引用本文: 張靜雯, 卓木清, 王林淦, 張宇昕, 陳志紅, 郭宇雄, 翟瓊香. 癲癇與異染性腦白質營養不良——附一個晚嬰型異染性腦白質營養不良家系臨床資料及基因突變分析. 癲癇雜志, 2016, 2(2): 101-105. doi: 10.7507/2096-0247.20160018 復制
版權信息: ?四川大學華西醫院華西期刊社《癲癇雜志》版權所有,未經授權不得轉載、改編
異染性腦白質營養不良(Metalchromatic leukodystrophy,MLD)是一種少見的溶酶體疾病,是由于芳基硫酸酯酶A(Arylsulfatase A,ARSA)或神經鞘脂激活蛋白B(Sphingolipid activator protein B,SAP-B)缺陷,從而導致腦硫苷脂沉積在少突膠質細胞和施萬細胞所致病的。國外報道的發病率為1/40 000~1/120 000活產嬰兒[1]。目前分子遺傳學研究已證實MLD發病與基因缺陷有關,其中以ARSA基因突變被廣泛關注[2]。本研究應用PCR直接測序的方法對一個晚嬰型MLD家系進行了ARSA基因突變分析,并分析MLD家系ARSA基因型與臨床表型的特點。
資料與方法
1 研究對象
先證者(圖 1 Ⅱ2)男,32個月。第2胎第2產,足月順產。孕期母親反復患支氣管炎,曾住院治療,具體不詳。患兒生后6個月會抬頭,7個月會翻身,9個月會坐,14個月可扶站,13個月會叫“媽媽”、“爸爸”等簡易的字語,但24個月仍不會走路。曾在外院診斷“腦性癱瘓”,予營養神經治療療效欠佳,并出現反復的抽搐,表現為四肢強直,牙關緊閉,意識不清,發作持續時間約1~2 min;診斷為癲癇,予左乙拉西坦片治療后發作有所減少,但半年后發作加重,每天均有發作,癥狀同前,加用丙戊酸鈉緩釋片治療后發作次數有所減少,但仍有發作,并逐漸出現語言能力倒退和雙下肢強直,雙下肢腳
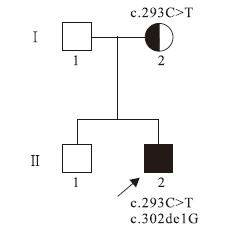
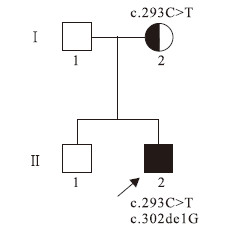 圖1
家系圖
Figure1.
Pedigree
圖1
家系圖
Figure1.
Pedigree
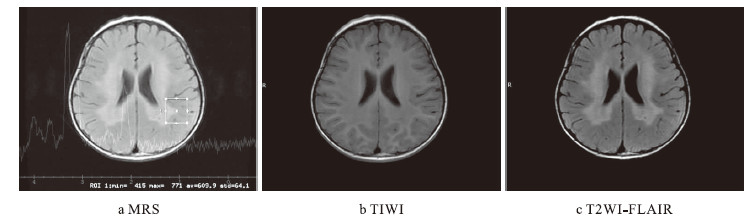 圖2
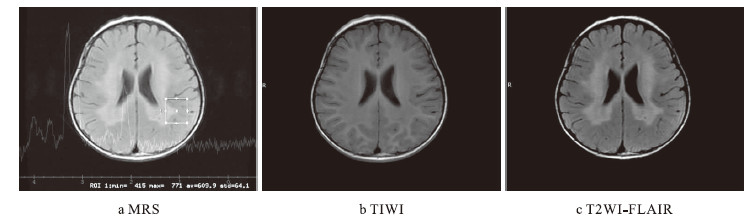
先證者頭顱MRI+MRS改變a.雙側側腦室周圍及胼胝體壓部白質區見對稱性大片狀異常信號區;b.病灶信號不均、T1WI呈等低信號;c.T2WI及T2-FLAIR呈稍高信號,可見“虎紋征”,其周弓形纖維未見累及、增強未見明顯強化。選取雙側枕葉為感興趣區,TE值分別為35ms和144ms。譜線顯示雙側枕葉NAA峰明顯降低,Cho峰明顯升高,Cr峰位置、高度未見明顯異常,左側枕葉NAA/Cr值減低(約=0.89),右側枕葉NAA/Cr值減低(約=0.86);雙側枕葉Cho/Cr值升高(約1.76)
Figure2.
Cranial MRI + MRS changes of proband: a.bilateral periventricular and corpus callosum white area showed large symmetrical abnormal signal,which was uneven; b.T1WI showed low-signal intensity; c. T2WI and T2-FLAIR showed high signal intensity,“tigroid pattern” was visible,the peripheral arcuate fiber were not involved,no significant strengthening when enhanced. Selected the bilateral occipital as region of interest,TE values were 35ms and 144ms respectively. Spectrum showed NAA peak decreased in bilateral occipital,Cho peak significantly increased,the position and height of Cr peak were not obviously abnormal,NAA/Cr value decreased in left occipital lobe (approximately=0.89),NAA / Cr value decreased in the right occipital lobe (approximately=0.86); Cho/Cr value increased in bilateral occipital lobe(approximately=1.76)
圖2
先證者頭顱MRI+MRS改變a.雙側側腦室周圍及胼胝體壓部白質區見對稱性大片狀異常信號區;b.病灶信號不均、T1WI呈等低信號;c.T2WI及T2-FLAIR呈稍高信號,可見“虎紋征”,其周弓形纖維未見累及、增強未見明顯強化。選取雙側枕葉為感興趣區,TE值分別為35ms和144ms。譜線顯示雙側枕葉NAA峰明顯降低,Cho峰明顯升高,Cr峰位置、高度未見明顯異常,左側枕葉NAA/Cr值減低(約=0.89),右側枕葉NAA/Cr值減低(約=0.86);雙側枕葉Cho/Cr值升高(約1.76)
Figure2.
Cranial MRI + MRS changes of proband: a.bilateral periventricular and corpus callosum white area showed large symmetrical abnormal signal,which was uneven; b.T1WI showed low-signal intensity; c. T2WI and T2-FLAIR showed high signal intensity,“tigroid pattern” was visible,the peripheral arcuate fiber were not involved,no significant strengthening when enhanced. Selected the bilateral occipital as region of interest,TE values were 35ms and 144ms respectively. Spectrum showed NAA peak decreased in bilateral occipital,Cho peak significantly increased,the position and height of Cr peak were not obviously abnormal,NAA/Cr value decreased in left occipital lobe (approximately=0.89),NAA / Cr value decreased in the right occipital lobe (approximately=0.86); Cho/Cr value increased in bilateral occipital lobe(approximately=1.76)
患兒父母非近親結婚,父親近期確診急性淋巴瘤,未行ARSA基因及被白細胞ASA活性檢測。患兒母親及先證者之兄(圖 1 Ⅱ1)均體健,患兒的母親白細胞ASA活性為3.0 nmol/(mg·h),患兒之兄的ASA活性正常。家族中其他人無類似病史。
2 方法
2.1 頭顱 MRI+MRS檢查
采用飛利浦MRI掃描儀,掃描范圍為全腦.掃描方法及參數如下:飛利浦Achieva 3.0T超導MRI成像系統。8通道頭顱相控線圈。患兒行橫斷位快速自旋同波(FSE)T2WI、TIWI,液體衰減反轉恢復(FLAIR)T2WI以及橫斷位、冠狀位、矢狀位自旋同波(SE)T1WI增強檢查。橫斷位T2WI/FSE:TR 4 114 ms,TE 110 ms,T1Wi/SE:TR 488 ms.TE 15 ms,T2-FLAIR:TR 6 000 ms。TE 120ms,TI 2 000 ms,視野24 cm×24 cm,矩陣384×381,掃描層厚5.0 mm,層間距1.0 mm。MRS序列為點解析波譜法,TR 1 000 ms,TE 144 ms。取病變區進行分析,自動勻場與水抑制。采用Function Tool后處理軟件包進行波譜后處理測量NAA(2.0 ppm)、 Cr(3.0 ppm)、Cho(3.2 ppm)和Lac(1.33 ppm)等改變,計算各代謝物波峰下面積比值NAA/(Cho+Cr)。
2.2 ARSA基因測序方法
采用QIAGEN Blood DNA Mini kit提取外周血DNA 5mL然后采用2×PCR MasterMix聚合酶(TIANGEN)進行對ARSA基因的8個外顯子編碼區進行PCR擴增,擴增引物、退火溫度見表 1。對擴增產物進行直接測序(ABI3500xl測序儀,美國Life Technology公司)。測序結果與參考序列(NG_009260.2與NM_000487.5)進行比較,從而發現可能存在的基因突變。
2.3 溶酶體酶活性測定
ASA活性測定采用改良的Baum法:采集研究對象的外周血5 mL肝素抗凝,按Shoog法提取白細 胞,白細胞進行超聲粉碎后,以對-硝基兒茶酚胺硫酸鹽為底物,焦磷酸鈉/醋酸鹽(pH5.0)為底物緩沖液,按改良后Baum法測定外周血白細胞ASA活性,ASA活性單位以每毫克蛋白每小時的納摩爾數表示[nmol/(h·mg) 蛋白],正常參考值范圍為38.9~ 98.3 nmol/(h·mg) 蛋白。
結 果
1 ARSA基因測序結果
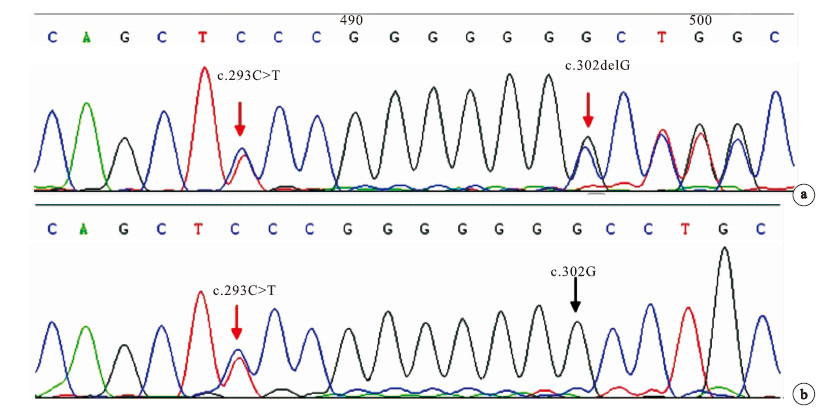
在先證者的ARSA基因的第2外顯子發現c.293C>T雜合突變和c.302de1G移碼突變(圖 3a),從而導致了第98位的絲氨酸被苯丙氨酸替代(Ser98Phe)和第101位的甘氨酸之后出現移碼(Gly101A1afs),患兒母親的ARSA基因的第2外顯子存在與先證者相同的雜合突變c.293C>T(圖 3b)。 先證者之兄ARSA基因檢測未發現突變。
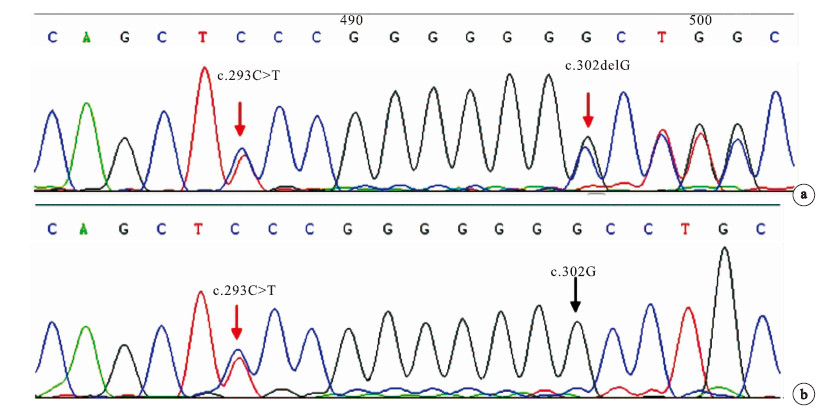 圖3
先證者及其母親的ARSA基因DNA直接測序結果
圖3
先證者及其母親的ARSA基因DNA直接測序結果
a 先證者:第2外顯子的c.293C>T雜合突變和c.302delG移碼突變;b 先證者之母:第2外顯子的c.293C>T雜合突變 (紅色箭頭示
a. proband: heterozygous missense mutation c.293C>T and frameshift mutation c.302delG in exon 2 of ARSA gene; b. proband’s mother: heterozygous missense mutation c.293C>T in exon 2 of
2 c.302de1G移碼突變位點分析
通過Mutation Taster數據庫分析(http://www.mutationtaster.org/),該突變位點位于chr22: 51065758_51065758delC,使第101位氨基酸之后出現移碼,導致在突變位點氨基酸與ARSA基因正常翻譯序列不一致,并且提前出現了終止密碼子,即移碼突變之前的基因序列可將101個氨基酸正常翻譯,而從第102位氨基酸開始出現移碼,且提前在第107位氨基酸出現翻譯終止(G101Afs*7),見
討 論
MLD是一種常染色體隱性遺傳性疾病,多見于ARSA基因突變而致ASA活性缺乏[3, 4],導致溶酶體內腦硫酯水解受阻而沉積在少突膠質細胞和施萬細胞[5],其特征性病理改變是在中樞神經系統少突膠質細胞和周圍神經施萬細胞內可見甲苯胺藍或甲酚紫染色呈現褐色或紅色的異染性物質[6]。MLD臨床表現為共濟失調、智能下降、四肢癱瘓、癲癇及精神癥狀等[7],其中,反復的癲癇發作在MLD中很常見,而且隨著病程的延長,發作頻率越高[8]。MLD通常按發病年齡可分為晚嬰型,青少年型及成人型,其中晚嬰型最常見[4, 9],約占50%~60%,同時病情程度最重。晚嬰型的MLD患兒早期可表現為行走困難、膝過伸、智力低下、易激惹、肌張力降低、腱反射減弱,后期出現廢用性肌萎縮、四肢痙攣性癱瘓、全身性強直陣攣性癲癇發作、眼震、視神經萎縮、失語等[10],病情常進行性發展,一般在5歲前死亡。本研究中的先證者生后出現精神運動發育遲緩、反復癲癇發作,逐漸出現語言能力倒退、運動障礙至四肢痙攣性癱瘓。頭顱MRI有典型的腦白質脫髓鞘改變,EEG表現異常,白細胞的ASA活性明顯缺乏,且病情呈進行性惡化,臨床癥狀符合MLD晚嬰型的診斷。
目前MLD的確診主要依賴于ASA活性檢測,但ASA活性降低并非MLD特異性表現,也可出現在ASA假性缺乏、多種硫酸酯酶缺乏癥、神經鞘脂激活蛋白B缺乏癥(saposinBdeficiency)、ARSA基因假性缺乏等位基因的雜合突變及22q13缺失綜合癥[11, 12]。ARSA基因假性缺乏是導致ASA活性下降常見的多態現象,在正常人群中約有10%~20%假缺陷基因,最常見的基因改變是Asn350Ser和c.2723A>G[3]。因此,基因診斷可作為明確診斷手段之一。ARSA基因突變是MLD主要致病原因,位于染色體22q13.33[9],含有9個外顯子,表達產物為含510個氨基酸的多肽。迄今為止,人類基因突變數據庫已有150余個被發現的ARSA基因突變[12]。MLD患者常見的ARSA基因突變位點在種族分布上存在差異。據報道,歐洲人群常見的ARSA基因突變位點多為c.459+1G>A、p.Pro426Leu、c.1204+1G>A和p.Ile179Ser,然而日本常見的ARSA基因突變多為p.Gly99Asp、p.Gly245Arg和p.Thr409Ile[4]。因此,中國人群的ARSA基因或許也存在一定的種族差異。
本研究采用PCR和DNA直接測序方法對這一家系進行ARSA突變檢測,在先證者及其母親的ARSA基因的第2外顯子均發現雜合突變c.293C>T,導致了第98位的絲氨酸被苯丙氨酸替代(Ser98Phe),先證者之兄未發現此突變。c.293C>T(rs74315456)此突變位點早已被發現可導致SmaI限制位點的缺失[13],但患者的癥狀表現為ASA假性缺乏,即突變位點可造成ASA活性下降,甚至達到MLD水平,但一般不造成MLD疾病的發生。本研究中先證者及其母親均發現c.293C>T雜合突變,但先證者之母的表型正常,白細胞ASA活性稍偏低,這與既往的研究結果相符。我們在先證者的ARSA基因的第2外顯子新發現了一個移碼突變c.302de1G,該突變導致第101位的甘氨酸之后出現移碼(Gly101A1afs)。目前,c.302delG移碼突變在國內外尚未見報道。我們通過Mutation Taster數據庫分析,c.302delG移碼突變使第101位氨基酸之后出現移碼,導致在突變后氨基酸序列與ARSA基因正常翻譯序列不一致,并且提前出現了終止密碼子。換之而言,移碼突變之前的氨基酸序列可正常翻譯,但從第102位氨基酸開始出現移碼翻譯,并且在第107位氨基酸出現翻譯終止,導致突變后的氨基酸多肽的長度比正常翻譯產物明顯縮短。該突變將嚴重影響蛋白質構象變化,從而導致ASA缺乏或失活。因此,我們推測移碼突變(c.302delG)為其致病位點。由于先證者之母及哥哥的ARSA基因未發現此突變,我們推測該移碼突變可能來自于父親。遺憾的是因先證者之父病危而未采集到DNA血樣。
綜上所述,本研究對一個晚嬰型MLD家系進行了臨床表型分析,先證者為難治性癲癇,并逐漸出現語言能力倒退、運動障礙至四肢痙攣性癱瘓,經頭顱MRI+MRS及ASA活性測定等檢查發現其難治性癲癇與異染性腦白質營養不良相關。進一步完善基因篩查發現了其存在ARSA基因c.293C>T雜合錯義突變和新發現c.302delG移碼突變,我們推測此移碼突變可能為MLD的新致病位點。這在一定程度上豐富了人類的MLD致病基因突變譜,有利于MLD患者相關家庭的遺傳咨詢,且為ARSA基因突變的功能、發病機制及尋找新的治療打下堅實基礎。
異染性腦白質營養不良(Metalchromatic leukodystrophy,MLD)是一種少見的溶酶體疾病,是由于芳基硫酸酯酶A(Arylsulfatase A,ARSA)或神經鞘脂激活蛋白B(Sphingolipid activator protein B,SAP-B)缺陷,從而導致腦硫苷脂沉積在少突膠質細胞和施萬細胞所致病的。國外報道的發病率為1/40 000~1/120 000活產嬰兒[1]。目前分子遺傳學研究已證實MLD發病與基因缺陷有關,其中以ARSA基因突變被廣泛關注[2]。本研究應用PCR直接測序的方法對一個晚嬰型MLD家系進行了ARSA基因突變分析,并分析MLD家系ARSA基因型與臨床表型的特點。
資料與方法
1 研究對象
先證者(圖 1 Ⅱ2)男,32個月。第2胎第2產,足月順產。孕期母親反復患支氣管炎,曾住院治療,具體不詳。患兒生后6個月會抬頭,7個月會翻身,9個月會坐,14個月可扶站,13個月會叫“媽媽”、“爸爸”等簡易的字語,但24個月仍不會走路。曾在外院診斷“腦性癱瘓”,予營養神經治療療效欠佳,并出現反復的抽搐,表現為四肢強直,牙關緊閉,意識不清,發作持續時間約1~2 min;診斷為癲癇,予左乙拉西坦片治療后發作有所減少,但半年后發作加重,每天均有發作,癥狀同前,加用丙戊酸鈉緩釋片治療后發作次數有所減少,但仍有發作,并逐漸出現語言能力倒退和雙下肢強直,雙下肢腳
圖1
家系圖
Figure1.
Pedigree
圖2
先證者頭顱MRI+MRS改變a.雙側側腦室周圍及胼胝體壓部白質區見對稱性大片狀異常信號區;b.病灶信號不均、T1WI呈等低信號;c.T2WI及T2-FLAIR呈稍高信號,可見“虎紋征”,其周弓形纖維未見累及、增強未見明顯強化。選取雙側枕葉為感興趣區,TE值分別為35ms和144ms。譜線顯示雙側枕葉NAA峰明顯降低,Cho峰明顯升高,Cr峰位置、高度未見明顯異常,左側枕葉NAA/Cr值減低(約=0.89),右側枕葉NAA/Cr值減低(約=0.86);雙側枕葉Cho/Cr值升高(約1.76)
Figure2.
Cranial MRI + MRS changes of proband: a.bilateral periventricular and corpus callosum white area showed large symmetrical abnormal signal,which was uneven; b.T1WI showed low-signal intensity; c. T2WI and T2-FLAIR showed high signal intensity,“tigroid pattern” was visible,the peripheral arcuate fiber were not involved,no significant strengthening when enhanced. Selected the bilateral occipital as region of interest,TE values were 35ms and 144ms respectively. Spectrum showed NAA peak decreased in bilateral occipital,Cho peak significantly increased,the position and height of Cr peak were not obviously abnormal,NAA/Cr value decreased in left occipital lobe (approximately=0.89),NAA / Cr value decreased in the right occipital lobe (approximately=0.86); Cho/Cr value increased in bilateral occipital lobe(approximately=1.76)
患兒父母非近親結婚,父親近期確診急性淋巴瘤,未行ARSA基因及被白細胞ASA活性檢測。患兒母親及先證者之兄(圖 1 Ⅱ1)均體健,患兒的母親白細胞ASA活性為3.0 nmol/(mg·h),患兒之兄的ASA活性正常。家族中其他人無類似病史。
2 方法
2.1 頭顱 MRI+MRS檢查
采用飛利浦MRI掃描儀,掃描范圍為全腦.掃描方法及參數如下:飛利浦Achieva 3.0T超導MRI成像系統。8通道頭顱相控線圈。患兒行橫斷位快速自旋同波(FSE)T2WI、TIWI,液體衰減反轉恢復(FLAIR)T2WI以及橫斷位、冠狀位、矢狀位自旋同波(SE)T1WI增強檢查。橫斷位T2WI/FSE:TR 4 114 ms,TE 110 ms,T1Wi/SE:TR 488 ms.TE 15 ms,T2-FLAIR:TR 6 000 ms。TE 120ms,TI 2 000 ms,視野24 cm×24 cm,矩陣384×381,掃描層厚5.0 mm,層間距1.0 mm。MRS序列為點解析波譜法,TR 1 000 ms,TE 144 ms。取病變區進行分析,自動勻場與水抑制。采用Function Tool后處理軟件包進行波譜后處理測量NAA(2.0 ppm)、 Cr(3.0 ppm)、Cho(3.2 ppm)和Lac(1.33 ppm)等改變,計算各代謝物波峰下面積比值NAA/(Cho+Cr)。
2.2 ARSA基因測序方法
采用QIAGEN Blood DNA Mini kit提取外周血DNA 5mL然后采用2×PCR MasterMix聚合酶(TIANGEN)進行對ARSA基因的8個外顯子編碼區進行PCR擴增,擴增引物、退火溫度見表 1。對擴增產物進行直接測序(ABI3500xl測序儀,美國Life Technology公司)。測序結果與參考序列(NG_009260.2與NM_000487.5)進行比較,從而發現可能存在的基因突變。
2.3 溶酶體酶活性測定
ASA活性測定采用改良的Baum法:采集研究對象的外周血5 mL肝素抗凝,按Shoog法提取白細 胞,白細胞進行超聲粉碎后,以對-硝基兒茶酚胺硫酸鹽為底物,焦磷酸鈉/醋酸鹽(pH5.0)為底物緩沖液,按改良后Baum法測定外周血白細胞ASA活性,ASA活性單位以每毫克蛋白每小時的納摩爾數表示[nmol/(h·mg) 蛋白],正常參考值范圍為38.9~ 98.3 nmol/(h·mg) 蛋白。
結 果
1 ARSA基因測序結果
在先證者的ARSA基因的第2外顯子發現c.293C>T雜合突變和c.302de1G移碼突變(圖 3a),從而導致了第98位的絲氨酸被苯丙氨酸替代(Ser98Phe)和第101位的甘氨酸之后出現移碼(Gly101A1afs),患兒母親的ARSA基因的第2外顯子存在與先證者相同的雜合突變c.293C>T(圖 3b)。 先證者之兄ARSA基因檢測未發現突變。
圖3
先證者及其母親的ARSA基因DNA直接測序結果
a 先證者:第2外顯子的c.293C>T雜合突變和c.302delG移碼突變;b 先證者之母:第2外顯子的c.293C>T雜合突變 (紅色箭頭示
a. proband: heterozygous missense mutation c.293C>T and frameshift mutation c.302delG in exon 2 of ARSA gene; b. proband’s mother: heterozygous missense mutation c.293C>T in exon 2 of
2 c.302de1G移碼突變位點分析
通過Mutation Taster數據庫分析(http://www.mutationtaster.org/),該突變位點位于chr22: 51065758_51065758delC,使第101位氨基酸之后出現移碼,導致在突變位點氨基酸與ARSA基因正常翻譯序列不一致,并且提前出現了終止密碼子,即移碼突變之前的基因序列可將101個氨基酸正常翻譯,而從第102位氨基酸開始出現移碼,且提前在第107位氨基酸出現翻譯終止(G101Afs*7),見
討 論
MLD是一種常染色體隱性遺傳性疾病,多見于ARSA基因突變而致ASA活性缺乏[3, 4],導致溶酶體內腦硫酯水解受阻而沉積在少突膠質細胞和施萬細胞[5],其特征性病理改變是在中樞神經系統少突膠質細胞和周圍神經施萬細胞內可見甲苯胺藍或甲酚紫染色呈現褐色或紅色的異染性物質[6]。MLD臨床表現為共濟失調、智能下降、四肢癱瘓、癲癇及精神癥狀等[7],其中,反復的癲癇發作在MLD中很常見,而且隨著病程的延長,發作頻率越高[8]。MLD通常按發病年齡可分為晚嬰型,青少年型及成人型,其中晚嬰型最常見[4, 9],約占50%~60%,同時病情程度最重。晚嬰型的MLD患兒早期可表現為行走困難、膝過伸、智力低下、易激惹、肌張力降低、腱反射減弱,后期出現廢用性肌萎縮、四肢痙攣性癱瘓、全身性強直陣攣性癲癇發作、眼震、視神經萎縮、失語等[10],病情常進行性發展,一般在5歲前死亡。本研究中的先證者生后出現精神運動發育遲緩、反復癲癇發作,逐漸出現語言能力倒退、運動障礙至四肢痙攣性癱瘓。頭顱MRI有典型的腦白質脫髓鞘改變,EEG表現異常,白細胞的ASA活性明顯缺乏,且病情呈進行性惡化,臨床癥狀符合MLD晚嬰型的診斷。
目前MLD的確診主要依賴于ASA活性檢測,但ASA活性降低并非MLD特異性表現,也可出現在ASA假性缺乏、多種硫酸酯酶缺乏癥、神經鞘脂激活蛋白B缺乏癥(saposinBdeficiency)、ARSA基因假性缺乏等位基因的雜合突變及22q13缺失綜合癥[11, 12]。ARSA基因假性缺乏是導致ASA活性下降常見的多態現象,在正常人群中約有10%~20%假缺陷基因,最常見的基因改變是Asn350Ser和c.2723A>G[3]。因此,基因診斷可作為明確診斷手段之一。ARSA基因突變是MLD主要致病原因,位于染色體22q13.33[9],含有9個外顯子,表達產物為含510個氨基酸的多肽。迄今為止,人類基因突變數據庫已有150余個被發現的ARSA基因突變[12]。MLD患者常見的ARSA基因突變位點在種族分布上存在差異。據報道,歐洲人群常見的ARSA基因突變位點多為c.459+1G>A、p.Pro426Leu、c.1204+1G>A和p.Ile179Ser,然而日本常見的ARSA基因突變多為p.Gly99Asp、p.Gly245Arg和p.Thr409Ile[4]。因此,中國人群的ARSA基因或許也存在一定的種族差異。
本研究采用PCR和DNA直接測序方法對這一家系進行ARSA突變檢測,在先證者及其母親的ARSA基因的第2外顯子均發現雜合突變c.293C>T,導致了第98位的絲氨酸被苯丙氨酸替代(Ser98Phe),先證者之兄未發現此突變。c.293C>T(rs74315456)此突變位點早已被發現可導致SmaI限制位點的缺失[13],但患者的癥狀表現為ASA假性缺乏,即突變位點可造成ASA活性下降,甚至達到MLD水平,但一般不造成MLD疾病的發生。本研究中先證者及其母親均發現c.293C>T雜合突變,但先證者之母的表型正常,白細胞ASA活性稍偏低,這與既往的研究結果相符。我們在先證者的ARSA基因的第2外顯子新發現了一個移碼突變c.302de1G,該突變導致第101位的甘氨酸之后出現移碼(Gly101A1afs)。目前,c.302delG移碼突變在國內外尚未見報道。我們通過Mutation Taster數據庫分析,c.302delG移碼突變使第101位氨基酸之后出現移碼,導致在突變后氨基酸序列與ARSA基因正常翻譯序列不一致,并且提前出現了終止密碼子。換之而言,移碼突變之前的氨基酸序列可正常翻譯,但從第102位氨基酸開始出現移碼翻譯,并且在第107位氨基酸出現翻譯終止,導致突變后的氨基酸多肽的長度比正常翻譯產物明顯縮短。該突變將嚴重影響蛋白質構象變化,從而導致ASA缺乏或失活。因此,我們推測移碼突變(c.302delG)為其致病位點。由于先證者之母及哥哥的ARSA基因未發現此突變,我們推測該移碼突變可能來自于父親。遺憾的是因先證者之父病危而未采集到DNA血樣。
綜上所述,本研究對一個晚嬰型MLD家系進行了臨床表型分析,先證者為難治性癲癇,并逐漸出現語言能力倒退、運動障礙至四肢痙攣性癱瘓,經頭顱MRI+MRS及ASA活性測定等檢查發現其難治性癲癇與異染性腦白質營養不良相關。進一步完善基因篩查發現了其存在ARSA基因c.293C>T雜合錯義突變和新發現c.302delG移碼突變,我們推測此移碼突變可能為MLD的新致病位點。這在一定程度上豐富了人類的MLD致病基因突變譜,有利于MLD患者相關家庭的遺傳咨詢,且為ARSA基因突變的功能、發病機制及尋找新的治療打下堅實基礎。