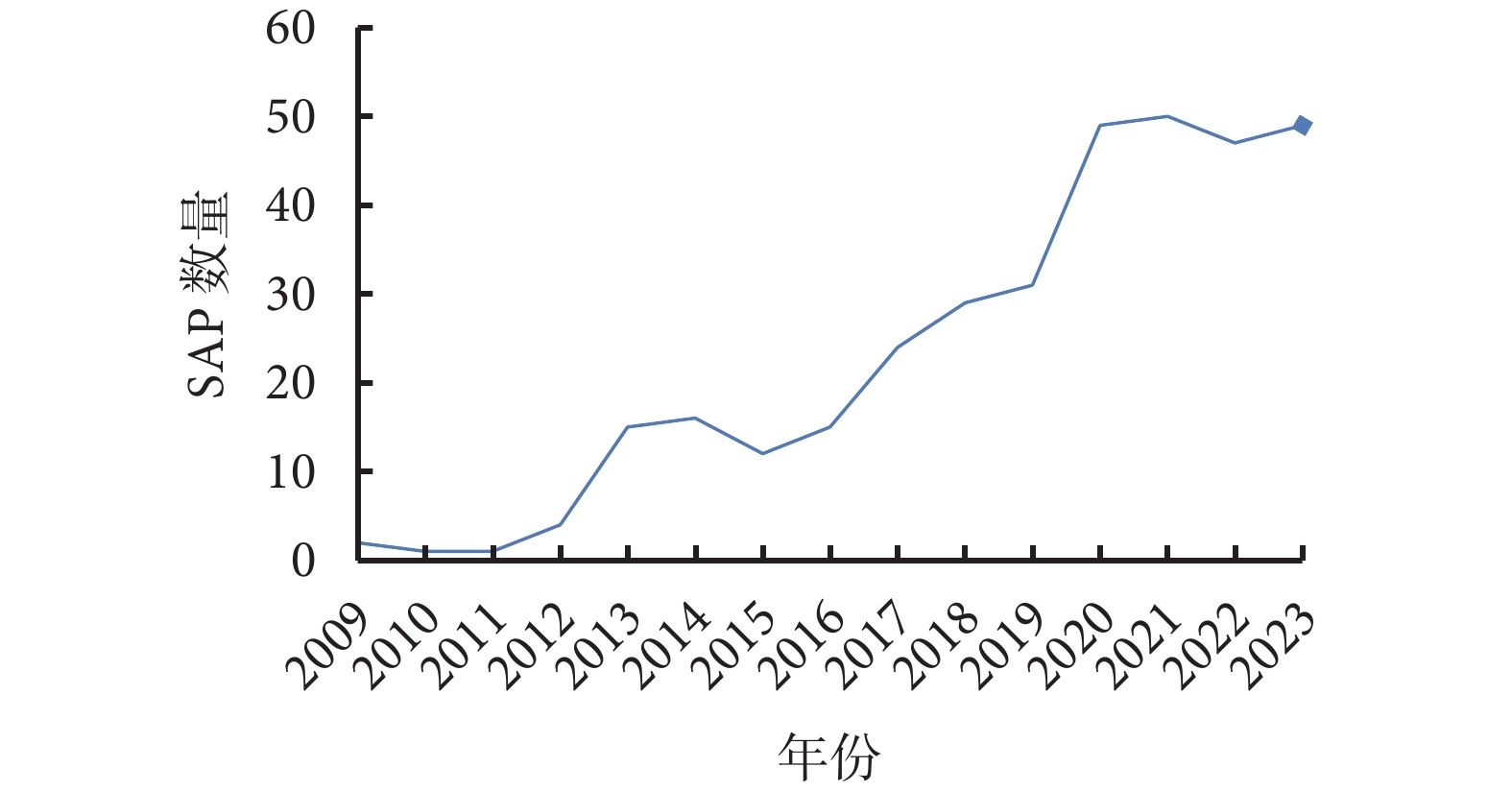
2017年12月《指南》的制訂,對SAP的發表有著促進作用,增強了臨床試驗統計分析的透明度。在PubMed上以“statistical analysis plan”進行標題檢索,截至2023年12月31日,檢索到含有SAP的文獻共345篇,發文量年度趨勢見圖1。
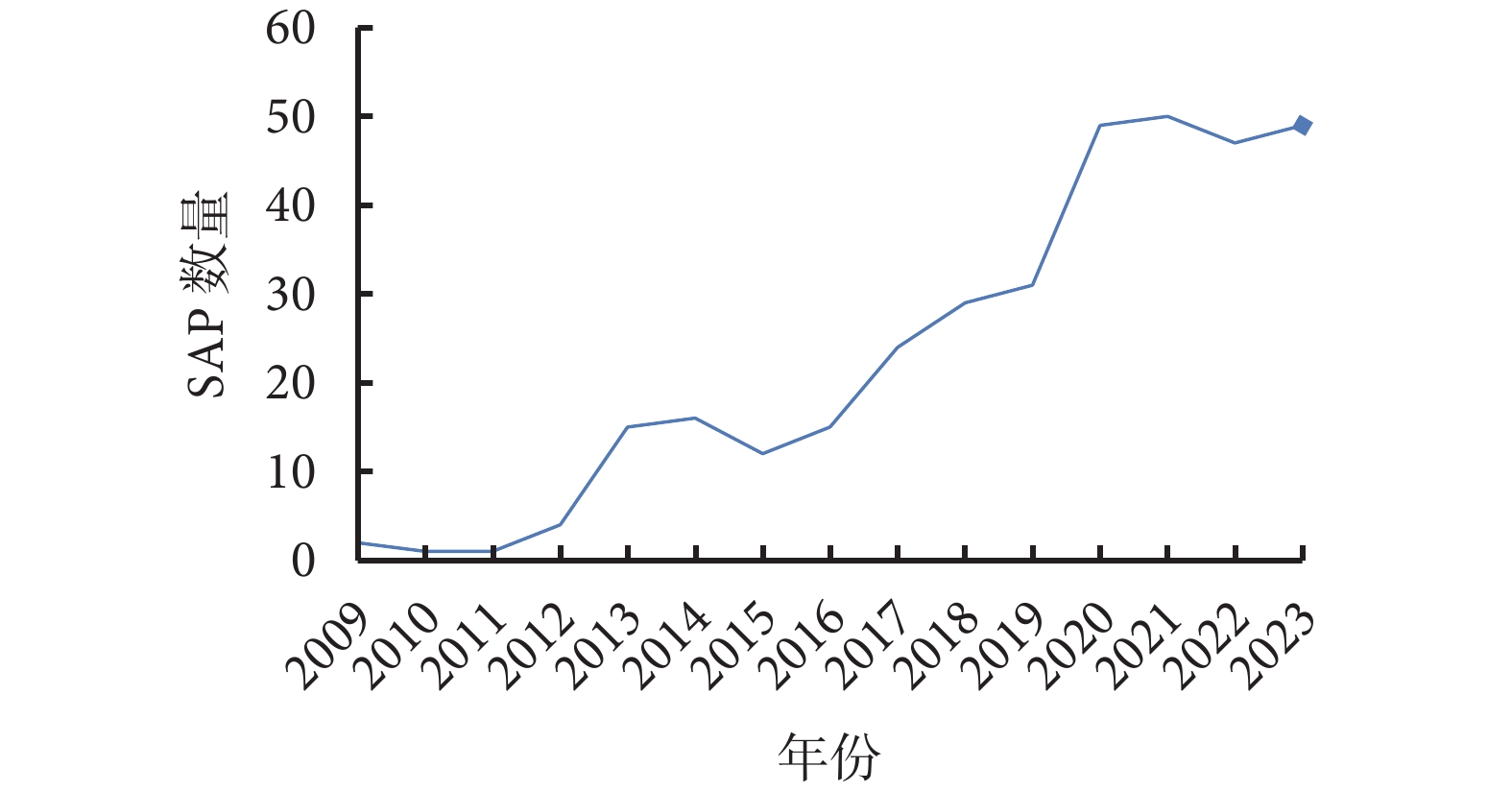 圖1
2009—2023年標題含SAP文獻發文量的年度趨勢
圖1
2009—2023年標題含SAP文獻發文量的年度趨勢
從SAP發文量的年度趨勢圖可看出,在2017年《指南》發布前SAP的發表數量以相對較為緩慢速度增長,在《指南》發布后其每年發表數量迅速提升,提示《指南》的發布,使得SAP的透明化報告得到了越來越多的重視。而SAP的發表使得試驗透明化,在將來進行數據分析、撰寫研究報告時可以與過去發表的SAP進行對照,減少選擇性報告、偏離研究方案等情況的發生。
2017版《指南》適用于后期隨機臨床試驗,包含Ⅱ期隨機臨床試驗以及Ⅲ期隨機臨床試驗。鑒于藥物的開發途徑,通過了早期臨床試驗的藥物才有機會參加后期臨床試驗。早期臨床試驗的應用比后期臨床試驗的應用更為普遍。一項發表在JAMA的研究對2000—2019年發表在ClinicalTrials.gov的臨床試驗進行分析,發現Ⅰ~Ⅱ期臨床試驗數量上總體高于Ⅲ~Ⅳ期臨床試驗[4]。
早期臨床試驗的結果對后期臨床試驗有著決定作用。首先,早期臨床試驗是新藥從臨床前動物實驗過渡到人體研究的重要階段,起到承上啟下的關鍵作用,設計和實施關系到藥物研發的成敗。其次,后期臨床試驗是基于早期臨床試驗準確而穩健的結論之上開展的,如藥物劑量和給藥間隔的選擇。如果早期臨床試驗的設計、分析上存在缺陷,會對后續試驗產生影響,Ⅲ期臨床試驗也不能很好地展示干預措施是否有益。因此,早期臨床試驗也應當在嚴格的高標準下進行。
早期臨床試驗與后期臨床試驗在試驗目的方面有所不同。例如,在I期臨床試驗中,確定試驗藥物的在人體的最大耐受劑量是主要目標之一,通過劑量遞增試驗來探究劑量與藥效(或毒性)的關系是常用的設計。這種研究具有高風險性,在研究方案中應當事先設定預期的最大耐受劑量,并說明設定依據。此外,藥動學研究也需要評價不同劑量的藥物與人體吸收藥物的關系,與后期臨床試驗不同,需要提前設計好起始劑量、最高劑量、劑量水平數量和劑量梯度等[5]。
由于I期臨床試驗是創新藥首次用于人體,是初步的臨床藥理學及人體安全性評價試驗,可以根據臨床耐受性試驗階段和臨床藥動學試驗階段分步設計,也可以不同階段交叉融合進行復合型研究方案設計。這一點和后期臨床試驗有著明顯區別,因此,2022年的《擴展版指南》在原《指南》SAP的報告內容上進行了擴展,適用于早期臨床試驗,包含I期臨床試驗以及Ⅱ期非隨機臨床試驗。
此外,由國際人用藥品注冊技術協調會(International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)的專家工作組制訂的統計原則指南(ICH-E9)主要用于新藥研發后期階段的臨床試驗,其大多數都是用于確認療效的試驗,但也表示ICH-E9對早期臨床試驗的SAP與后期臨床試驗有著相同要求[6]。一項研究提出,在早期臨床試驗中使用基于模型的設計更有優勢,也建議更多地使用基于模型的設計[7]。使用這種設計會引入額外的統計參數,對SAP的質量有著更高的需求。針對臨床早期試驗對《指南》進行擴展,是遵循了ICH-E9早期臨床試驗也應有SAP的要求,并考慮到了與后期臨床試驗的差異,使得《擴展版指南》覆蓋范圍更加廣泛,更加通用。
2 制訂過程及條目變化概要
2018年4月,在英國臨床研究合作(UK Clinical Research Collaboration)注冊的臨床試驗單位(Clinical Trials Unit,CTU)網絡的統計師業務小組會議上對《指南》進行了討論,認為有必要針對早期臨床試驗進行擴展。經過對現有SAP指南的全面檢索,對臨床試驗資助者及監管者的調查,對CTU的調查以及嚴格評價、專家評審會議后,進行了《擴展版指南》的試點,最后形成《擴展版指南》的最終版,于2022年1月發表在BMJ[3]。
《指南》含有55個條目,經過本次修改,30個條目保持不變,為了更好地貼合早期臨床試驗,對25個條目進行了修改,增加了11個新條目。新增及有較大改動的條目包括:① 針對早期臨床試驗的設計方法差異,增加關于統計設計方法細節的報告,以及模型選擇的細節(如果適用)。② 由于ICH-E9(R1)[8]的廣泛應用,對“結果(outcome)”的定義進行更新,納入了估計目標的定義,與ICH-E9(R1)概述的原則保持一致。③ 納入了含有操作特征(operating characteristics)的數據模擬報告,對不同情況下模型的運行狀況進行評估。④ 納入了新設計方法所使用的模型代碼。⑤ 在使用劑量遞增試驗的設計時,納入劑量轉變路徑(dose transition pathways)。⑥ 修改措辭,使用語在頻率論和貝葉斯方法間更加中立,以反映一些早期臨床試驗設計,特別是在I期試驗中使用貝葉斯方法的情況。
3 條目變化的具體闡述
《擴展版指南》中條目的變化可以主要分為幾大原因,主要包括試驗目的、設計、貝葉斯統計、數據模擬、樣本量和ICH-E9(R1)的應用等。《擴展版指南》的條目見附件表1。
3.1 試驗目的
試驗目的反映了試驗要回答的科學問題。與后期臨床試驗更關注療效相比,早期臨床試驗有著更多的目標,如應當明確最終的試驗結論是否基于毒性、療效、藥代動力學、藥效動力學或上述因素的某種組合。在設計聯合評估毒性和功效時,還需要規定如果得出不同的結論,應以哪一個為優先目的。因此條目8添加了要明確研究“關鍵”目的,要明確主要目的和次要目的,以貼合早期臨床試驗的多目標性。
3.2 試驗設計
由于I期臨床試驗需要確定新藥的毒性和安全劑量范圍,常使用劑量遞增試驗來檢驗人體藥物耐受性。在劑量遞增試驗中研究者需要確定最大推薦起始劑量,即基于安全性考慮推薦使用的最大劑量,預期中這個劑量不會產生毒性反應[5],試驗將從這個劑量開始。之后劑量遞增的決策可以基于兩種不同的設計進行,一種是基于規則的設計,如傳統3+3設計、快速滴定設計,這種設計要求按照一定的規則提前制定劑量隊列,以及制定做出劑量上升、下降的規則,后續在試驗實施時,嚴格按照規則進行。另一種是基于模型的設計,如連續重評估法(continual reassessment method,CRM),這是一種貝葉斯框架的適應性方法,在研究前制定好劑量-毒性關系假設統計模型,在試驗中將先完成試驗的患者數據納入模型中進行模型更新,由此計算得出下一個試驗劑量。此外,還有一種模型輔助(model-assisted)設計,將模型設計與規則設計結合使用。
在早期臨床試驗中,研究者需要在SAP中報告試驗階段和設計方法,如Ⅱ期單臂試驗或劑量遞增試驗,因為根據不同的試驗階段和試驗設計,研究者需要報告的內容有所不同,見條目9a。此處條目的變化主要集中在劑量遞增試驗部分,研究者需要報告起始劑量水平的來源,針對不同的設計,研究者需要對劑量隊列、劑量變化的規則、過量用藥的劃分、期中分析的劑量決定、模型參數、公式及更新時間等信息進行說明,這些在條目9b、9c、9d、9e、13a、20中有所體現。在分析方法中,基于模型設計需要預先指定替代模型以及公式和數學說明(27d說明)。
劑量遞增試驗中,有時會有復雜的劑量遞增/下降機制,或是使用新的設計方法,此時使用劑量轉變途徑(dose transition pathways)可以有效輔助決策。應當使用表格或樹狀/圖說明不同劑量限制性毒性(dose limiting toxicity,DLT)下的劑量轉變途徑,見條目34。
3.3 貝葉斯方法
早期臨床試驗中有許多研究是基于貝葉斯方法下進行的,如劑量遞增試驗中的CRM法。貝葉斯方法與通常的頻率統計的假設檢驗法有所不同,假設檢驗是針對樣本進行假設以及統計推斷,以拒絕零假設的方式來反證備擇假設的正確(即一般根據P<0.05進行判斷);而貝葉斯方法需要先確定模型先驗分布(prior distribution),得到樣本信息后計算先驗分布得到當前樣本的概率似然函數,最后再綜合考慮先驗分布和似然函數,得出模型的后驗分布(posterior distribution),即根據新數據對模型進行調整。因此,SAP也需要說明研究是在頻率統計的假設檢驗還是貝葉斯框架下進行的,見條目12。
貝葉斯方法由于沒有進行假設檢驗,也不需指定檢驗顯著性水準,結果的呈現也不存在P值。為了加入對貝葉斯方法的考慮,《擴展版指南》中將原標題“可信區間與P值”更改為“不確定標識(indications of uncertainty)”,條目16也對解釋部分進行更改,以體現P值的報告是可選的。根據研究者使用的方法,也要選擇報告置信區間(confidence interval,CI)還是貝葉斯可信區間(credible interval,CrI)。
3.4 統計模擬
當使用基于模型或是模型輔助的劑量遞增試驗設計時,可以使用統計模擬的方式對估計目標以及不同假設下模型的運行情況進行評估。此時應當在SAP中報告進行模型設計、進行模擬和統計分析的統計軟件包(條目31),模擬運行的特征(條目33),完整的模型詳情以及程序代碼(條目35),使得劑量遞增的決策過程透明化和可復現化。
3.5 樣本量及缺失數據
I期臨床試驗是新藥首次用于人體,受試人群的樣本量往往因藥物的作用機制、生物學效應的類型和程度、動物實驗的安全劑量范圍不同等有差異。所以樣本量的計算具有不確定性。例如,一般情況下,耐受性試驗設計時考慮的原則是起始劑量組的例數較少[“哨兵試驗(sentinel trail)”,甚至只有1例受試者]。而藥動學試驗則會考慮滿足統計學要求,達到8~12例/組。考慮到這個問題,《擴展版指南》不要求報告樣本量計算的完整細節,而是“樣本量確定或證明合理”的完整細節。對于I期試驗,通過每個隊列的患者數量以及預期入組的隊列總數可能足以證明試驗樣本量的合理性;對于劑量遞增試驗,還需要詳細說明在未觀測到DLT情況下預計招募的最小樣本量(條目11)。由于樣本量有限,對于缺失數據也不建議填補,包括多重填補等常規方式(條目28說明)。
3.6 ICH-E9(R1)的考量
2017年ICH公布的ICH-E9的附錄,即ICH-E9(R1)中提出了統計分析的不同策略[8]。首先是對伴發事件(intercurrent events)的定義,指的是在治療開始后發生的,會妨礙變量的觀測或影響對變量解釋的事件,如需要對受試者進行搶救時額外服用的藥物或因為毒性事件而停止治療等。ICH-E9(R1)中針對伴發事件提供了五種策略,分別是:① 療法策略(treatment policy strategy),無論是否發生伴發事件,都使用事先計劃的指標進行分析。這種策略與ICH-E9中的ITT原則相似,使用該策略時研究的是混合了伴發事件時的治療效應。但當伴發事件導致關注的結局指標消失時,不能采用該策略。② 復合策略(composite strategy),將伴發事件與一個或多個其他測量結局合并作為一個新的關注變量,如此分析新的變量便可以同時考慮到臨床指標和伴發事件。③ 假想策略(hypothetical strategy),假設不會發生伴發事件,所研究的是試驗藥物在不發生所定義的伴發事件下的療效。該策略的關鍵是對假設進行精準的描述,以反映所研究的科學問題。④ 主層策略(principal stratum strategy),將潛在的可能發生(或不發生)伴發事件的人群定義為主層人群,所研究的問題將針對主層人群進行分析。主層與亞組有區別,亞組是根據已發生、已明確的協變量進行分類的,而主層要根據潛在的發生(未發生)伴發事件進行區分,這在研究前是無法得出的,需要根據協變量推測出主層人群。⑤ 在治策略(while on treatment strategy),只關注伴發事件發生前的數據,根據伴發事件發生前的數據進行評價。
ICH-E9(R1)中為了將治療效果量化,對治療效應進行精確描述,提出了估計目標的概念。估計目標是用于將試驗目標轉化為研究者所關心的科學問題而產生的,其包括四個方面,分別是目標人群即研究人群、目標變量(終點)即評價指標、伴發事件、目標變量在目標人群水平的效果(population-level summary for the variable)。目標變量在目標人群水平的效果需要通過比較得出,以率差、均值差等形式體現。
為了使臨床試驗有更強的一致性和清晰度,ICH-E9(R1)基于估計目標提出了一個結構性框架。首先根據試驗目標定義出估計目標,由此將試驗目標通過對估計目標的定義轉化為研究者關心的科學問題。之后在估計目標下根據數據類型選擇主要估計方法(main estimator),根據試驗數據計算得出估計值(estimate)。對于每一個估計方法,都建議使用敏感性分析,評估在偏離檢驗假設的情況下采用當前估計方法的穩健性。
由于ICH-E9(R1)的廣泛應用,《擴展版指南》中根據ICH-E9(R1)的概念將標題“結局定義”改為“估計目標定義”,并要求報告干預措施的細節(26a)和估計目標定義中的四個部分(26b~26e)。
此外,在“分析方法”部分對條目或是說明用詞進行了調整,加入了估計目標、估計方法等概念(27a~27f)。
4 《擴展版指南》對早期臨床試驗設計的啟示
《擴展版指南》針對ICH-E9(R1)的發布以及臨床試驗早期的特殊性進行了擴展,對于早期臨床試驗透明化以及SAP報告規范化有著重要作用。在早期臨床試驗設計階段應注重以下內容:① 明確研究關鍵目的。若存在多個研究目的并行,應當分清主次,應當規定得出不同的結論時以哪一個為優先目的。② 重視《擴展版指南》要求報告的設計細節。需要報告的內容往往都會對試驗的結果產生影響,為了使試驗透明化并可復現才要求報告,因此應當在這些方面更加重視。早期臨床試驗在設計階段需要重視劑量水平、模型等設計細節對結果可能產生的影響。③ 將ICH-E9(R1)中的伴發事件納入設計考量。早期臨床試驗以探索性目的居多,主要的伴發事件為劑量調整、受試者依從性差、終止治療或者脫落等。早期臨床試驗中,干預組的凈效應對后期臨床試驗有著更重要的參考意義,所以假想策略是此階段的主要策略。例如,針對劑量調整和依從性差的情況,導致實際藥物暴露水平發生了變化,可以假設所有受試者都依從試驗方案情況下,對初步的劑量-反應關系的進行探索。同時,進行敏感性分析對結果的穩健性進行估計。④ 考慮使用貝葉斯的方法進行早期臨床試驗設計。貝葉斯方法在早期臨床研究領域的應用有著特殊優勢。在使用貝葉斯方法的過程中,也需要注重模型的模擬與模擬報告,可以驗證模型在不同條件下的穩健性,并使得該結果更加可信。
《指南》和《擴展版指南》的提出對從早期到后期的臨床研究質量的提升將起到關鍵作用。建議未來的臨床研究在設計階段,同時參考兩版指南,進行縱向前瞻性的規劃。使得更多早期臨床試驗能夠進入后期驗證階段,也促進后期臨床試驗有更高的成功率。
統計分析過程是臨床研究的重要組成部分,其統計結果對最終的研究結論有著直接的影響。非透明的統計分析過程可能會出現大量的事后分析(post-hoc analysis),并由此做出偏倚風險較高的臨床結論。例如,揭盲后仍修改統計分析方法、不完整報告全部統計方法導致統計過程無法復現等。因此,統計分析方法的透明化報告在臨床研究中越來越得到研究者的重視。統計分析計劃(statistical analysis plan,SAP)可以對臨床研究的統計分析方法進行事先約束,全面地對臨床研究的統計分析過程和內容進行描述,有利于增加臨床研究的可信度、透明度,其統計結果以及結論也有更強的說服力。2017年12月,針對后期(late phase)臨床研究的SAP報告指南(下文簡稱《指南》)在JAMA發布[1]。2019年,對《指南》的解讀文章發表[2],該文章對《指南》進行了翻譯并進行解讀,說明了發表SAP的重要性。2022年1月,該指南的開發團隊針對早期(early phase)臨床研究,即I期臨床試驗和Ⅱ期的非隨機對照試驗,在BMJ發布了SAP早期臨床試驗擴展版報告指南(下文簡稱《擴展版指南》)[3]。本研究分析了在《指南》發表后SAP的發表現狀,對《擴展版指南》的發布緣起以及主要修訂內容進行介紹和解讀,并討論了《擴展版指南》對早期臨床研究的借鑒意義。
1 SAP的發表現狀及《指南》擴展動因
2017年12月《指南》的制訂,對SAP的發表有著促進作用,增強了臨床試驗統計分析的透明度。在PubMed上以“statistical analysis plan”進行標題檢索,截至2023年12月31日,檢索到含有SAP的文獻共345篇,發文量年度趨勢見圖1。
圖1
2009—2023年標題含SAP文獻發文量的年度趨勢
從SAP發文量的年度趨勢圖可看出,在2017年《指南》發布前SAP的發表數量以相對較為緩慢速度增長,在《指南》發布后其每年發表數量迅速提升,提示《指南》的發布,使得SAP的透明化報告得到了越來越多的重視。而SAP的發表使得試驗透明化,在將來進行數據分析、撰寫研究報告時可以與過去發表的SAP進行對照,減少選擇性報告、偏離研究方案等情況的發生。
2017版《指南》適用于后期隨機臨床試驗,包含Ⅱ期隨機臨床試驗以及Ⅲ期隨機臨床試驗。鑒于藥物的開發途徑,通過了早期臨床試驗的藥物才有機會參加后期臨床試驗。早期臨床試驗的應用比后期臨床試驗的應用更為普遍。一項發表在JAMA的研究對2000—2019年發表在ClinicalTrials.gov的臨床試驗進行分析,發現Ⅰ~Ⅱ期臨床試驗數量上總體高于Ⅲ~Ⅳ期臨床試驗[4]。
早期臨床試驗的結果對后期臨床試驗有著決定作用。首先,早期臨床試驗是新藥從臨床前動物實驗過渡到人體研究的重要階段,起到承上啟下的關鍵作用,設計和實施關系到藥物研發的成敗。其次,后期臨床試驗是基于早期臨床試驗準確而穩健的結論之上開展的,如藥物劑量和給藥間隔的選擇。如果早期臨床試驗的設計、分析上存在缺陷,會對后續試驗產生影響,Ⅲ期臨床試驗也不能很好地展示干預措施是否有益。因此,早期臨床試驗也應當在嚴格的高標準下進行。
早期臨床試驗與后期臨床試驗在試驗目的方面有所不同。例如,在I期臨床試驗中,確定試驗藥物的在人體的最大耐受劑量是主要目標之一,通過劑量遞增試驗來探究劑量與藥效(或毒性)的關系是常用的設計。這種研究具有高風險性,在研究方案中應當事先設定預期的最大耐受劑量,并說明設定依據。此外,藥動學研究也需要評價不同劑量的藥物與人體吸收藥物的關系,與后期臨床試驗不同,需要提前設計好起始劑量、最高劑量、劑量水平數量和劑量梯度等[5]。
由于I期臨床試驗是創新藥首次用于人體,是初步的臨床藥理學及人體安全性評價試驗,可以根據臨床耐受性試驗階段和臨床藥動學試驗階段分步設計,也可以不同階段交叉融合進行復合型研究方案設計。這一點和后期臨床試驗有著明顯區別,因此,2022年的《擴展版指南》在原《指南》SAP的報告內容上進行了擴展,適用于早期臨床試驗,包含I期臨床試驗以及Ⅱ期非隨機臨床試驗。
此外,由國際人用藥品注冊技術協調會(International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)的專家工作組制訂的統計原則指南(ICH-E9)主要用于新藥研發后期階段的臨床試驗,其大多數都是用于確認療效的試驗,但也表示ICH-E9對早期臨床試驗的SAP與后期臨床試驗有著相同要求[6]。一項研究提出,在早期臨床試驗中使用基于模型的設計更有優勢,也建議更多地使用基于模型的設計[7]。使用這種設計會引入額外的統計參數,對SAP的質量有著更高的需求。針對臨床早期試驗對《指南》進行擴展,是遵循了ICH-E9早期臨床試驗也應有SAP的要求,并考慮到了與后期臨床試驗的差異,使得《擴展版指南》覆蓋范圍更加廣泛,更加通用。
2 制訂過程及條目變化概要
2018年4月,在英國臨床研究合作(UK Clinical Research Collaboration)注冊的臨床試驗單位(Clinical Trials Unit,CTU)網絡的統計師業務小組會議上對《指南》進行了討論,認為有必要針對早期臨床試驗進行擴展。經過對現有SAP指南的全面檢索,對臨床試驗資助者及監管者的調查,對CTU的調查以及嚴格評價、專家評審會議后,進行了《擴展版指南》的試點,最后形成《擴展版指南》的最終版,于2022年1月發表在BMJ[3]。
《指南》含有55個條目,經過本次修改,30個條目保持不變,為了更好地貼合早期臨床試驗,對25個條目進行了修改,增加了11個新條目。新增及有較大改動的條目包括:① 針對早期臨床試驗的設計方法差異,增加關于統計設計方法細節的報告,以及模型選擇的細節(如果適用)。② 由于ICH-E9(R1)[8]的廣泛應用,對“結果(outcome)”的定義進行更新,納入了估計目標的定義,與ICH-E9(R1)概述的原則保持一致。③ 納入了含有操作特征(operating characteristics)的數據模擬報告,對不同情況下模型的運行狀況進行評估。④ 納入了新設計方法所使用的模型代碼。⑤ 在使用劑量遞增試驗的設計時,納入劑量轉變路徑(dose transition pathways)。⑥ 修改措辭,使用語在頻率論和貝葉斯方法間更加中立,以反映一些早期臨床試驗設計,特別是在I期試驗中使用貝葉斯方法的情況。
3 條目變化的具體闡述
《擴展版指南》中條目的變化可以主要分為幾大原因,主要包括試驗目的、設計、貝葉斯統計、數據模擬、樣本量和ICH-E9(R1)的應用等。《擴展版指南》的條目見附件表1。
3.1 試驗目的
試驗目的反映了試驗要回答的科學問題。與后期臨床試驗更關注療效相比,早期臨床試驗有著更多的目標,如應當明確最終的試驗結論是否基于毒性、療效、藥代動力學、藥效動力學或上述因素的某種組合。在設計聯合評估毒性和功效時,還需要規定如果得出不同的結論,應以哪一個為優先目的。因此條目8添加了要明確研究“關鍵”目的,要明確主要目的和次要目的,以貼合早期臨床試驗的多目標性。
3.2 試驗設計
由于I期臨床試驗需要確定新藥的毒性和安全劑量范圍,常使用劑量遞增試驗來檢驗人體藥物耐受性。在劑量遞增試驗中研究者需要確定最大推薦起始劑量,即基于安全性考慮推薦使用的最大劑量,預期中這個劑量不會產生毒性反應[5],試驗將從這個劑量開始。之后劑量遞增的決策可以基于兩種不同的設計進行,一種是基于規則的設計,如傳統3+3設計、快速滴定設計,這種設計要求按照一定的規則提前制定劑量隊列,以及制定做出劑量上升、下降的規則,后續在試驗實施時,嚴格按照規則進行。另一種是基于模型的設計,如連續重評估法(continual reassessment method,CRM),這是一種貝葉斯框架的適應性方法,在研究前制定好劑量-毒性關系假設統計模型,在試驗中將先完成試驗的患者數據納入模型中進行模型更新,由此計算得出下一個試驗劑量。此外,還有一種模型輔助(model-assisted)設計,將模型設計與規則設計結合使用。
在早期臨床試驗中,研究者需要在SAP中報告試驗階段和設計方法,如Ⅱ期單臂試驗或劑量遞增試驗,因為根據不同的試驗階段和試驗設計,研究者需要報告的內容有所不同,見條目9a。此處條目的變化主要集中在劑量遞增試驗部分,研究者需要報告起始劑量水平的來源,針對不同的設計,研究者需要對劑量隊列、劑量變化的規則、過量用藥的劃分、期中分析的劑量決定、模型參數、公式及更新時間等信息進行說明,這些在條目9b、9c、9d、9e、13a、20中有所體現。在分析方法中,基于模型設計需要預先指定替代模型以及公式和數學說明(27d說明)。
劑量遞增試驗中,有時會有復雜的劑量遞增/下降機制,或是使用新的設計方法,此時使用劑量轉變途徑(dose transition pathways)可以有效輔助決策。應當使用表格或樹狀/圖說明不同劑量限制性毒性(dose limiting toxicity,DLT)下的劑量轉變途徑,見條目34。
3.3 貝葉斯方法
早期臨床試驗中有許多研究是基于貝葉斯方法下進行的,如劑量遞增試驗中的CRM法。貝葉斯方法與通常的頻率統計的假設檢驗法有所不同,假設檢驗是針對樣本進行假設以及統計推斷,以拒絕零假設的方式來反證備擇假設的正確(即一般根據P<0.05進行判斷);而貝葉斯方法需要先確定模型先驗分布(prior distribution),得到樣本信息后計算先驗分布得到當前樣本的概率似然函數,最后再綜合考慮先驗分布和似然函數,得出模型的后驗分布(posterior distribution),即根據新數據對模型進行調整。因此,SAP也需要說明研究是在頻率統計的假設檢驗還是貝葉斯框架下進行的,見條目12。
貝葉斯方法由于沒有進行假設檢驗,也不需指定檢驗顯著性水準,結果的呈現也不存在P值。為了加入對貝葉斯方法的考慮,《擴展版指南》中將原標題“可信區間與P值”更改為“不確定標識(indications of uncertainty)”,條目16也對解釋部分進行更改,以體現P值的報告是可選的。根據研究者使用的方法,也要選擇報告置信區間(confidence interval,CI)還是貝葉斯可信區間(credible interval,CrI)。
3.4 統計模擬
當使用基于模型或是模型輔助的劑量遞增試驗設計時,可以使用統計模擬的方式對估計目標以及不同假設下模型的運行情況進行評估。此時應當在SAP中報告進行模型設計、進行模擬和統計分析的統計軟件包(條目31),模擬運行的特征(條目33),完整的模型詳情以及程序代碼(條目35),使得劑量遞增的決策過程透明化和可復現化。
3.5 樣本量及缺失數據
I期臨床試驗是新藥首次用于人體,受試人群的樣本量往往因藥物的作用機制、生物學效應的類型和程度、動物實驗的安全劑量范圍不同等有差異。所以樣本量的計算具有不確定性。例如,一般情況下,耐受性試驗設計時考慮的原則是起始劑量組的例數較少[“哨兵試驗(sentinel trail)”,甚至只有1例受試者]。而藥動學試驗則會考慮滿足統計學要求,達到8~12例/組。考慮到這個問題,《擴展版指南》不要求報告樣本量計算的完整細節,而是“樣本量確定或證明合理”的完整細節。對于I期試驗,通過每個隊列的患者數量以及預期入組的隊列總數可能足以證明試驗樣本量的合理性;對于劑量遞增試驗,還需要詳細說明在未觀測到DLT情況下預計招募的最小樣本量(條目11)。由于樣本量有限,對于缺失數據也不建議填補,包括多重填補等常規方式(條目28說明)。
3.6 ICH-E9(R1)的考量
2017年ICH公布的ICH-E9的附錄,即ICH-E9(R1)中提出了統計分析的不同策略[8]。首先是對伴發事件(intercurrent events)的定義,指的是在治療開始后發生的,會妨礙變量的觀測或影響對變量解釋的事件,如需要對受試者進行搶救時額外服用的藥物或因為毒性事件而停止治療等。ICH-E9(R1)中針對伴發事件提供了五種策略,分別是:① 療法策略(treatment policy strategy),無論是否發生伴發事件,都使用事先計劃的指標進行分析。這種策略與ICH-E9中的ITT原則相似,使用該策略時研究的是混合了伴發事件時的治療效應。但當伴發事件導致關注的結局指標消失時,不能采用該策略。② 復合策略(composite strategy),將伴發事件與一個或多個其他測量結局合并作為一個新的關注變量,如此分析新的變量便可以同時考慮到臨床指標和伴發事件。③ 假想策略(hypothetical strategy),假設不會發生伴發事件,所研究的是試驗藥物在不發生所定義的伴發事件下的療效。該策略的關鍵是對假設進行精準的描述,以反映所研究的科學問題。④ 主層策略(principal stratum strategy),將潛在的可能發生(或不發生)伴發事件的人群定義為主層人群,所研究的問題將針對主層人群進行分析。主層與亞組有區別,亞組是根據已發生、已明確的協變量進行分類的,而主層要根據潛在的發生(未發生)伴發事件進行區分,這在研究前是無法得出的,需要根據協變量推測出主層人群。⑤ 在治策略(while on treatment strategy),只關注伴發事件發生前的數據,根據伴發事件發生前的數據進行評價。
ICH-E9(R1)中為了將治療效果量化,對治療效應進行精確描述,提出了估計目標的概念。估計目標是用于將試驗目標轉化為研究者所關心的科學問題而產生的,其包括四個方面,分別是目標人群即研究人群、目標變量(終點)即評價指標、伴發事件、目標變量在目標人群水平的效果(population-level summary for the variable)。目標變量在目標人群水平的效果需要通過比較得出,以率差、均值差等形式體現。
為了使臨床試驗有更強的一致性和清晰度,ICH-E9(R1)基于估計目標提出了一個結構性框架。首先根據試驗目標定義出估計目標,由此將試驗目標通過對估計目標的定義轉化為研究者關心的科學問題。之后在估計目標下根據數據類型選擇主要估計方法(main estimator),根據試驗數據計算得出估計值(estimate)。對于每一個估計方法,都建議使用敏感性分析,評估在偏離檢驗假設的情況下采用當前估計方法的穩健性。
由于ICH-E9(R1)的廣泛應用,《擴展版指南》中根據ICH-E9(R1)的概念將標題“結局定義”改為“估計目標定義”,并要求報告干預措施的細節(26a)和估計目標定義中的四個部分(26b~26e)。
此外,在“分析方法”部分對條目或是說明用詞進行了調整,加入了估計目標、估計方法等概念(27a~27f)。
4 《擴展版指南》對早期臨床試驗設計的啟示
《擴展版指南》針對ICH-E9(R1)的發布以及臨床試驗早期的特殊性進行了擴展,對于早期臨床試驗透明化以及SAP報告規范化有著重要作用。在早期臨床試驗設計階段應注重以下內容:① 明確研究關鍵目的。若存在多個研究目的并行,應當分清主次,應當規定得出不同的結論時以哪一個為優先目的。② 重視《擴展版指南》要求報告的設計細節。需要報告的內容往往都會對試驗的結果產生影響,為了使試驗透明化并可復現才要求報告,因此應當在這些方面更加重視。早期臨床試驗在設計階段需要重視劑量水平、模型等設計細節對結果可能產生的影響。③ 將ICH-E9(R1)中的伴發事件納入設計考量。早期臨床試驗以探索性目的居多,主要的伴發事件為劑量調整、受試者依從性差、終止治療或者脫落等。早期臨床試驗中,干預組的凈效應對后期臨床試驗有著更重要的參考意義,所以假想策略是此階段的主要策略。例如,針對劑量調整和依從性差的情況,導致實際藥物暴露水平發生了變化,可以假設所有受試者都依從試驗方案情況下,對初步的劑量-反應關系的進行探索。同時,進行敏感性分析對結果的穩健性進行估計。④ 考慮使用貝葉斯的方法進行早期臨床試驗設計。貝葉斯方法在早期臨床研究領域的應用有著特殊優勢。在使用貝葉斯方法的過程中,也需要注重模型的模擬與模擬報告,可以驗證模型在不同條件下的穩健性,并使得該結果更加可信。
《指南》和《擴展版指南》的提出對從早期到后期的臨床研究質量的提升將起到關鍵作用。建議未來的臨床研究在設計階段,同時參考兩版指南,進行縱向前瞻性的規劃。使得更多早期臨床試驗能夠進入后期驗證階段,也促進后期臨床試驗有更高的成功率。