引用本文: 盧曉棟, 陳春智, 周淵峰, 邱甜, 王新華, 王佶, 柴毅明, 周水珍, 王藝, 吳冰冰. DNM1 基因變異致早發性嬰兒癲癇性腦病 31 型 三例并文獻復習. 癲癇雜志, 2021, 7(2): 104-111. doi: 10.7507/2096-0247.20210018 復制
版權信息: ?四川大學華西醫院華西期刊社《癲癇雜志》版權所有,未經授權不得轉載、改編
Dynamin 1 基因(DNM1;NM_004408)位于染色體 9q34.11,編碼位于 DNM1 蛋白,DNM1 基因變異致早發性嬰兒癲癇性腦病 31 型,主要特征性臨床表現為早發型難治性癲癇、發育落后和運動障礙,本文對在我院神經科登記的 3 例 DNM1 癲癇性腦病患兒的資料進行總結,并文獻檢索國內外報道的 DNM1 相關腦病病例,分析和探討臨床表型和基因型的相關性,以提高對該病的認識和診斷。
1 資料和方法
1.1 3 例病例資料
例 1 男,G1P1,足月,剖腹產,否認窒息搶救史。生后 8 月齡出現成串癲癇性痙攣發作,接受左乙拉西坦和丙戊酸鈉治療,發作頻率每周 1~3 串,7~8 余次/串。腦電圖(EEG)為高度失律,頭顱核磁正常。隨訪至 30 月齡時,不追聲不追物,不能抬頭和獨坐,無言語,肢體肌張力低下,平時能喝奶和吞咽稀飯,但不會咀嚼和吞咽固體食物。經家系全外顯子二代測序,發現在 DNM1 基因第 4 號外顯子區域一處雜合位點變異 NM_004408.4:c.415 G>A(P. Gly 139Arg),為錯義變異(新發變異,考慮為致病性變異,PVS1+PS2[1]),該錯義變異未見文獻報道。
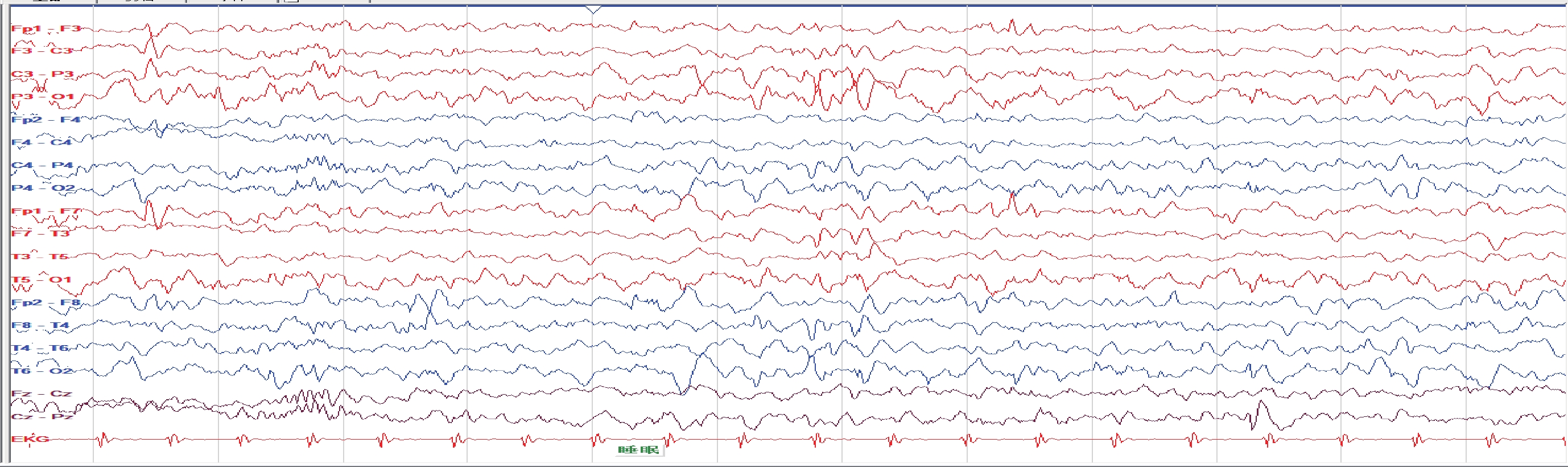
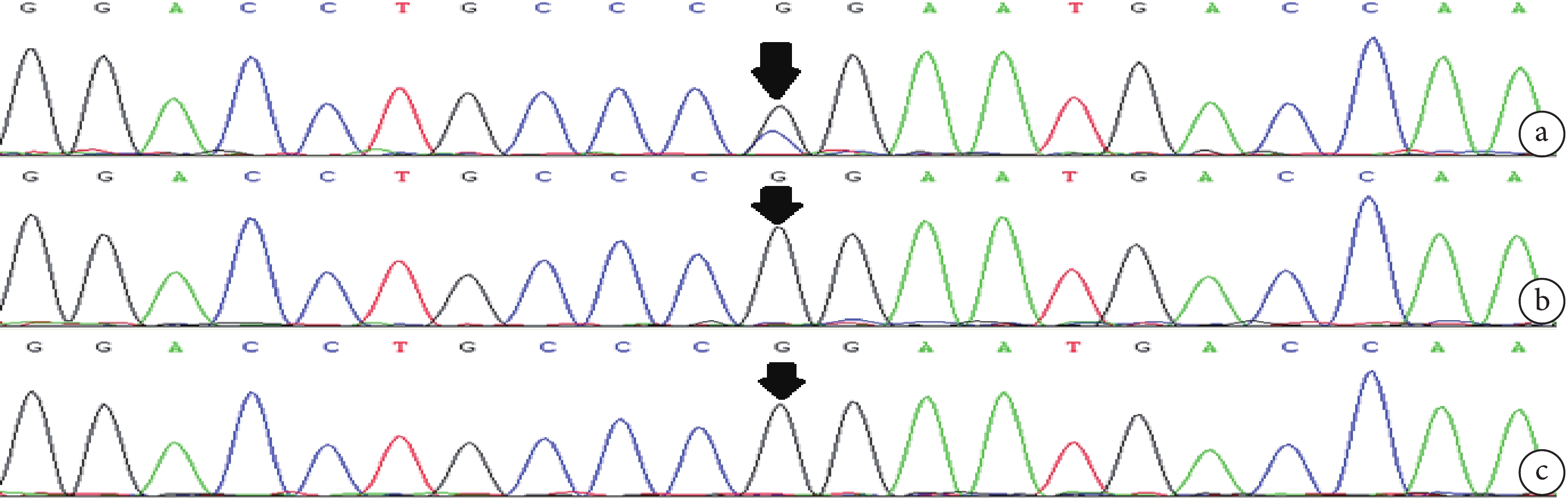
例 2 男,G1P1,足月,剖腹產,否認窒息搶救史。17 月齡首次發作,表現為局灶性運動性發作(一側肢體陣攣)或繼發雙側驚厥發作,接受左乙拉西坦和丙戊酸鈉治療,發作頻率 2~3 次/月,EEG 為多灶性癲癇樣放電(圖 1),頭部核磁共振成像(MRI)提示胼胝體發育不良。隨訪至 36 月齡時能翻身,不能獨坐,無言語,平時只能喝奶和稀飯,不會咀嚼和吞咽固體食物。經癲癇基因 panel 二代測序,發現在 DNM1 基因第 4 號外顯子區域一處雜合位點變異 NM_004408.4:c.415 G>A(P. Gly 139Arg)(圖 2),為錯義變異(新發變異,PM2+PM6+PP3[1])。
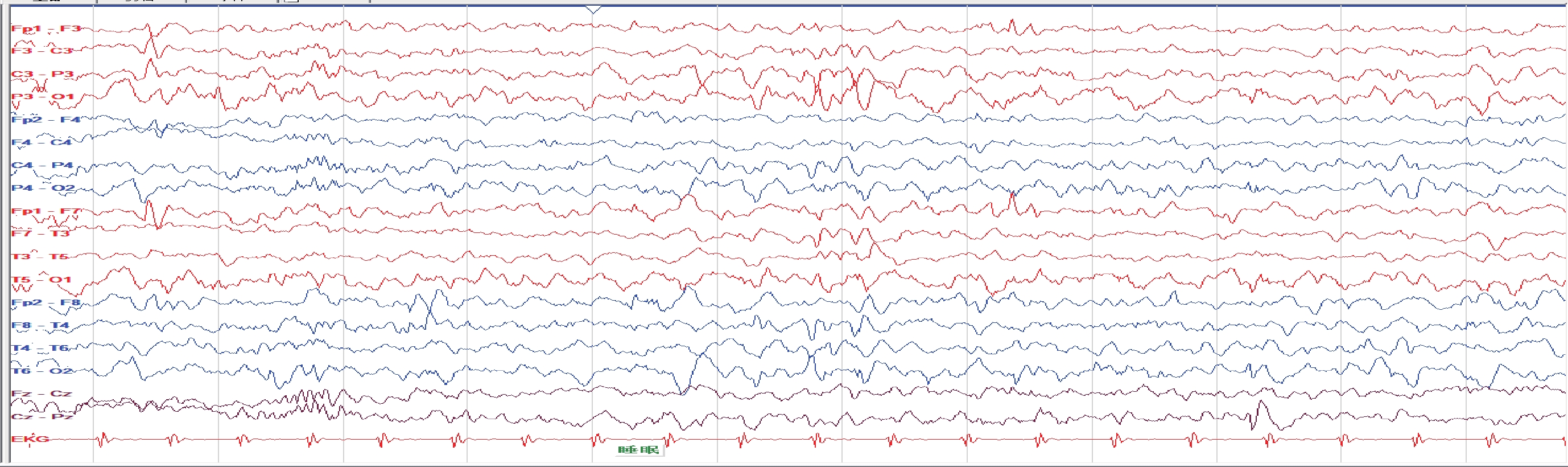 圖1
例 2 患兒腦電圖
圖1
例 2 患兒腦電圖
睡眠期多灶性癲癇樣放電
Figure1. EEG of case 2Multifocal epileptiform discharge during sleep
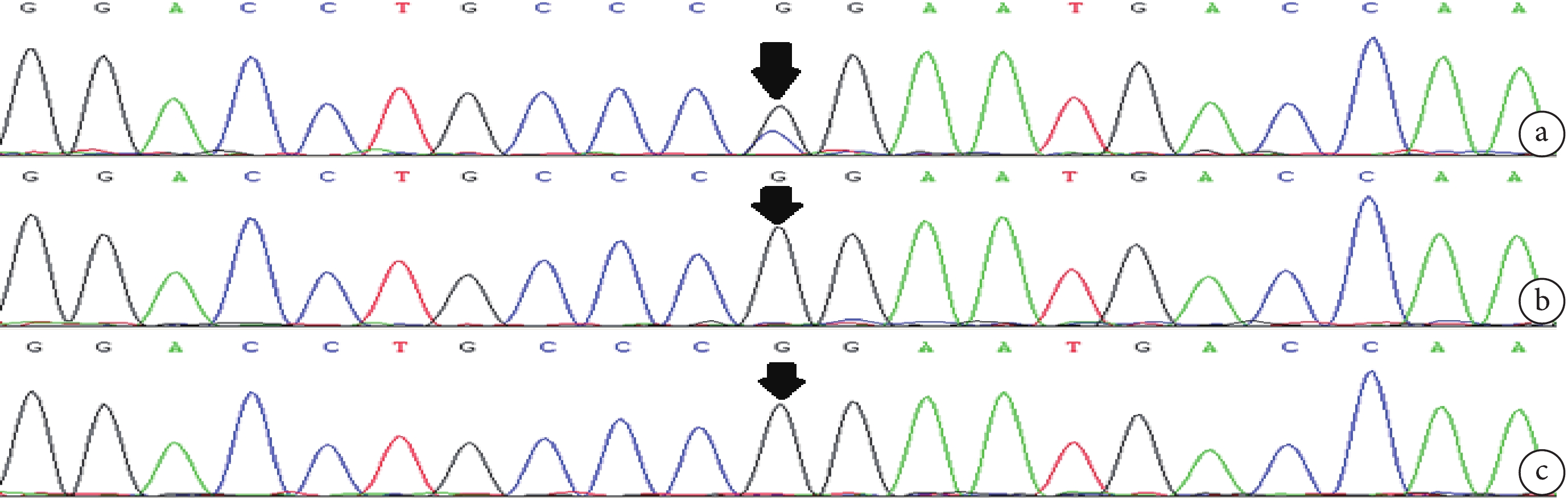 圖2
例 2 患兒以及父母 DNM1 基因測序
圖2
例 2 患兒以及父母 DNM1 基因測序
a. 患兒 DNM1 基因雜合位點變異 NM_004408.4:c.415 G>A(箭頭);b,c. 患兒父母相同位點未見此變異(箭頭)
Figure2. DNM1 gene sequencing of case 2 and his parentsa. Heterozygous mutation of DNM1 gene in children with NM_004408.4: c.415 G>A (arrow); b, c. the same site mutation is not seen in the parents (arrow)
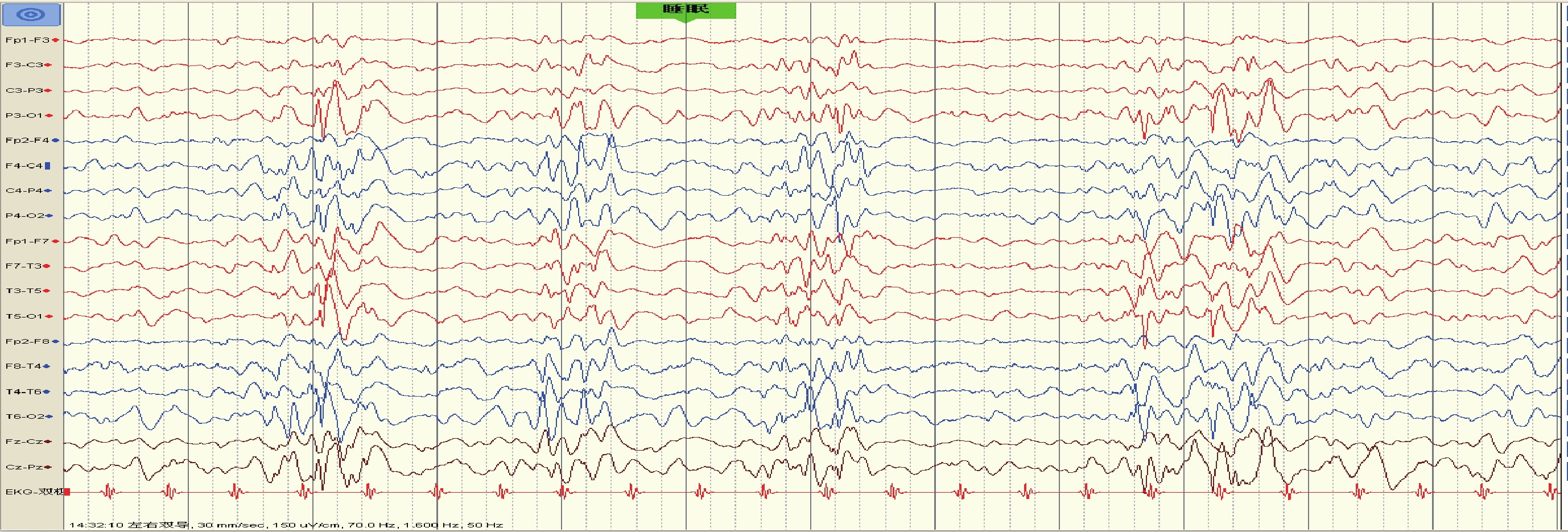
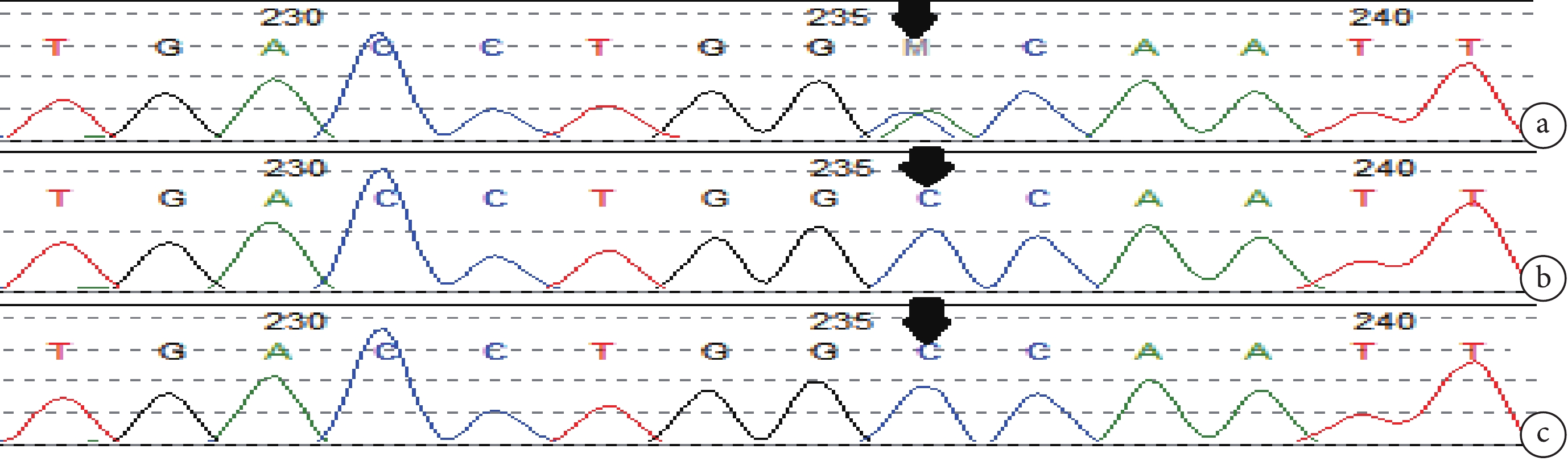
例 3 女,G4P2,足月,剖腹產,否認窒息搶救史(第 2 胎、第 3 胎人工流產)。出生后 2 月齡出現成串癲癇性痙攣發作,3 月齡起接受左乙拉西坦、丙戊酸鈉、托吡酯和氯巴占治療,發作頻率每天 5~8 串,10 余次/串,EEG 為高度失律(圖 3),頭部 MRI 正常。隨訪至 24 月齡時不追聲不追物,不能抬頭和獨坐,無言語,肢體肌張力低下,平時喝奶慢,不能咀嚼和吞咽米飯等固體食物。經家系全外顯子二代測序,發現在 DNM1 基因第 4 號外顯子區域一處雜合位點變異 NM_004408.4:c.545 C>A(P. Ala 182Asp),為錯義變異(新發變異,考慮為致病性變異,PVS1+PS2[1])(圖 4),該錯義變異未見文獻報道。
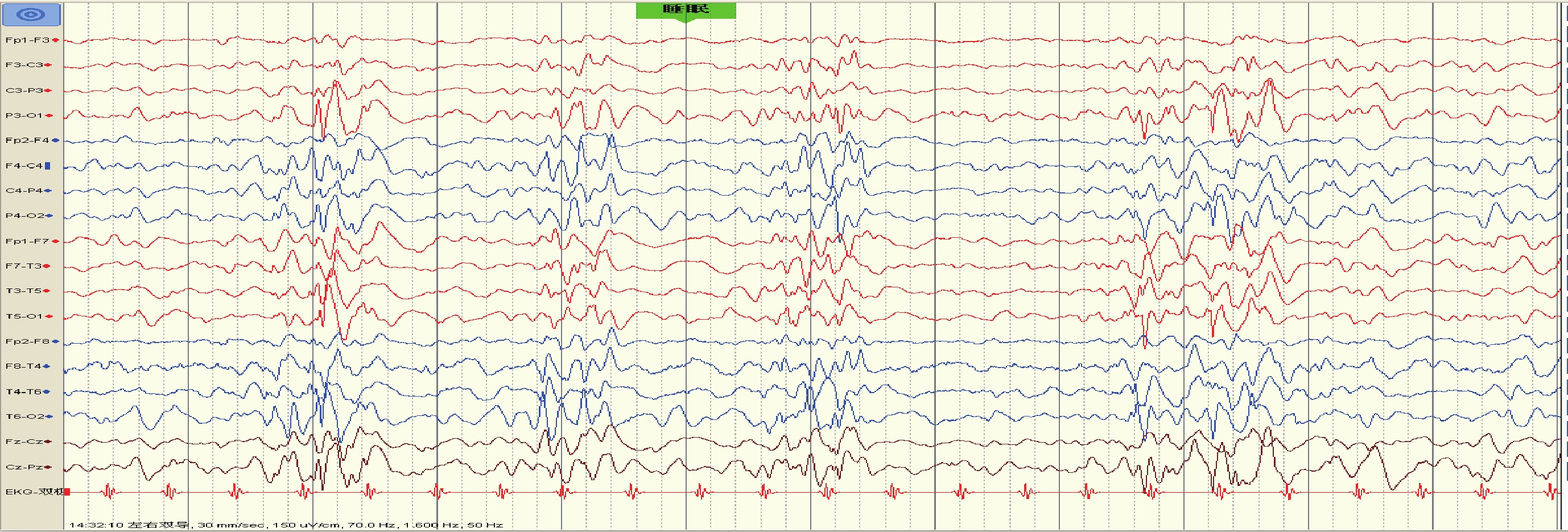 圖3
例 3 患兒腦電圖
圖3
例 3 患兒腦電圖
睡眠期高度失律
Figure3. EEG of case 3Highly irregular during sleep
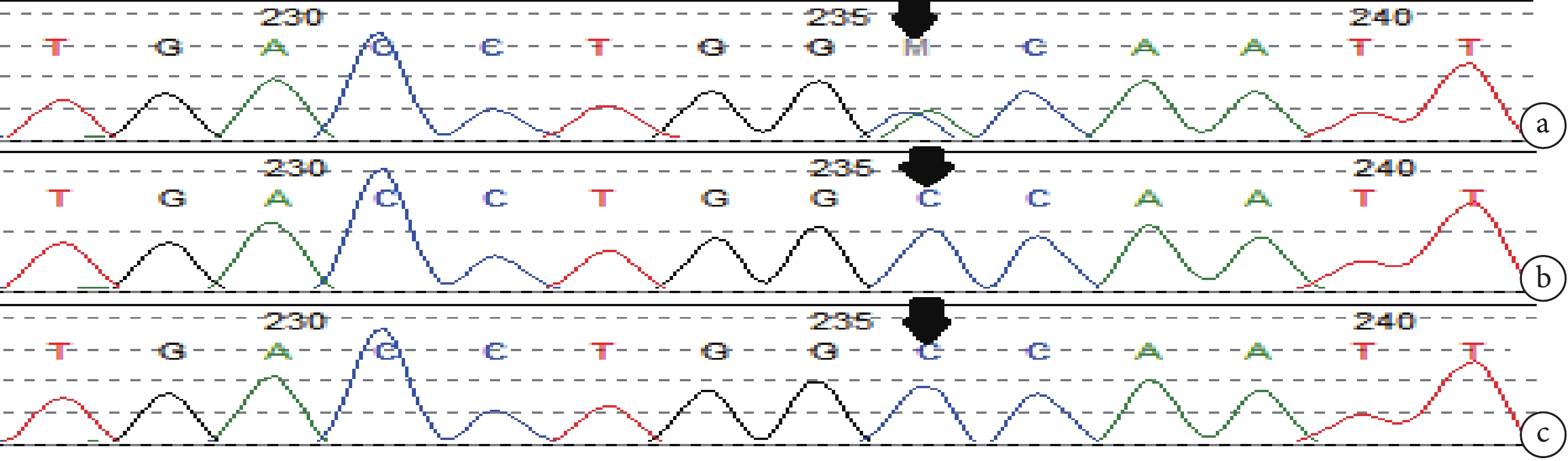 圖4
例 3 患兒以及父母 DNM1 基因測序
圖4
例 3 患兒以及父母 DNM1 基因測序
a. 患兒 DNM1 基因雜合位點變異 NM_004408.4:c.545 C>A(箭頭);b,c. 患兒父母相同位點未見此變異(箭頭)
Figure4. DNM1 gene sequencing of case 2 and her parentsa. Heterozygous mutation of DNM1 gene in children with NM_004408.4: c.545 C>A (arrow); b, c. the same site mutation is not seen in the parents (arrow)
1.2 文獻檢索
截止至 2020 年 12 月,以“Dynamin 1”“DNM1”為檢索詞查閱中國知網數據庫、萬方數據庫、在線人類孟德爾遺傳數據庫(OMIM)和 PubMed 數據庫,11 篇文章報道 36 例 DNM1 相關腦病[2-13],包括本組病例共計 39 例。我們總結并分析了 39 例 DNM1 相關腦病的基因變異位點和臨床表現,藥物治療反應和頭部影像改變。
1.3 統計學方法
采用 Stata 12.0 軟件行統計分析:31 例臨床資料完整的病例(包括起病年齡、癲癇發作形式和藥物治療反應)依據不同基因位點區分成 GTPase 酶結構域組和中間區域 2 組,計數資料的比較采用 Fisher 確切概率法。
2 結果
39 例 DNM1 相關腦病中,37 例(94.9%)為錯義變異,1 例為框內插入變異,1 例為剪接變異,9 例(23.1%)基因變異為 c.709C>T(p.Arg237Trp);基因變異位于 GTPase 酶結構域 28 例(71.8%),中間區域 9 例(23.1%),PH 結構域 2 例(5.1%);37 例 GTPase 酶結構域和中間區域變異中,嚴重或顯著智力發育障礙 36 例(92.3%),1 例不詳;輕-中度智力發育障礙 2 例,基因變異位于 PH 結構域。肌張力低下 36 例(92.3%),基因變異分別位于 GTPase 酶結構域、中間區域和 PH 結構域,3 例不詳;35 例(89.7%)診斷癲癇并接受抗癲癇藥物(AEDs)治療,GTPase 酶結構域和中間區域無癲癇發作各 1 例,PH 結構域 2 例均無癲癇發作,35 例中 28 例(80 %)多種癲癇發作形式,6 例單一癲癇發作形式,1 例不詳;頭部 MRI 異常 17 例,正常 15 例,7 例不詳(表 1)。
31 例診斷癲癇并接受 AEDs 且資料完整(包括起病年齡、癲癇發作形式和藥物治療反應),GTPase 酶結構域 25 例,中間區域 6 例,20 例男孩(64.5%),26 例(83.9%)1 歲前出現癲癇發作,癲癇性痙攣發作(74.2%),強直-陣攣發作(48.4%),失神發作(41.9%),局灶性發作(38.7%),強直發作(35.5%),肌陣攣發作(32.3%),失張力發作(22.6%)和癲癇持續狀態(19.4%),28 例(90.3%)為耐藥性癲癇,不同基因變異位點區域的兩組病例,失神發作具有統計學差異(P=0.02),在性別、起病年齡、其他癲癇發作形式和藥物治療反應上無統計學差異(表 2)。
3 討論
Dynamin 1 基因編碼 DNM1 蛋白,是一種鳥苷酸三磷酸酶(GTPase),主要在神經元表達,在突觸前受體介導的突觸囊泡內吞和循環發揮重要作用,同時在生后神經發育期間表達上調并參與突觸的發生發展。DNM1 蛋白有 5 個結構域,其中 GTPase 酶結構域和中間區域是主要的功能域,體外研究表明,位于 GTPase 酶結構域的基因變異會干擾突觸囊泡內吞、降低囊泡內吞的活性以及突觸傳遞,位于中間區域的基因變異會破壞 DNM1 蛋白形成更大的寡聚體,而位于 PH 結構域的基因變異會影響磷酸肌醇的結合和突觸囊泡再攝取[14-16]。
DNM1 基因變異致早發性嬰兒癲癇性腦病 31 型,臨床以早發型難治性癲癇、嚴重智力和語言發育障礙、運動障礙和肌張力低下為特征,綜合分析文獻報道的 39 例 DNM1 病例,肌張力低下與基因變異位點無相關性,提示肌張力低下是 DNM1 相關腦病的共有癥狀。GTPase 酶結構域和中間區域變異病例均有嚴重或顯著的智力、言語發育障礙和運動障礙,本組 3 例病例基因變異位點位于 GTPase 酶結構域,臨床表現均符合上述臨床特點,同時本組病例均不會咀嚼和吞咽固體食物,既往文獻均未提及,提示今后需關注 DNM1 相關腦病合并進食障礙的現象。
此外,文獻報道的基因變異位點位于 PH 結構域的 2 例單卵孿生姐妹,除無癲癇發作外,智力、言語和運動障礙的程度好于基因變異位點位于 GTPase 酶結構域和中間區域的病例,但病情嚴重程度不同的機制尚未明確[5],同時,也需要更多的 PH 結構域變異的病例來證實基因和表型之間的相關性。
35 例 DNM1 變異病例存在癲癇,80% 的病例具有多種癲癇發作形式,對 31 例資料完整的病例進一步統計學研究發現,男孩相對多見,80% 以上的病例在 1 歲前出現癲癇發作,最常見的癲癇發作類型是癲癇性痙攣發作,其次為強直-陣攣和失神發作,其他還包括局灶性、肌陣攣和強直發作等,90% 以上為耐藥性癲癇,本組 3 例病例,癲癇發病年齡、癲癇發作形式和 AEDs 治療反應與文獻報道基本相符,提示對于病因不明的早發型癲癇性腦病,尤其表現為癲癇性痙攣發作和肌張力低下,需通過基因檢測除外 DNM1 基因變異并明確病因。
有文獻報道,在起病初的癲癇發作形式,在 GTPase 酶結構域和中間區域變異之間無明顯差異[3],對 31 病例所有的癲癇發作形式進一步細分后發現,GTPase 酶結構域組失神發作明顯多于中間區域組,有統計學差異,但需更多的病例來證實失神發作和基因型之間的相關性,而在性別、起病年齡、其他癲癇發作形式和藥物治療反應上均無統計學差異,與文獻報道相符[3, 8],提示 GTPase 酶結構域和中間區域變異具有類似的臨床表現,也提示兩組變異具有相同的發病機制。
綜上,肌張力低下、嚴重智力和運動障礙以及早發型癲癇是 DNM1 相關腦病的主要表現,癲癇性痙攣發作是最常見的發作形式,除失神發作外,GTPase 酶結構域和中間區域變異的臨床和癲癇表型無明顯區別,本組病例尚合并進食障礙。
Dynamin 1 基因(DNM1;NM_004408)位于染色體 9q34.11,編碼位于 DNM1 蛋白,DNM1 基因變異致早發性嬰兒癲癇性腦病 31 型,主要特征性臨床表現為早發型難治性癲癇、發育落后和運動障礙,本文對在我院神經科登記的 3 例 DNM1 癲癇性腦病患兒的資料進行總結,并文獻檢索國內外報道的 DNM1 相關腦病病例,分析和探討臨床表型和基因型的相關性,以提高對該病的認識和診斷。
1 資料和方法
1.1 3 例病例資料
例 1 男,G1P1,足月,剖腹產,否認窒息搶救史。生后 8 月齡出現成串癲癇性痙攣發作,接受左乙拉西坦和丙戊酸鈉治療,發作頻率每周 1~3 串,7~8 余次/串。腦電圖(EEG)為高度失律,頭顱核磁正常。隨訪至 30 月齡時,不追聲不追物,不能抬頭和獨坐,無言語,肢體肌張力低下,平時能喝奶和吞咽稀飯,但不會咀嚼和吞咽固體食物。經家系全外顯子二代測序,發現在 DNM1 基因第 4 號外顯子區域一處雜合位點變異 NM_004408.4:c.415 G>A(P. Gly 139Arg),為錯義變異(新發變異,考慮為致病性變異,PVS1+PS2[1]),該錯義變異未見文獻報道。
例 2 男,G1P1,足月,剖腹產,否認窒息搶救史。17 月齡首次發作,表現為局灶性運動性發作(一側肢體陣攣)或繼發雙側驚厥發作,接受左乙拉西坦和丙戊酸鈉治療,發作頻率 2~3 次/月,EEG 為多灶性癲癇樣放電(圖 1),頭部核磁共振成像(MRI)提示胼胝體發育不良。隨訪至 36 月齡時能翻身,不能獨坐,無言語,平時只能喝奶和稀飯,不會咀嚼和吞咽固體食物。經癲癇基因 panel 二代測序,發現在 DNM1 基因第 4 號外顯子區域一處雜合位點變異 NM_004408.4:c.415 G>A(P. Gly 139Arg)(圖 2),為錯義變異(新發變異,PM2+PM6+PP3[1])。
圖1
例 2 患兒腦電圖
睡眠期多灶性癲癇樣放電
Figure1. EEG of case 2Multifocal epileptiform discharge during sleep
圖2
例 2 患兒以及父母 DNM1 基因測序
a. 患兒 DNM1 基因雜合位點變異 NM_004408.4:c.415 G>A(箭頭);b,c. 患兒父母相同位點未見此變異(箭頭)
Figure2. DNM1 gene sequencing of case 2 and his parentsa. Heterozygous mutation of DNM1 gene in children with NM_004408.4: c.415 G>A (arrow); b, c. the same site mutation is not seen in the parents (arrow)
例 3 女,G4P2,足月,剖腹產,否認窒息搶救史(第 2 胎、第 3 胎人工流產)。出生后 2 月齡出現成串癲癇性痙攣發作,3 月齡起接受左乙拉西坦、丙戊酸鈉、托吡酯和氯巴占治療,發作頻率每天 5~8 串,10 余次/串,EEG 為高度失律(圖 3),頭部 MRI 正常。隨訪至 24 月齡時不追聲不追物,不能抬頭和獨坐,無言語,肢體肌張力低下,平時喝奶慢,不能咀嚼和吞咽米飯等固體食物。經家系全外顯子二代測序,發現在 DNM1 基因第 4 號外顯子區域一處雜合位點變異 NM_004408.4:c.545 C>A(P. Ala 182Asp),為錯義變異(新發變異,考慮為致病性變異,PVS1+PS2[1])(圖 4),該錯義變異未見文獻報道。
圖3
例 3 患兒腦電圖
睡眠期高度失律
Figure3. EEG of case 3Highly irregular during sleep
圖4
例 3 患兒以及父母 DNM1 基因測序
a. 患兒 DNM1 基因雜合位點變異 NM_004408.4:c.545 C>A(箭頭);b,c. 患兒父母相同位點未見此變異(箭頭)
Figure4. DNM1 gene sequencing of case 2 and her parentsa. Heterozygous mutation of DNM1 gene in children with NM_004408.4: c.545 C>A (arrow); b, c. the same site mutation is not seen in the parents (arrow)
1.2 文獻檢索
截止至 2020 年 12 月,以“Dynamin 1”“DNM1”為檢索詞查閱中國知網數據庫、萬方數據庫、在線人類孟德爾遺傳數據庫(OMIM)和 PubMed 數據庫,11 篇文章報道 36 例 DNM1 相關腦病[2-13],包括本組病例共計 39 例。我們總結并分析了 39 例 DNM1 相關腦病的基因變異位點和臨床表現,藥物治療反應和頭部影像改變。
1.3 統計學方法
采用 Stata 12.0 軟件行統計分析:31 例臨床資料完整的病例(包括起病年齡、癲癇發作形式和藥物治療反應)依據不同基因位點區分成 GTPase 酶結構域組和中間區域 2 組,計數資料的比較采用 Fisher 確切概率法。
2 結果
39 例 DNM1 相關腦病中,37 例(94.9%)為錯義變異,1 例為框內插入變異,1 例為剪接變異,9 例(23.1%)基因變異為 c.709C>T(p.Arg237Trp);基因變異位于 GTPase 酶結構域 28 例(71.8%),中間區域 9 例(23.1%),PH 結構域 2 例(5.1%);37 例 GTPase 酶結構域和中間區域變異中,嚴重或顯著智力發育障礙 36 例(92.3%),1 例不詳;輕-中度智力發育障礙 2 例,基因變異位于 PH 結構域。肌張力低下 36 例(92.3%),基因變異分別位于 GTPase 酶結構域、中間區域和 PH 結構域,3 例不詳;35 例(89.7%)診斷癲癇并接受抗癲癇藥物(AEDs)治療,GTPase 酶結構域和中間區域無癲癇發作各 1 例,PH 結構域 2 例均無癲癇發作,35 例中 28 例(80 %)多種癲癇發作形式,6 例單一癲癇發作形式,1 例不詳;頭部 MRI 異常 17 例,正常 15 例,7 例不詳(表 1)。
31 例診斷癲癇并接受 AEDs 且資料完整(包括起病年齡、癲癇發作形式和藥物治療反應),GTPase 酶結構域 25 例,中間區域 6 例,20 例男孩(64.5%),26 例(83.9%)1 歲前出現癲癇發作,癲癇性痙攣發作(74.2%),強直-陣攣發作(48.4%),失神發作(41.9%),局灶性發作(38.7%),強直發作(35.5%),肌陣攣發作(32.3%),失張力發作(22.6%)和癲癇持續狀態(19.4%),28 例(90.3%)為耐藥性癲癇,不同基因變異位點區域的兩組病例,失神發作具有統計學差異(P=0.02),在性別、起病年齡、其他癲癇發作形式和藥物治療反應上無統計學差異(表 2)。
3 討論
Dynamin 1 基因編碼 DNM1 蛋白,是一種鳥苷酸三磷酸酶(GTPase),主要在神經元表達,在突觸前受體介導的突觸囊泡內吞和循環發揮重要作用,同時在生后神經發育期間表達上調并參與突觸的發生發展。DNM1 蛋白有 5 個結構域,其中 GTPase 酶結構域和中間區域是主要的功能域,體外研究表明,位于 GTPase 酶結構域的基因變異會干擾突觸囊泡內吞、降低囊泡內吞的活性以及突觸傳遞,位于中間區域的基因變異會破壞 DNM1 蛋白形成更大的寡聚體,而位于 PH 結構域的基因變異會影響磷酸肌醇的結合和突觸囊泡再攝取[14-16]。
DNM1 基因變異致早發性嬰兒癲癇性腦病 31 型,臨床以早發型難治性癲癇、嚴重智力和語言發育障礙、運動障礙和肌張力低下為特征,綜合分析文獻報道的 39 例 DNM1 病例,肌張力低下與基因變異位點無相關性,提示肌張力低下是 DNM1 相關腦病的共有癥狀。GTPase 酶結構域和中間區域變異病例均有嚴重或顯著的智力、言語發育障礙和運動障礙,本組 3 例病例基因變異位點位于 GTPase 酶結構域,臨床表現均符合上述臨床特點,同時本組病例均不會咀嚼和吞咽固體食物,既往文獻均未提及,提示今后需關注 DNM1 相關腦病合并進食障礙的現象。
此外,文獻報道的基因變異位點位于 PH 結構域的 2 例單卵孿生姐妹,除無癲癇發作外,智力、言語和運動障礙的程度好于基因變異位點位于 GTPase 酶結構域和中間區域的病例,但病情嚴重程度不同的機制尚未明確[5],同時,也需要更多的 PH 結構域變異的病例來證實基因和表型之間的相關性。
35 例 DNM1 變異病例存在癲癇,80% 的病例具有多種癲癇發作形式,對 31 例資料完整的病例進一步統計學研究發現,男孩相對多見,80% 以上的病例在 1 歲前出現癲癇發作,最常見的癲癇發作類型是癲癇性痙攣發作,其次為強直-陣攣和失神發作,其他還包括局灶性、肌陣攣和強直發作等,90% 以上為耐藥性癲癇,本組 3 例病例,癲癇發病年齡、癲癇發作形式和 AEDs 治療反應與文獻報道基本相符,提示對于病因不明的早發型癲癇性腦病,尤其表現為癲癇性痙攣發作和肌張力低下,需通過基因檢測除外 DNM1 基因變異并明確病因。
有文獻報道,在起病初的癲癇發作形式,在 GTPase 酶結構域和中間區域變異之間無明顯差異[3],對 31 病例所有的癲癇發作形式進一步細分后發現,GTPase 酶結構域組失神發作明顯多于中間區域組,有統計學差異,但需更多的病例來證實失神發作和基因型之間的相關性,而在性別、起病年齡、其他癲癇發作形式和藥物治療反應上均無統計學差異,與文獻報道相符[3, 8],提示 GTPase 酶結構域和中間區域變異具有類似的臨床表現,也提示兩組變異具有相同的發病機制。
綜上,肌張力低下、嚴重智力和運動障礙以及早發型癲癇是 DNM1 相關腦病的主要表現,癲癇性痙攣發作是最常見的發作形式,除失神發作外,GTPase 酶結構域和中間區域變異的臨床和癲癇表型無明顯區別,本組病例尚合并進食障礙。