引用本文: 李梅, 常捷, 吳晗晗, 徐靜, 杜霄燁, 崔金剛, 張騰, 陳瑜. 黃芪甲苷干預碘酸鈉誘導的光感受器退行性病變的效應及相關機制. 中華眼底病雜志, 2024, 40(6): 454-462. doi: 10.3760/cma.j.cn511434-20240108-00011 復制
版權信息: ?四川大學華西醫院華西期刊社《中華眼底病雜志》版權所有,未經授權不得轉載、改編
視網膜光感受器細胞是負責啟動視覺的一級神經元,其不可逆死亡是導致年齡相關性黃斑變性(AMD)和視網膜色素變性等光感受器退行性疾病患者視力損害的核心原因。但目前尚無有效干預光感受器細胞死亡的措施[1]。本課題組前期研究發現,傳統明目中藥黃芪的主要活性成分黃芪甲苷(AS-A)具有光感受器細胞保護作用[2]。AS-A能夠顯著抑制光誘導的視網膜氧化應激和光感受器細胞死亡,減輕光感受器細胞DNA損傷、多聚ADP核糖聚合酶活化和核蛋白高遷移率族蛋白B1的釋放。在分子水平,AS-A干預可拮抗光誘導的視網膜全基因表達譜改變,特別是抑制了光損傷條件下視網膜壞死性凋亡和炎癥反應相關基因表達的上調。同時,AS-A對光損傷視網膜小膠質細胞活化具有顯著的抑制作用。此外,AS-A可減輕DNA烷化劑甲磺酸甲酯(MMS)誘導的DNA損傷介導的光感受器細胞退行性改變,抑制MMS誘導的視網膜壞死性凋亡和促炎基因表達上調以及小膠質細胞的活化[2]。碘酸鈉(NaIO3)可選擇性誘導視網膜色素上皮(RPE)氧化損傷,繼而導致光感受器退行性改變的發生。因此,為更全面理解AS-A在干預光感受器退行性病變的藥理學作用,本研究進一步在NaIO3誘導RPE損傷介導的光感受器細胞退行性病變模型中探究其作用。現將結果報道如下。
1 材料和方法
本研究經上海中醫藥大學岳陽中西醫結合醫院動物保護與使用委員會審核通過(動物倫理批號:YYLAC-2019-021-2)。
1.1 實驗動物及主要材料
健康雄性C57BL/6J小鼠60只,6~8周齡,體重(24±2)g,無特定病原體級,購自上海斯萊克實驗動物有限公司。飼養于光照12 h/12 h晝夜明暗交替,溫度(20±2)℃,濕度35%~55%條件下,自由進食和飲水。
AS-A(B20564,上海源葉生物科技有限公司);NaIO3(71702,美國Sigma公司);緊密連接蛋白(ZO-1)抗體(ab221547,美國Abcam公司);離子鈣接頭蛋白1(Iba1)抗體(019-19741,日本FUJIFILM Wako Pure Chemical公司);Müller細胞特異性標志物神經膠質纖維酸性蛋白(GFAP)抗體(Z0334,美國DAKO公司);綿羊抗兔Cy3二抗(C2306,美國Sigma公司);原位末端標記(TUNEL)試劑盒(美國Promega公司);TRIzol(美國Invitrogen公司);PrimeScript RT Master Mix(日本TaKaRa公司);LightCycler 480 SYBR Green I Master(德國Roche公司);光相干斷層掃描(OCT)儀(美國Phoenix Research Labs公司);光學顯微鏡、熒光顯微鏡(德國Leica公司);實時熒光定量聚合酶鏈反應(qPCR)系統(德國Roche公司)。
1.2 方法
分組、建模及給藥。采用隨機數字表法將小鼠分為正常對照組(NC組)、NaIO3模型組(NaIO3組)、AS-A組,每組20只。AS-A、NaIO3分別溶于30%二甲基亞砜(DMSO)溶液、生理鹽水中。建模前30 min,AS-A組小鼠參照課題組前期研究結果按100 mg/kg的劑量腹腔注射100 μl AS-A;NC組、NaIO3組小鼠腹腔注射等體積30% DMSO作為溶劑對照。30 min后,NaIO3組、AS-A組小鼠按30 mg/kg的劑量腹腔注射100 μl NaIO3溶液;NC組給予生理鹽水作為溶劑對照。其后,AS-A組小鼠每天早晚間隔12 h給藥2次,直到實驗結束。
眼底彩色照相、OCT檢查。建模后4 d,各組小鼠腹腔注射鹽酸氯胺酮(82.5 mg/kg)和塞拉嗪(8.25 mg/kg)混合溶液麻醉,1%托吡卡胺散瞳,行眼底彩色照相和OCT檢查,采集眼底圖像和視網膜橫截面圖像并測量顳側、鼻側視網膜外核層(ONL)厚度。
蘇木精-伊紅(HE)染色觀察各組小鼠視網膜形態結構。建模后4 d,小鼠過量麻醉處死,摘除眼球,置于4%多聚甲醛中固定24 h,常規脫水、浸蠟后包埋,按4 μm厚度切片。制備好的石蠟切片脫蠟,水化后,浸于HE染液中染色,重新脫水,透明,中性樹脂封片。光學顯微鏡下觀察拍照。
ZO-1染色觀察RPE細胞結構。建模后1 d,小鼠過量麻醉處死,摘除眼球并去除前房、晶狀體、神經視網膜,100%甲醇室溫固定30 min,含10%綿羊血清抗原封閉液室溫孵育30 min,ZO-1抗體(1∶500)4℃孵育過夜,綿羊抗兔Cy3二抗(1:1 000)37 ℃孵育30 min,鋪片,甘油明膠封片。
免疫組織化學染色觀察視網膜中Iba1、GFAP表達;TUNEL法檢測光感受器細胞死亡情況。建模后3 d,小鼠過量麻醉處死,摘除眼球,常規制作石蠟切片。石蠟切片檸檬酸鈉溶液加熱修復抗原30 min,封閉非特異性抗原,一抗GFAP(1:500)4 ℃孵育過夜,綿羊抗兔Cy3二抗(1:1 000)37 ℃孵育30 min;眼球去除角膜、晶狀體制成視杯,4%多聚甲醛中固定2 h,蔗糖梯度脫水,OCT包埋劑包埋,按12 μm厚度制備冰凍切片,一抗Iba1(1:200)和綿羊抗兔Cy3二抗(1:1 000)染色,4′, 6-二氨基-2-苯基吲哚(DAPI)復染,甘油明膠封片。石蠟切片按說明書進行原位TUNEL染色。
qPCR檢測各組小鼠視網膜趨化因子配體2(Ccl2)、白細胞介素-1β(IL-1β)、混合譜系激酶結構域樣蛋白(Mlkl)、受體相互作用蛋白激酶3(Ripk3)、腫瘤壞死因子(Tnf)等基因的mRNA相對表達量。建模后1 d,過量麻醉處死小鼠,摘除眼球剝離視網膜,Trizol提取總RNA,反轉錄試劑逆轉錄成cDNA,按相應基因配置SYBR Green qPCR擴增反應體系進行qPCR檢測。應用Express 3.0軟件設計引物序列(表1)。以18S rRNA作為內參照,根據2?[Ct(目標)?Ct(18S rRNA)]計算基因表達的倍數變化。
1.3 統計學方法
采用GraphPad Prism 10軟件行統計學分析。經正態性檢驗和方差齊性檢驗后,符合正態分布的計量資料以均數±標準差(x±s)表示。兩組間比較采用獨立樣本t檢驗;多組間比較采用單因素方差分析;事后多重比較采用Tukey檢驗。P<0.05為差異有統計學意義。
2 結果
2.1 AS-A抑制NaIO3誘導的視網膜結構損傷
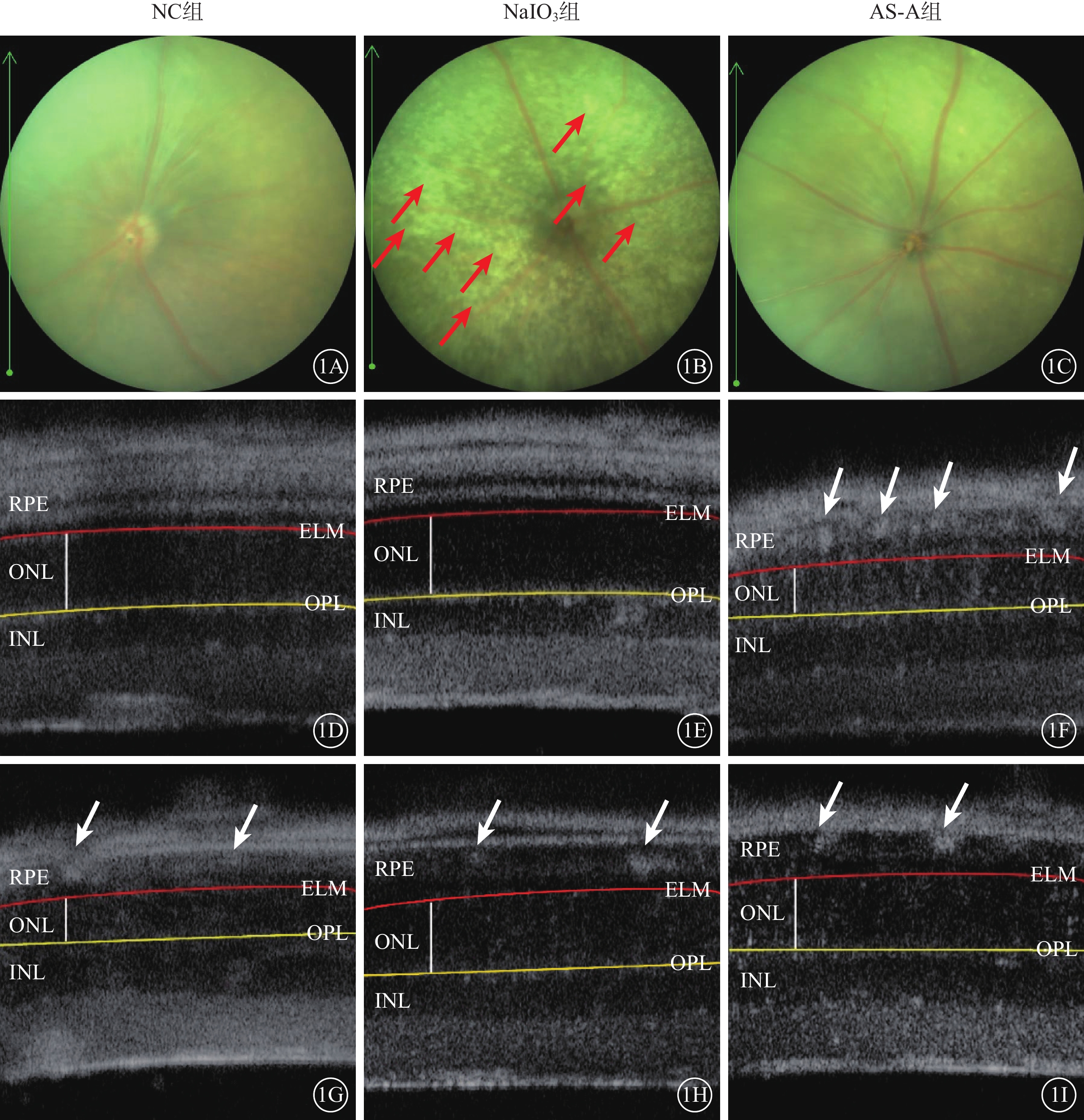
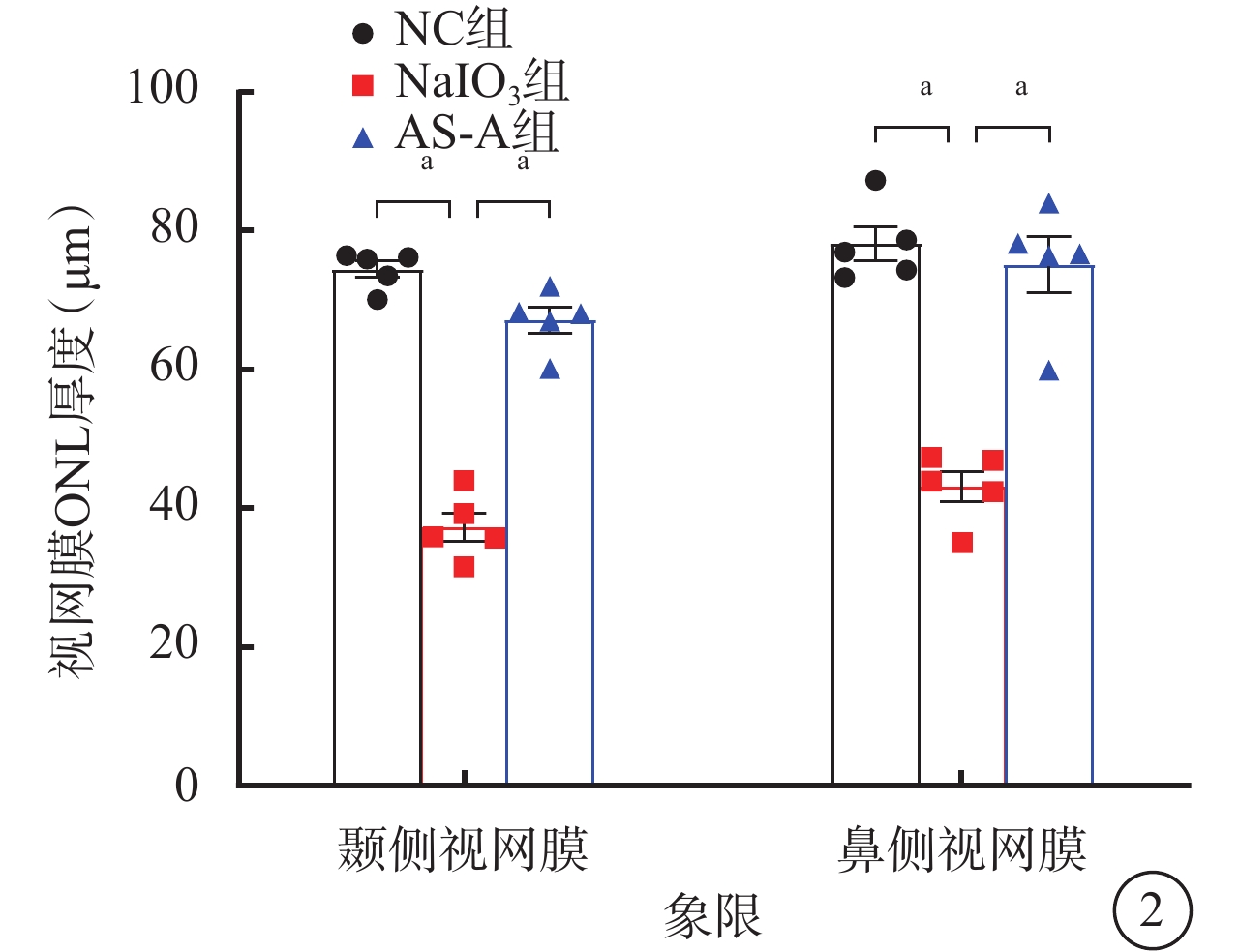
建模后4 d,NC組、AS-A組小鼠眼底未見明顯異常;NaIO3組小鼠眼底可見大量散在黃白色玻璃膜疣樣結構(圖1A~1C)。與NC組比較,NaIO3組小鼠顳側和鼻側RPE層出現大量強反射區;與NaIO3組比較,AS-A組小鼠顳側和鼻側RPE層中強反射區數量減少(圖1D~1I)。與NC組、AS-A組比較,NaIO3組小鼠視網膜顳側、鼻側ONL厚度均顯著變薄,差異有統計學意義(F=123.00、41.37,P<0.001)(圖2)。
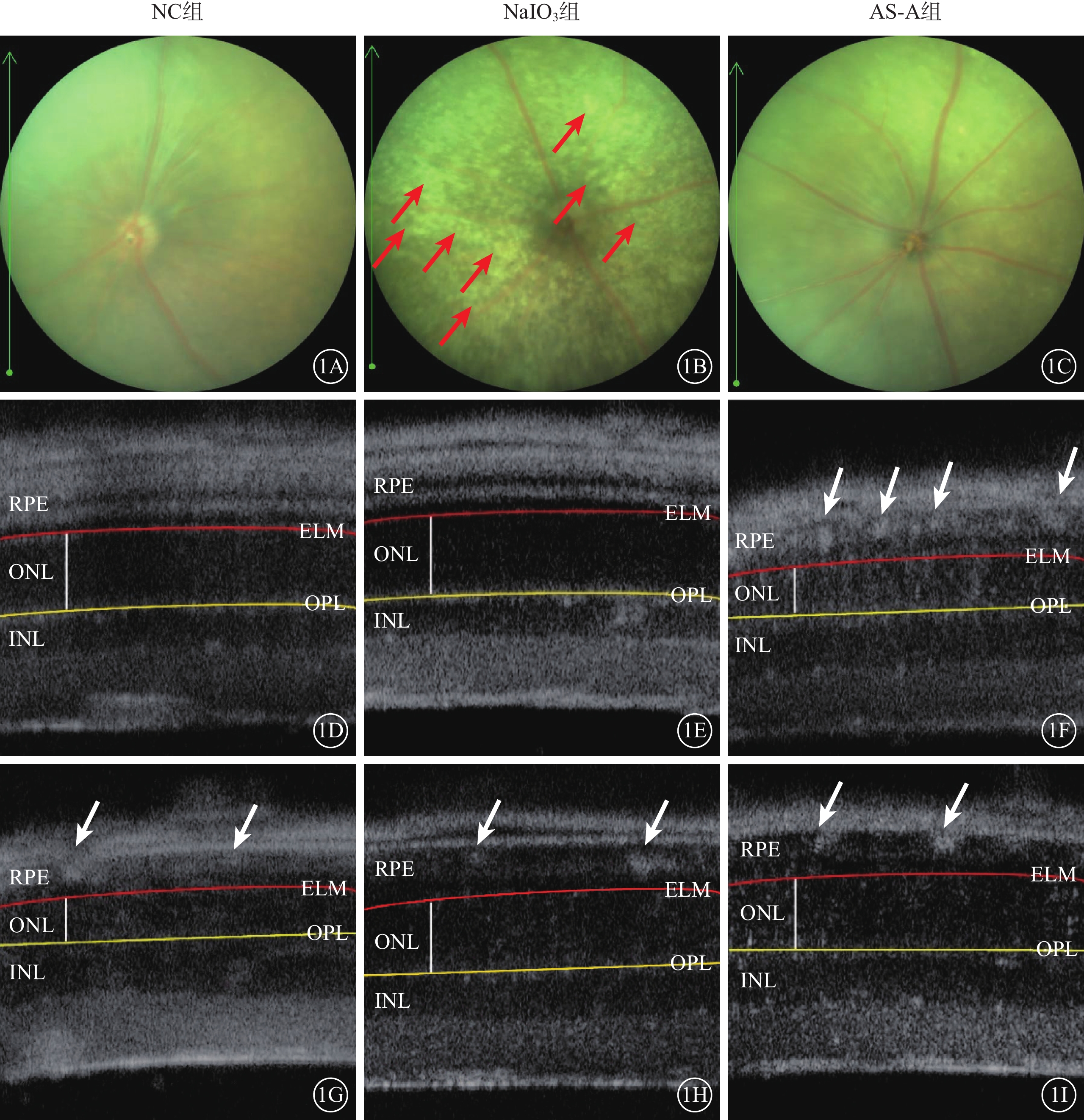 圖1
AS-A抑制NaIO3誘導的小鼠視網膜結構損傷
圖1
AS-A抑制NaIO3誘導的小鼠視網膜結構損傷
1A~1C分別示NC組、NaIO3組、AS-A組小鼠彩色眼底像,建模后4 d,與NC組、AS-A組比較,NaIO3組小鼠眼底可見大量散在黃白色玻璃膜疣樣結構(紅箭)。1D、1E分別示NC組小鼠顳側、鼻側視網膜OCT像;1F、1G示NaIO3組小鼠顳側、鼻側視網膜OCT像;1H、1I示AS-A組小鼠顳側、鼻側視網膜OCT像。與NC組比較,NaIO3組小鼠顳側和鼻側RPE層出現大量強反射區(白箭);NaIO3組、AS-A組 與NaIO3組比較,AS-A組小鼠顳側和鼻側RPE層中強反射區數量減少(白箭) NC:正常對照;NaIO3:碘酸鈉;AS-A:黃芪甲苷;OCT:光相干斷層掃描;RPE:視網膜色素上皮;ELM:外界膜(紅線所在處);INL:內核層(黃線以下弱反射區);ONL:外核層(紅、黃線之間白線所示弱反射區);OPL:外叢狀層(黃線所在處)
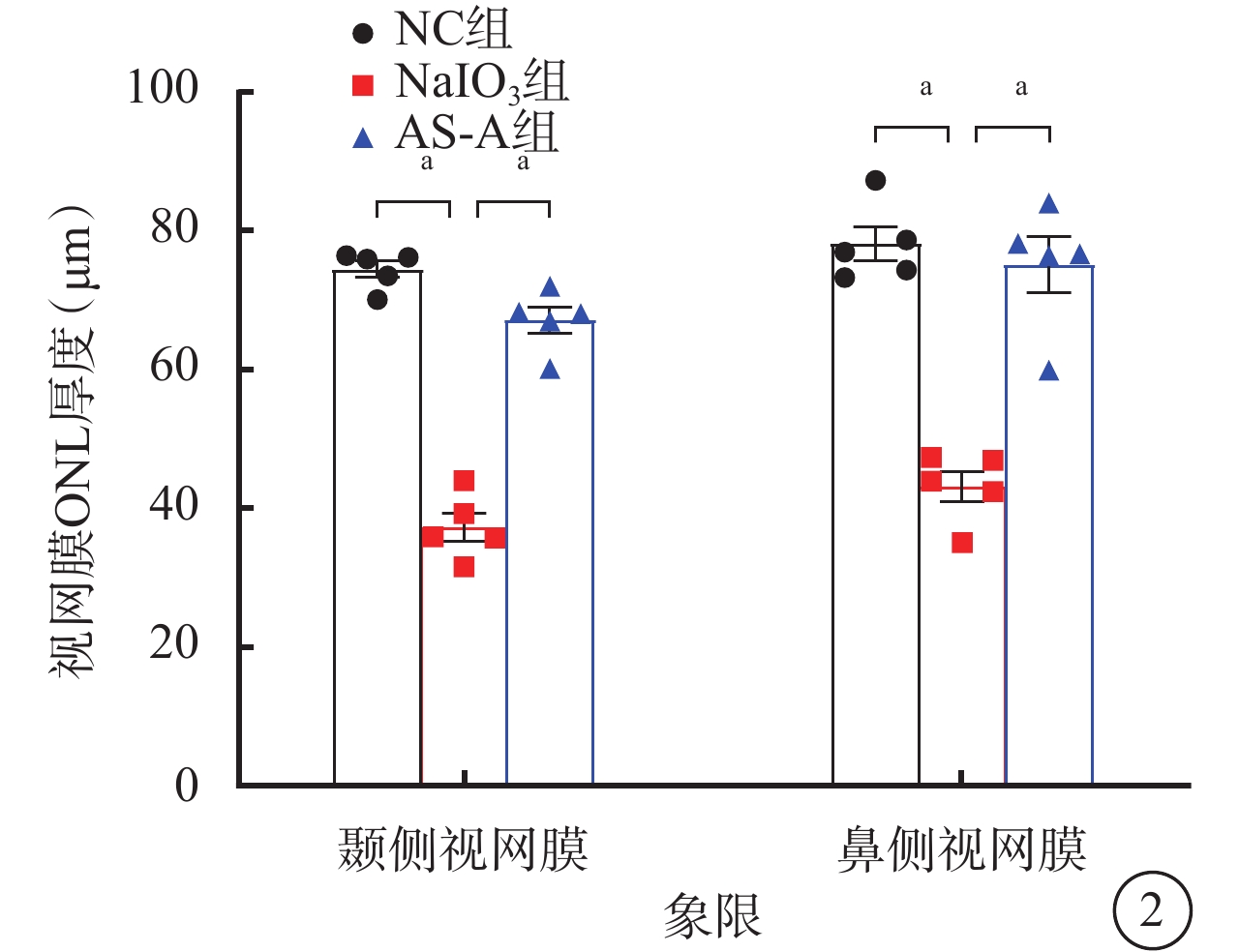 圖2
各組小鼠視網膜外核層厚度比較(n=5),aP<0.001
圖2
各組小鼠視網膜外核層厚度比較(n=5),aP<0.001
NC:正常對照;NaIO3:碘酸鈉;AS-A:黃芪甲苷;ONL:外核層
2.2 AS-A保護RPE細胞形態結構
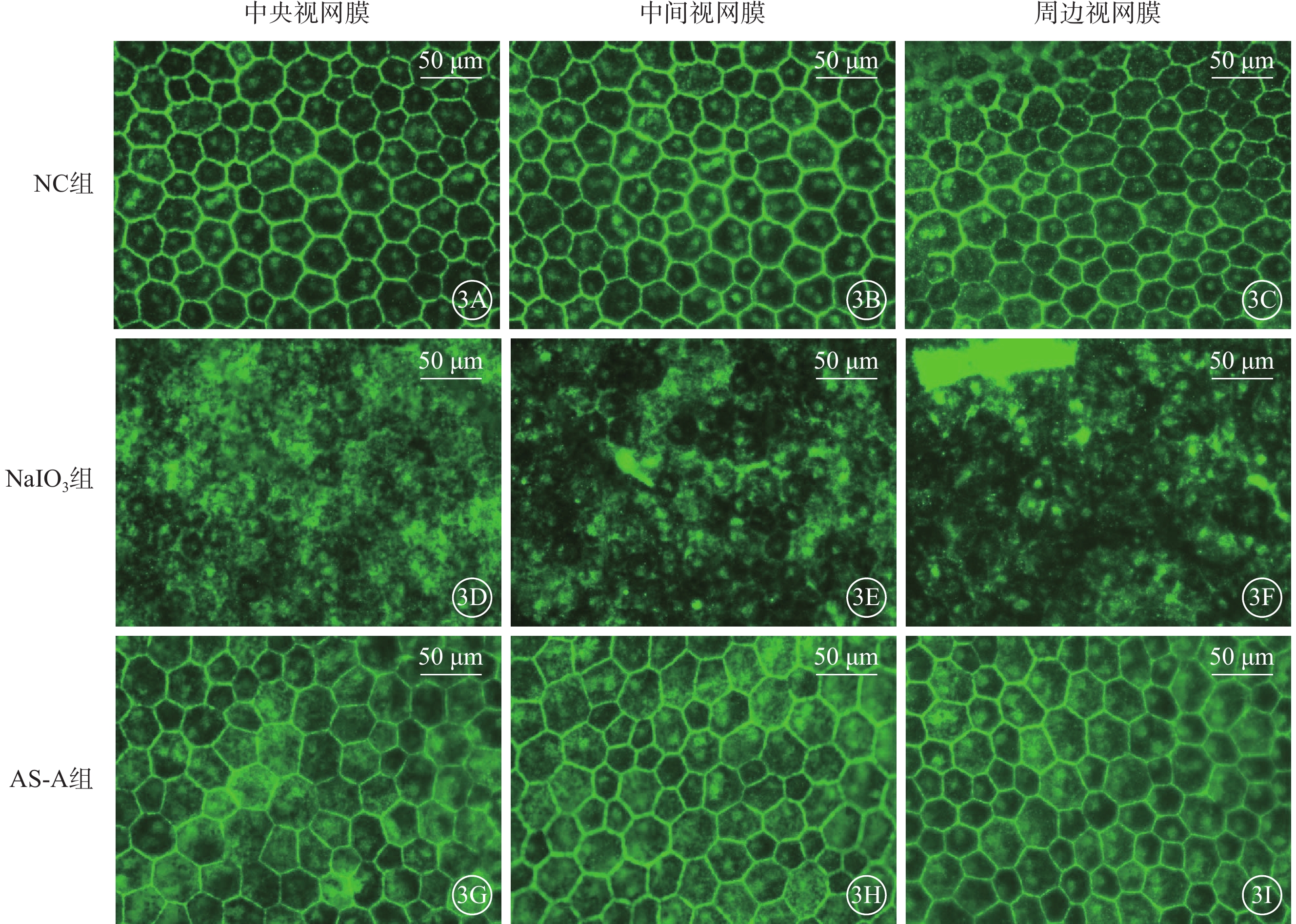
建模后1 d,NC組、AS-A組小鼠RPE細胞呈六邊形結構,ZO-1均勻分布于其膜上。NaIO3組小鼠視網膜中央、中間、周邊RPE細胞膜上ZO-1大量丟失,RPE細胞明顯萎縮(圖3)。
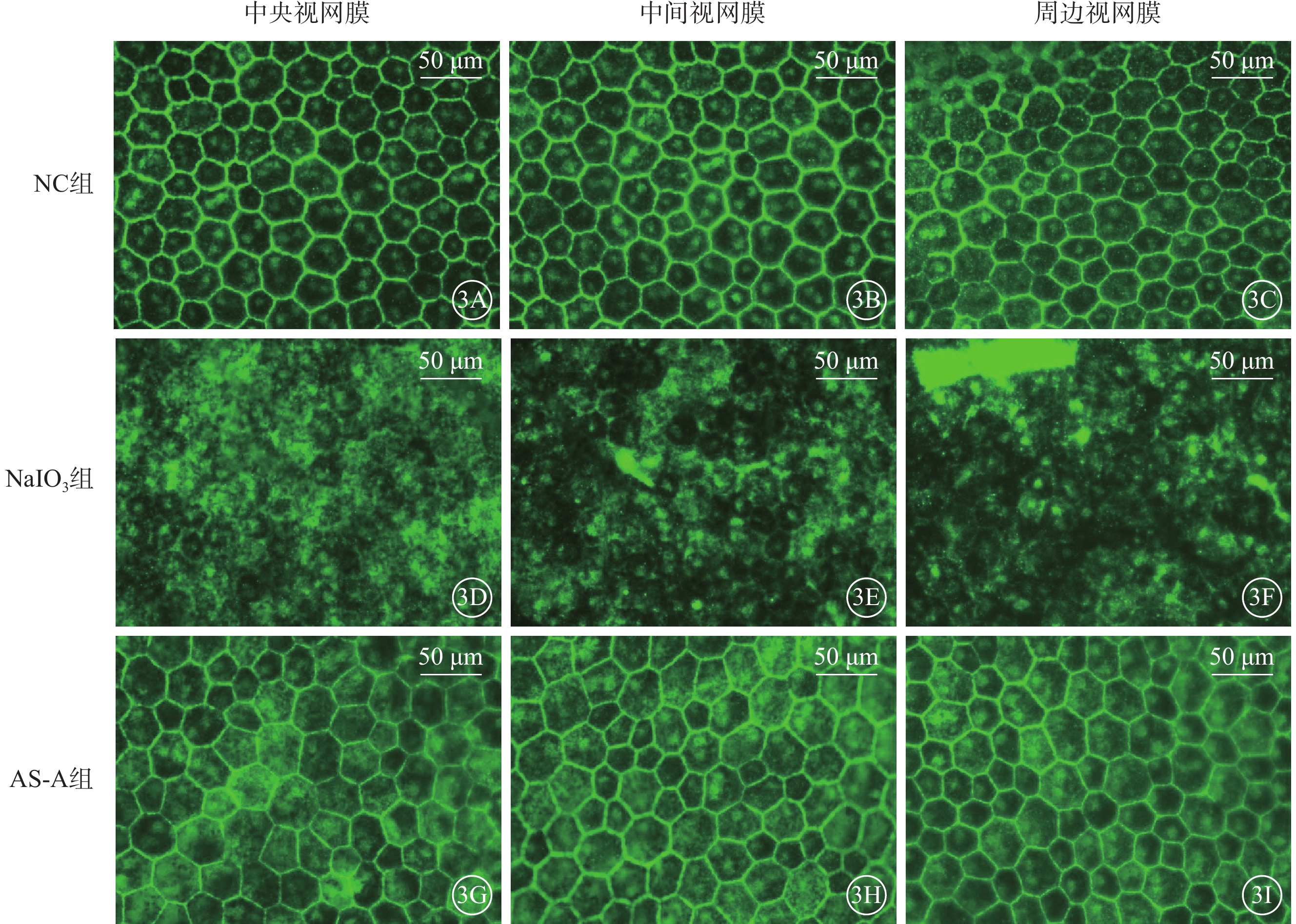 圖3
AS-A抑制NaIO3誘導RPE細胞結構損傷
圖3
AS-A抑制NaIO3誘導RPE細胞結構損傷
3A~3C、3D~3F、3G~3I分別示NC組、NaIO3組、AS-A組小鼠視網膜中央、中間、周邊RPE細胞熒光顯微鏡像(標尺:50 μm),ZO-1呈綠色熒光表達。NC組(3A~3C)、AS-A組(3G~3I),RPE細胞呈六邊形結構,ZO-1均勻分布于RPE細胞膜上;NaIO3組建模后1 d,小鼠視網膜中央、中間、周邊ZO-1大量丟失,RPE細胞明顯萎縮(3D~3F) NC:正常對照;NaIO3:碘酸鈉;AS-A:黃芪甲苷;RPE:視網膜色素上皮;ZO-1:緊密連接蛋白-1
2.3 AS-A抑制NaIO3誘導光感受器細胞丟失
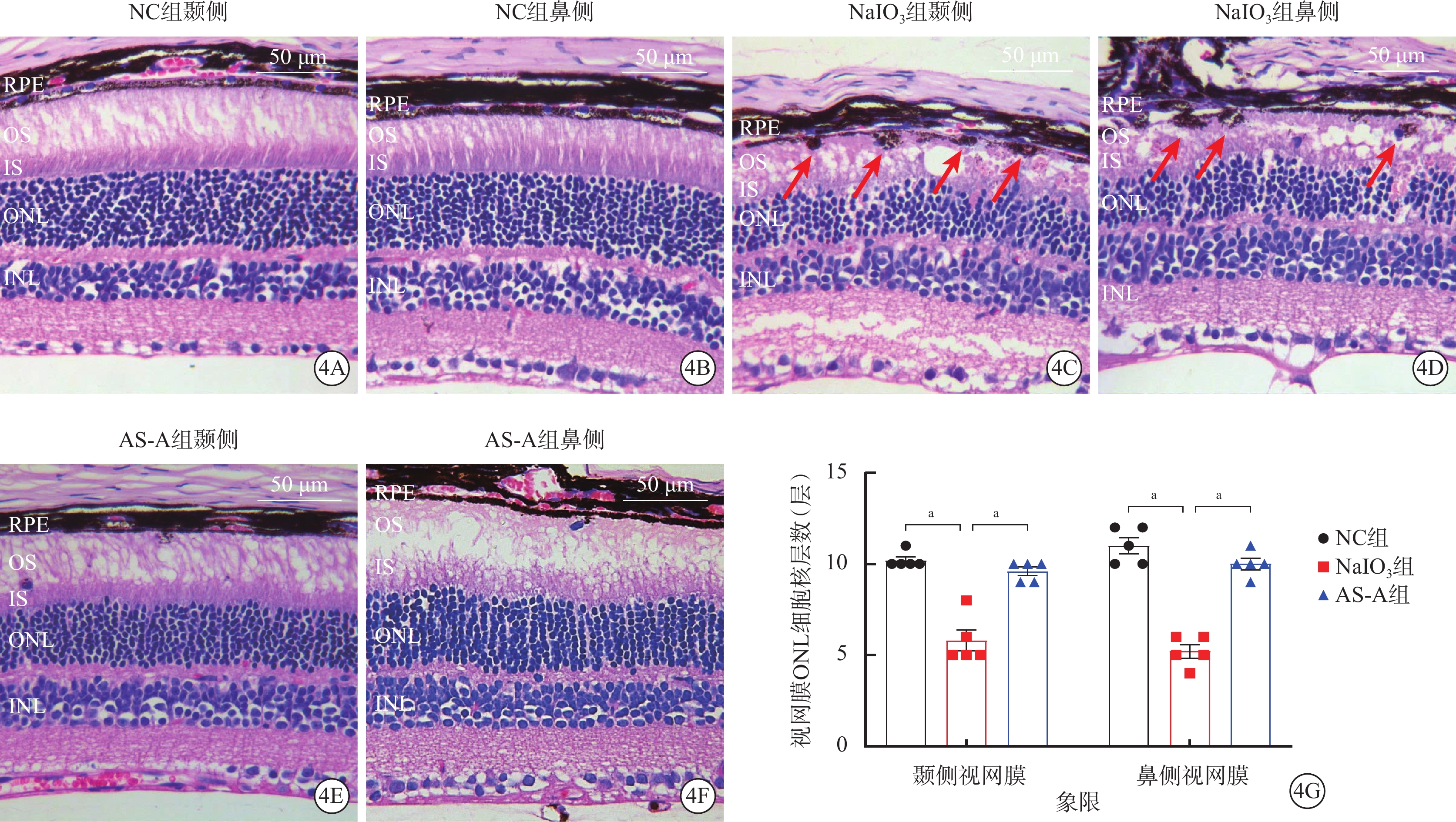
NC組小鼠視網膜各層形態結構完整(圖4A,4B);AS-A組小鼠RPE層色素整齊,光感受器內外節結構完整,ONL細胞核層數顯著增加(圖4C,4D);NaIO3組小鼠RPE層可見圓形黑色沉積物,光感受器內外節結構嚴重受損,ONL細胞核層數顯著減少(圖4E,4F)。與NC組、AS-A組比較,NaIO3組小鼠視網膜顳側、鼻側ONL細胞核層數顯著降低,差異有統計學意義(F=38.82、65.55,P<0.001)(圖4G)。
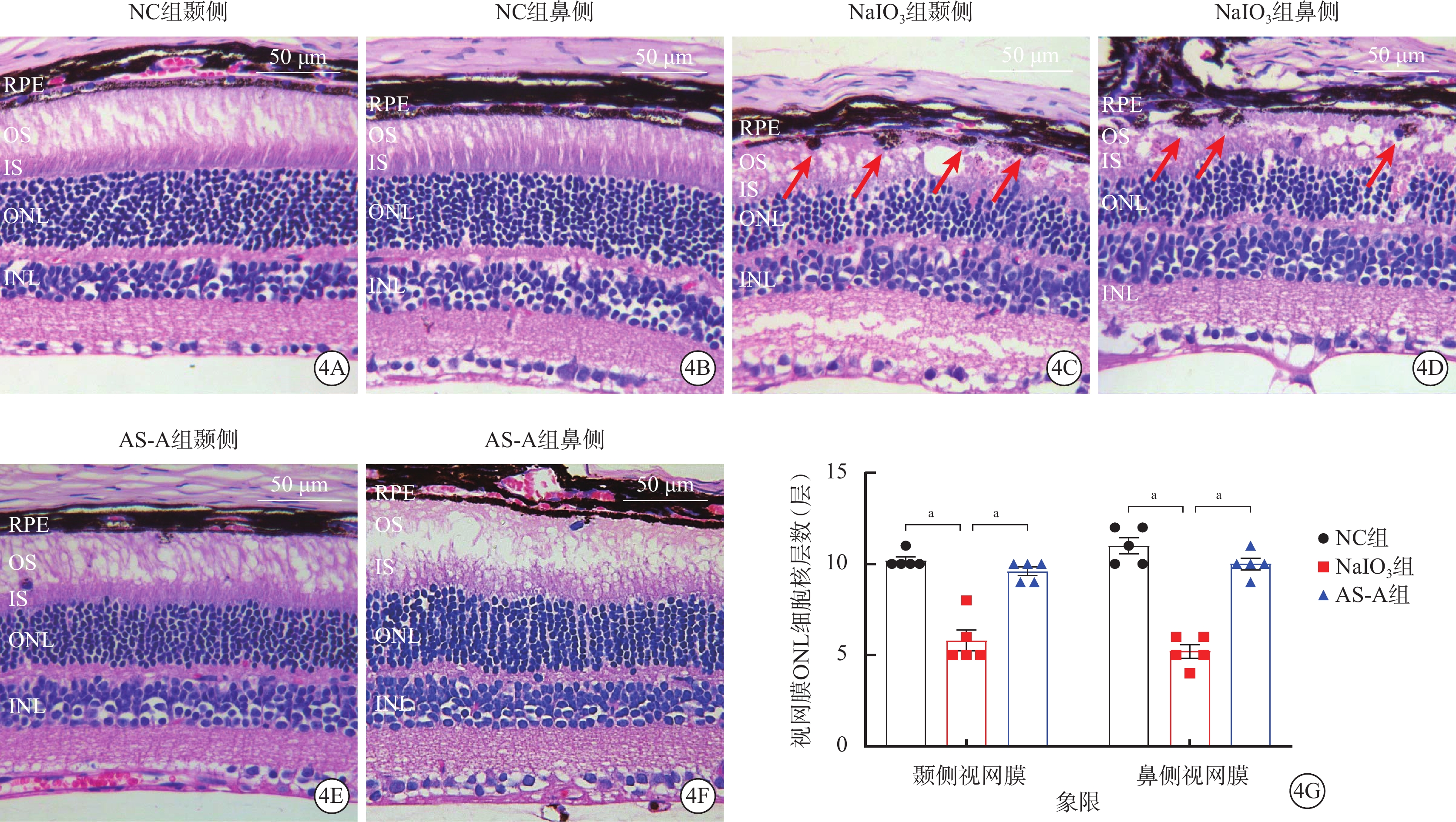 圖4
AS-A抑制NaIO3誘導的光感受器細胞丟失
圖4
AS-A抑制NaIO3誘導的光感受器細胞丟失
4A、4B,4C、4D、4E、4F分別示NC組、NaIO3組、AS-A組建模后4 d小鼠視網膜光學顯微鏡像(蘇木精-伊紅染色,標尺:50 μm),NC組小鼠視網膜各層形態結構完整;NaIO3組RPE層可見圓形黑色沉積物(紅箭),IS、OS結構嚴重受損,ONL細胞核層數顯著減少;AS-A組小鼠RPE層色素整齊,IS、OS結構完整,ONL細胞核層數顯著增加。4G示NC組、AS-A組、NaIO3組小鼠視網膜ONL細胞核層數比較(
2.4 AS-A抑制NaIO3誘導的光感受器細胞死亡
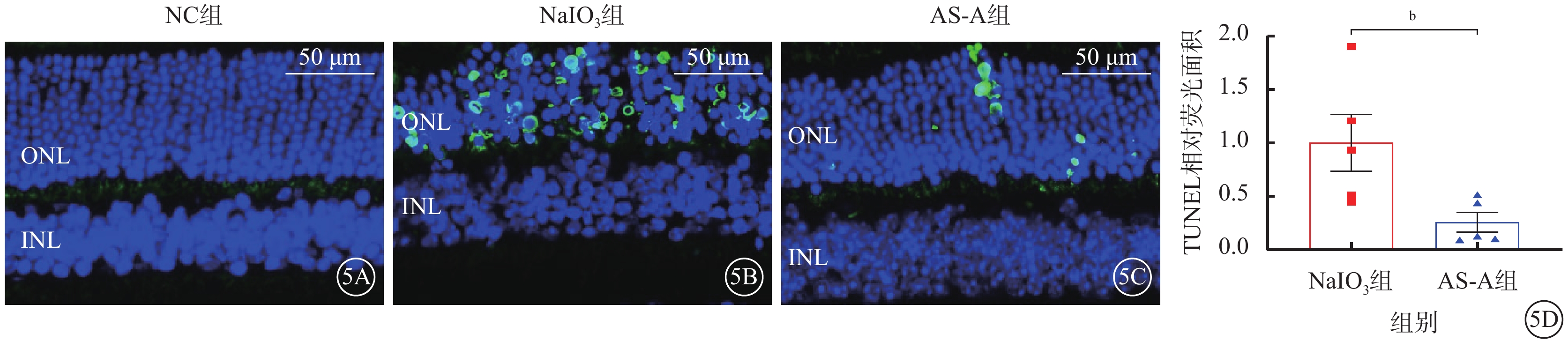
建模后3 d,與NC組比較,NaIO3組小鼠視網膜ONL可見大量TUNEL陽性細胞核;與NaIO3組比較,AS-A組小鼠視網膜ONL中TUNEL陽性細胞核數量顯著減少,差異有統計學意義(t=2.657,P<0.05)(圖5)。
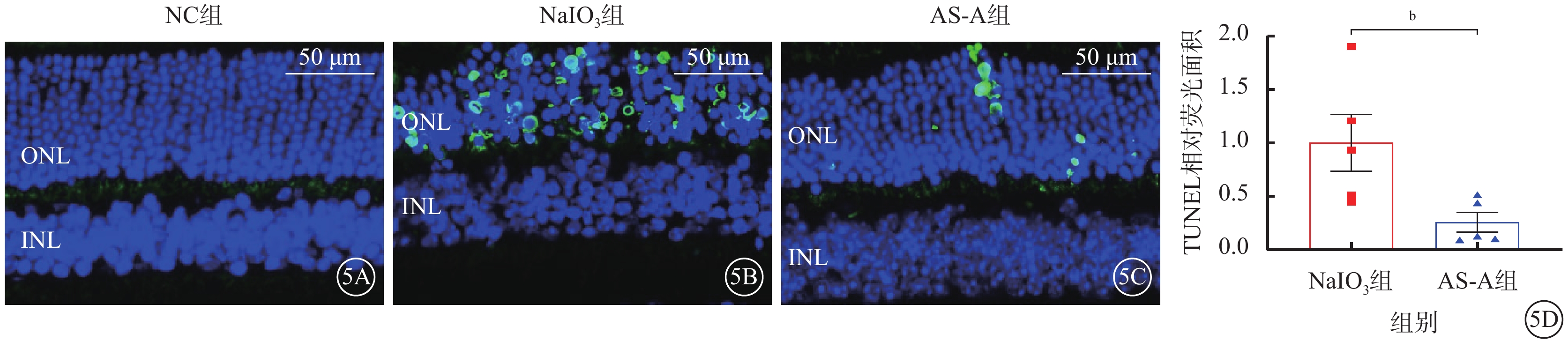 圖5
AS-A抑制NaIO3誘導的光感受器細胞死亡
圖5
AS-A抑制NaIO3誘導的光感受器細胞死亡
5A~5C分別示NC組、NaIO3組、AS-A組熒光顯微鏡像(標尺:50 μm),DAPI標記的細胞核呈藍色熒光;TUNEL染色的光感受器死亡細胞呈綠色熒光。5D示NaIO3組、AS-A組陽性細胞核相對熒光面積比較(
2.5 AS-A抑制NaIO3誘導的視網膜壞死性凋亡和炎癥相關基因的表達上調
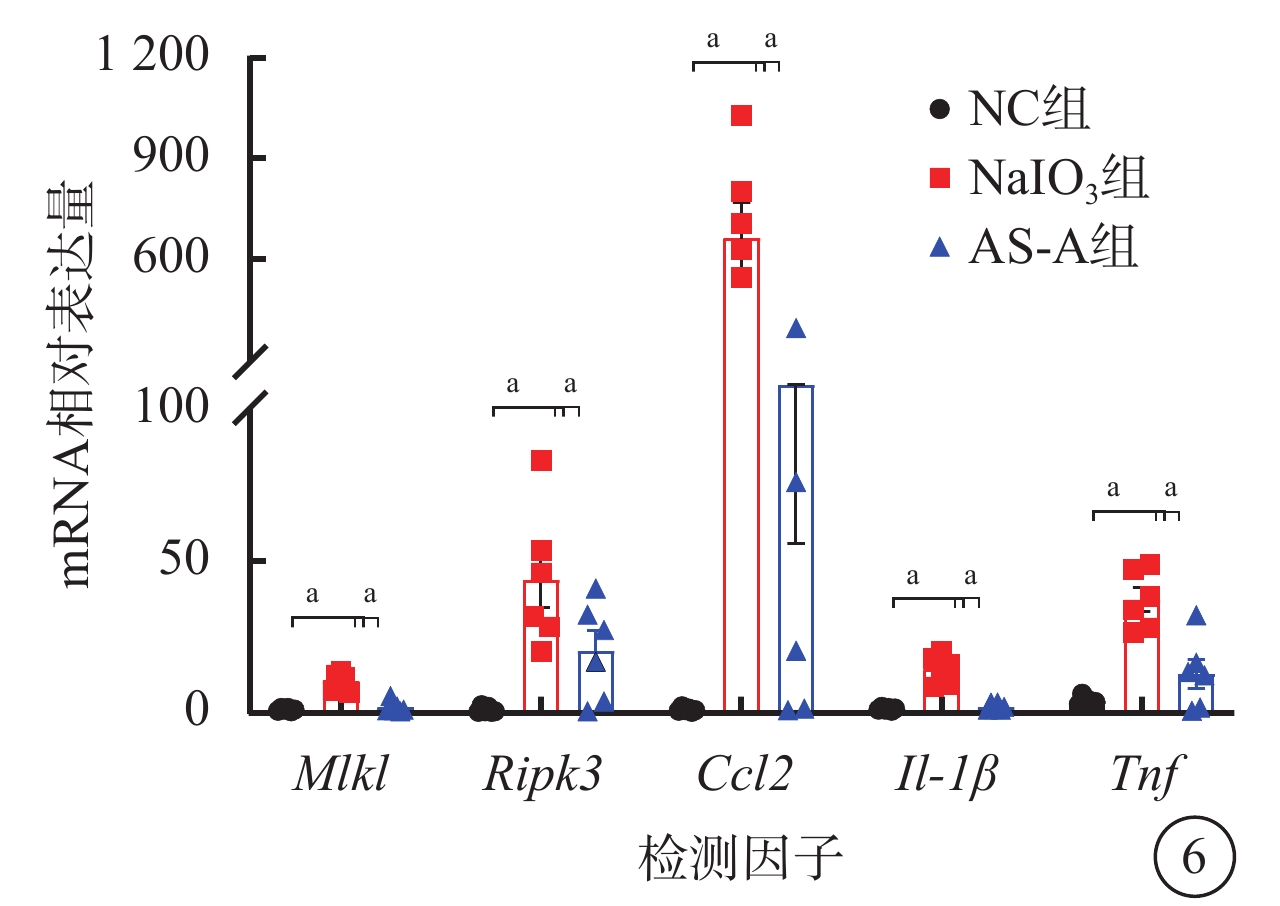
建模后1 d,與NC組、AS-A組比較,NaIO3組視網膜中Mlkl、Ripk3、Ccl2、Il-1β、Tnf mRNA相對表達量顯著升高,差異有統計學意義(F=39.18、10.66、53.51、41.40、24.13,P<0.001)(圖6)。
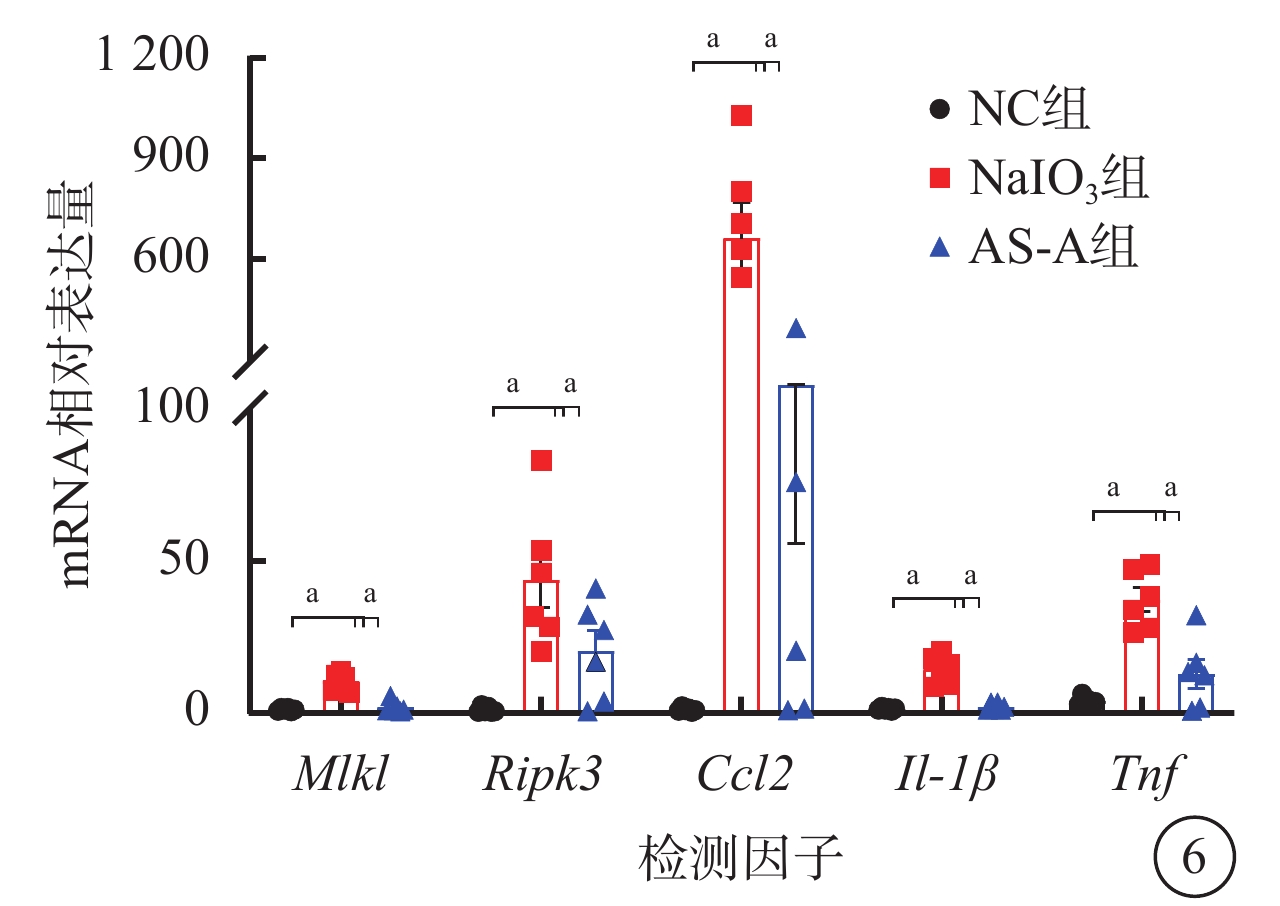 圖6
NC組、NaIO3組、AS-A組小鼠視網膜中Mlkl、Ripk3、Ccl2、Il-1β、Tnf mRNA相對表達量比較(n=5),aP<0.001
圖6
NC組、NaIO3組、AS-A組小鼠視網膜中Mlkl、Ripk3、Ccl2、Il-1β、Tnf mRNA相對表達量比較(n=5),aP<0.001
NC:正常對照;NaIO3:碘酸鈉;AS-A:黃芪甲苷;
2.6 AS-A抑制NaIO3誘導的小膠質細胞激活
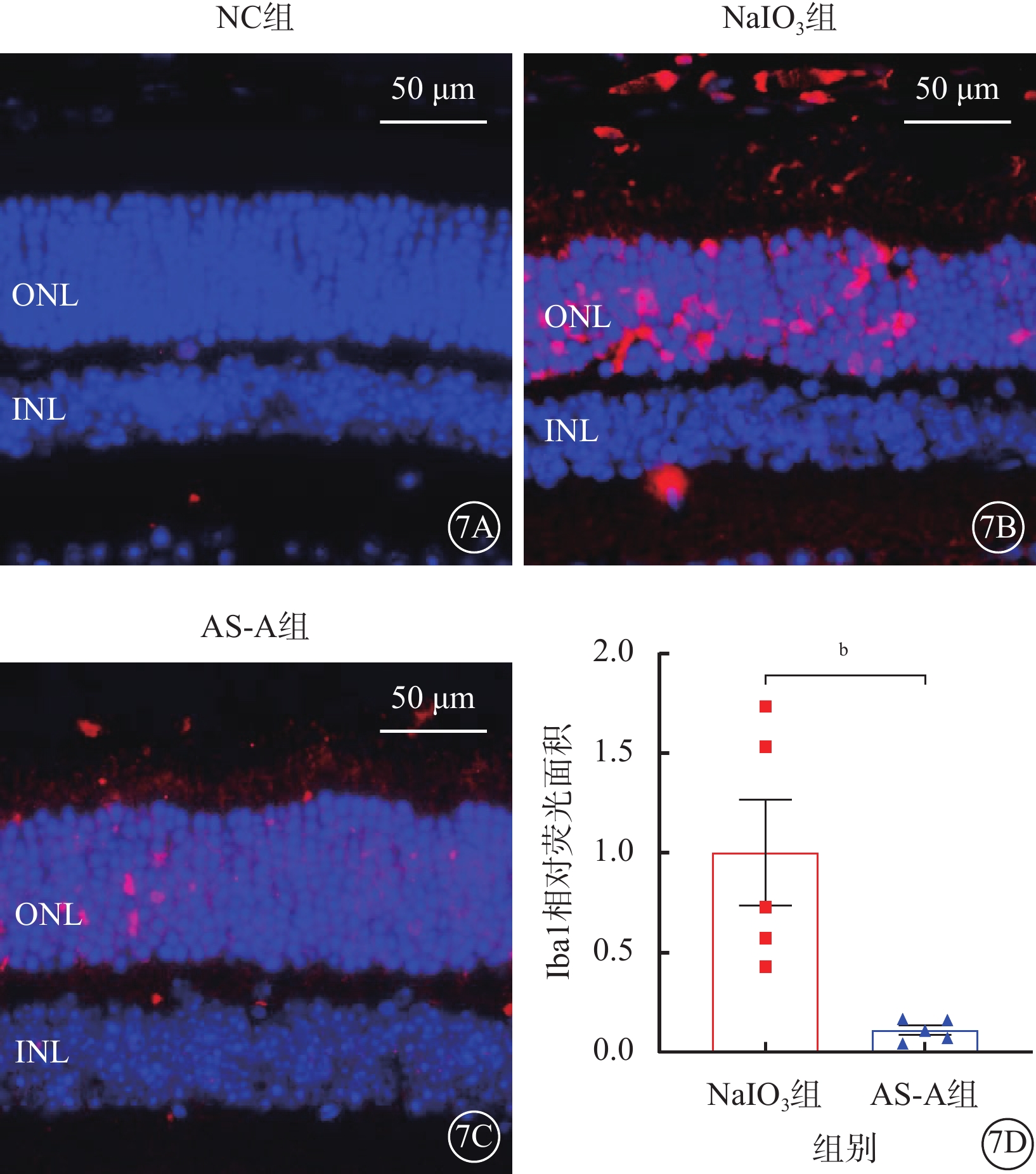
建模后3 d,NC組小鼠僅內層視網膜可見少量Iba1陽性表達;NaIO3組小鼠ONL和視網膜下間隙處見大量Iba1陽性表達;AS-A組小鼠ONL和視網膜下間隙處可見少量Iba1陽性表達(圖7A~7C)。與NaIO3組比較,AS-A組小鼠ONL和視網膜下間隙處Iba1陽性表達顯著降低,差異有統計學意義(t=3.34,P<0.05)(圖7D)。
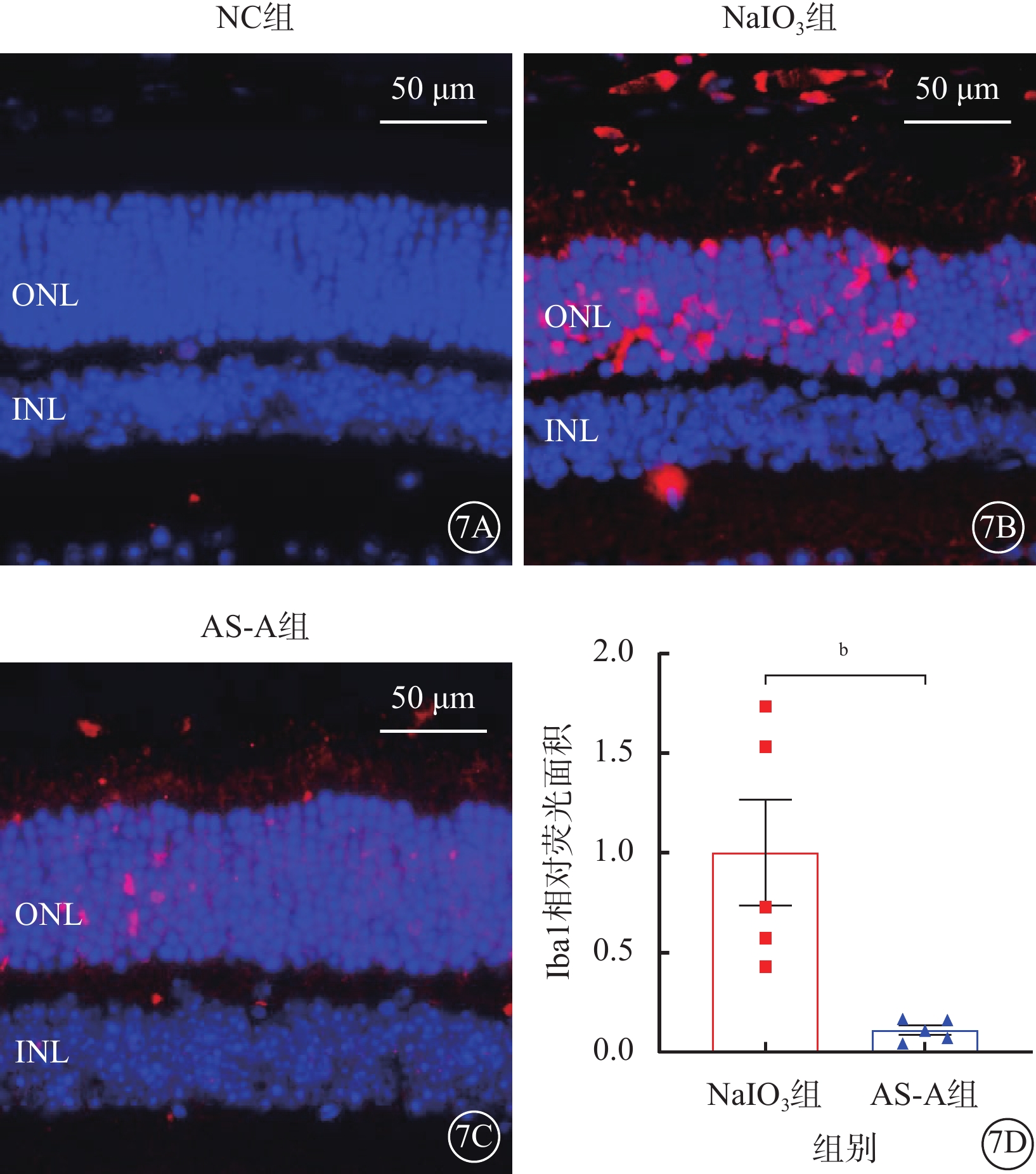 圖7
AS-A抑制NaIO3誘導的小膠質細胞激活
圖7
AS-A抑制NaIO3誘導的小膠質細胞激活
7A~7C分別示NC組、NaIO3組、AS-A組小鼠視網膜免疫熒光顯微鏡像(標尺:50 μm),DAPI標記細胞核呈藍色熒光,Iba1標記的陽性細胞呈紅色熒光;7D示NaIO3組、AS-A組Iba1相對熒光面積比較(
2.7 AS-A抑制NaIO3誘導的Müller細胞反應性膠質增生
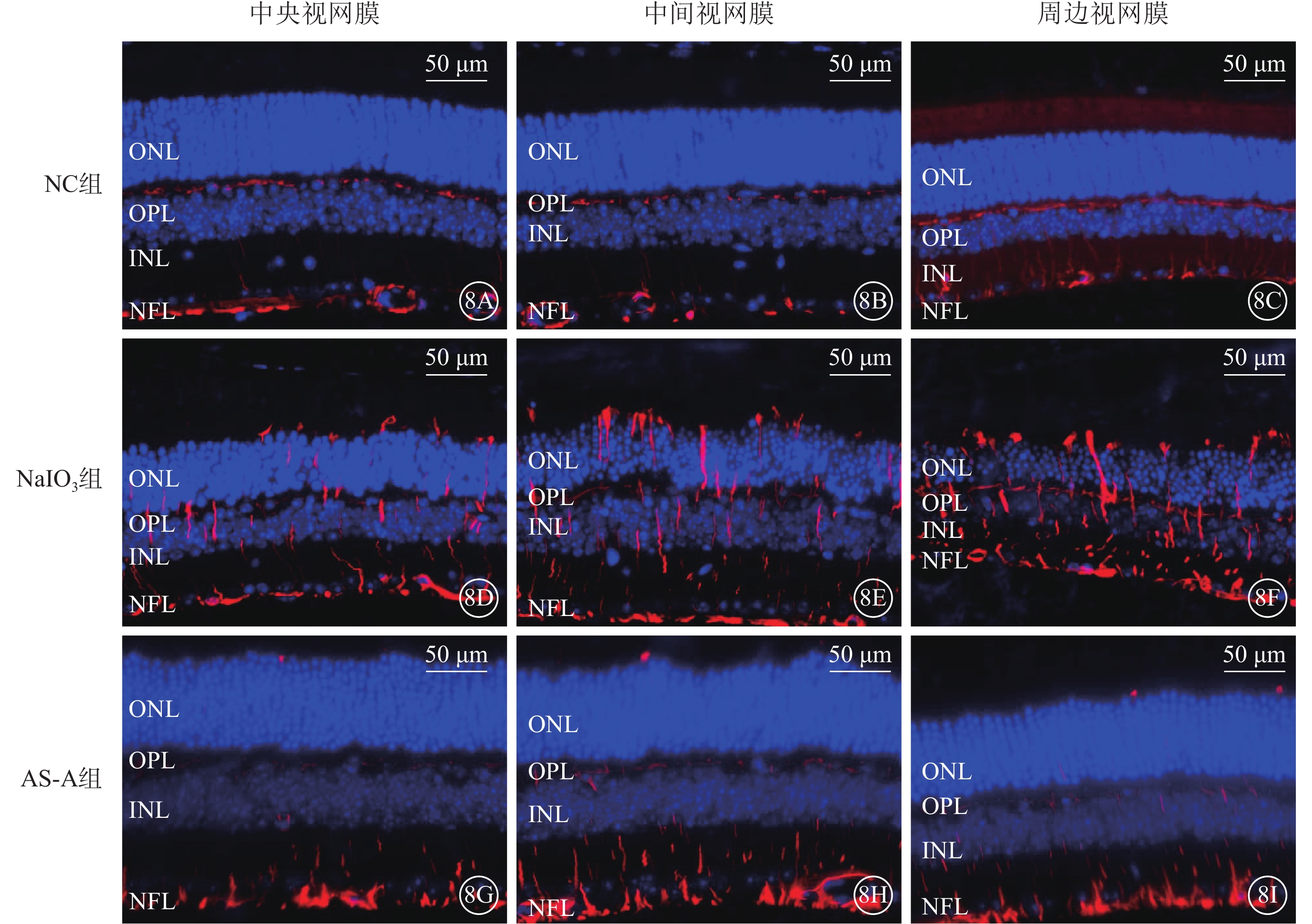
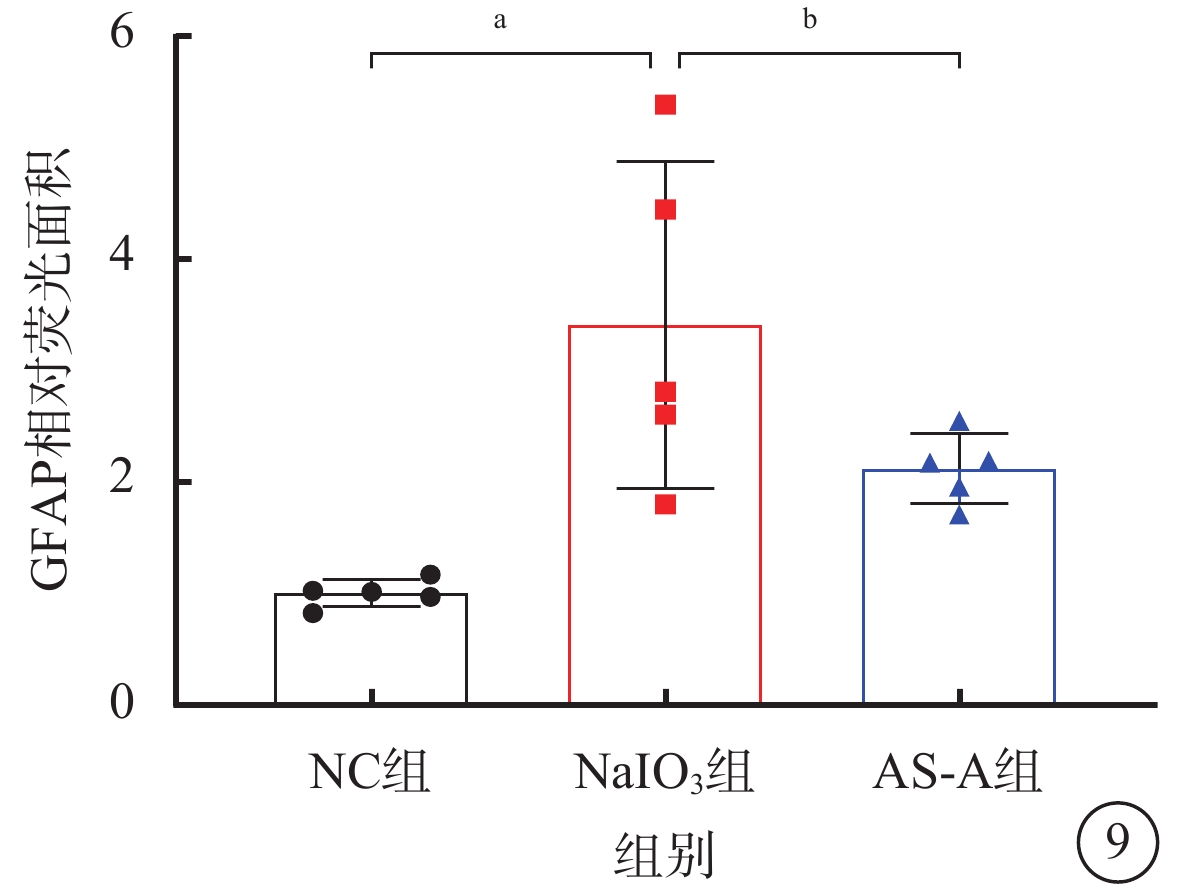
建模后3 d,NC組小鼠視網膜外從狀層、神經纖維層可見少量GFAP陽性表達(圖8A~8C);NaIO3組小鼠中央、中間、周邊視網膜GFAP陽性表達顯著增加(圖8D~8F);AS-A組小鼠視網膜GFAP陽性表達顯著減少,僅局限于外從狀層和神經纖維層(圖8G~8I)。與NC組、AS-A組比較,NaIO3組小鼠視網膜GFAP陽性表達顯著升高,差異有統計學意義(F=9.62,P<0.05)(圖9)。
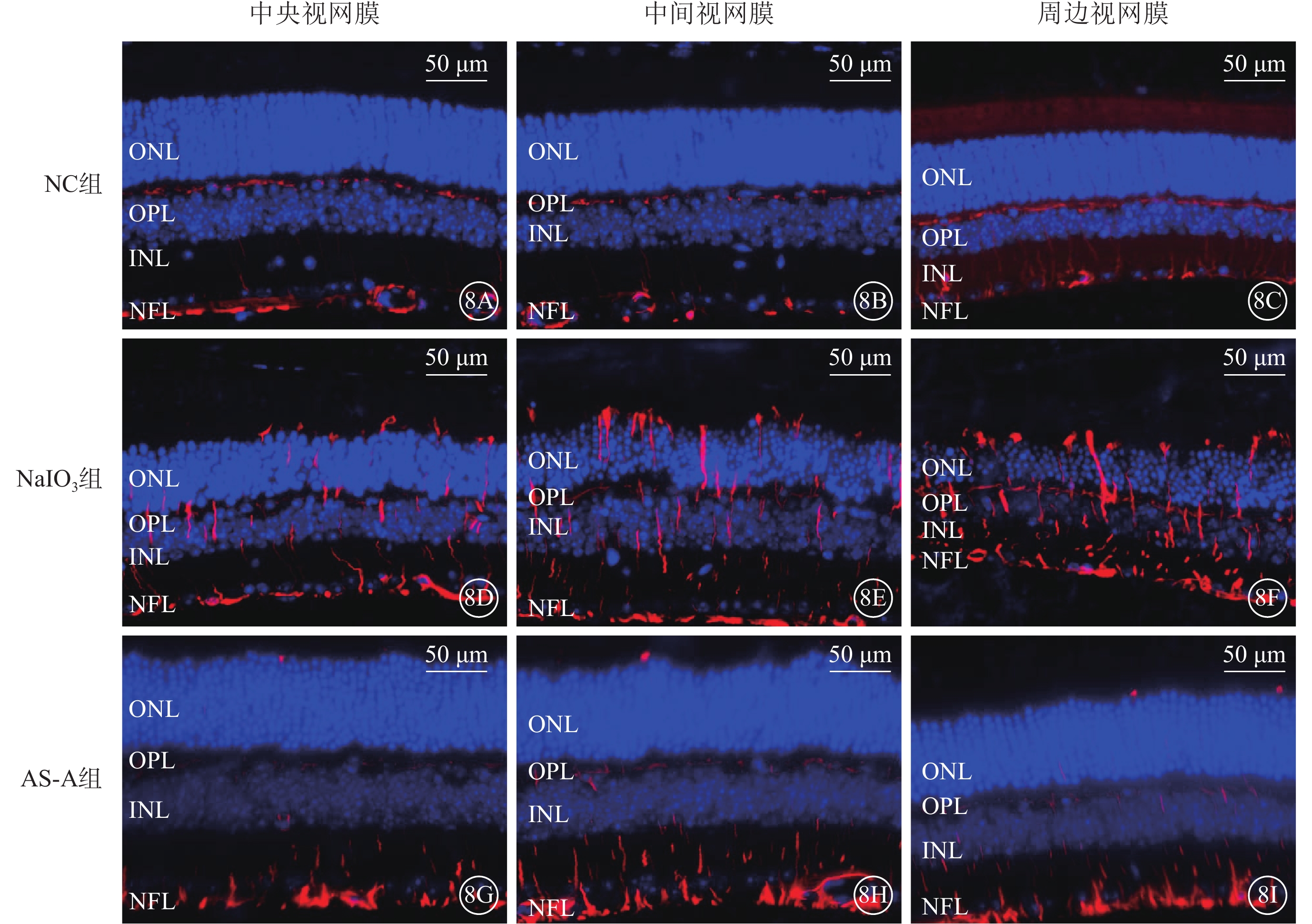 圖8
AS-A抑制NaIO3誘導Müller細胞反應性膠質增生
圖8
AS-A抑制NaIO3誘導Müller細胞反應性膠質增生
8A~8C、8D~8F、8G~8I分別示NC組、NaIO3組、AS-A組小鼠視網膜中央、中間、周邊熒光顯微鏡像(標尺:50 μm),DAPI標記的細胞核呈藍色熒光,GFAP標記的陽性細胞呈紅色熒光 NC:正常對照;NaIO3:碘酸鈉;AS-A:黃芪甲苷;INL:內核層;ONL:外核層;OPL:外從狀層;NFL:神經纖維層;DAPI:4',6-二脒基-2-苯基吲哚;GFAP:神經膠質纖維酸性蛋白
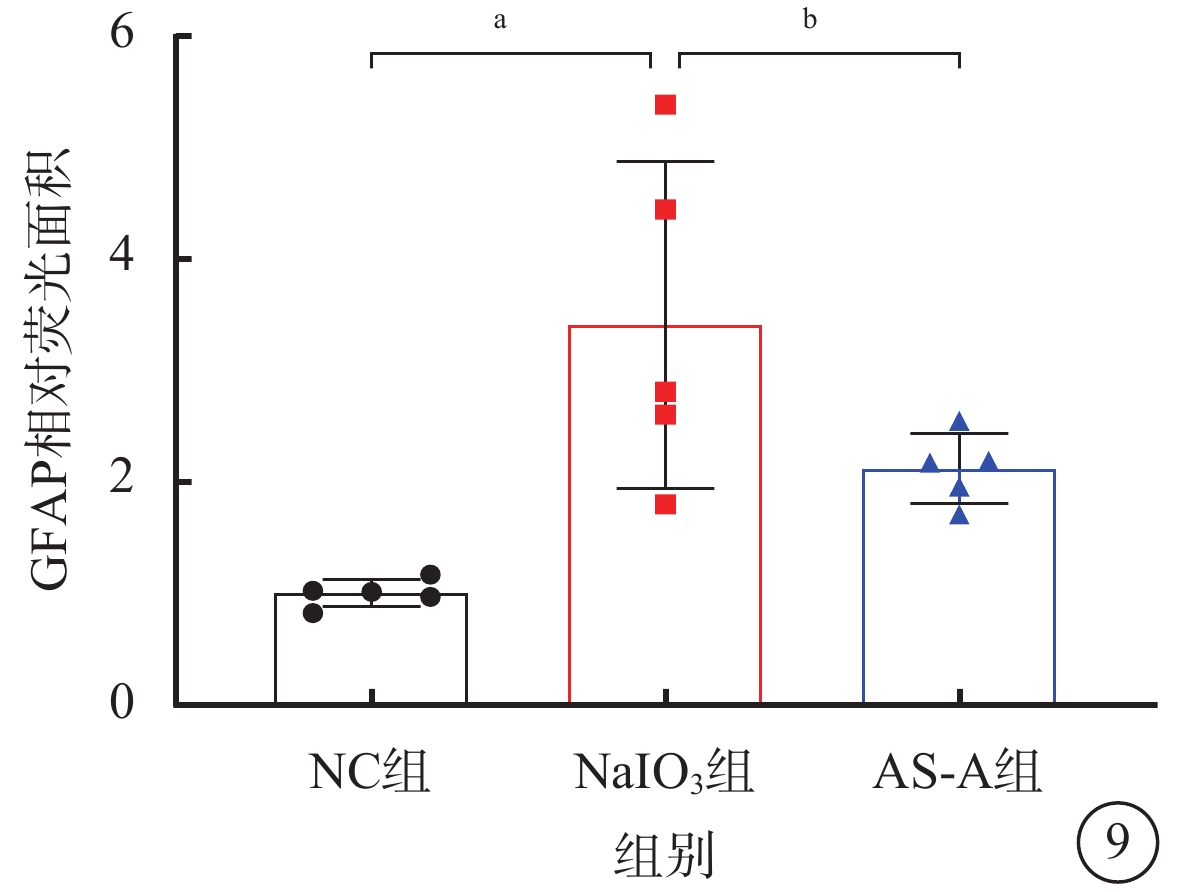 圖9
NC組、NaIO3組、AS-A組GFAP相對熒光面積比較(n=5),aP<0.001,bP<0.05
圖9
NC組、NaIO3組、AS-A組GFAP相對熒光面積比較(n=5),aP<0.001,bP<0.05
NC:正常對照;NaIO3:碘酸鈉;AS-A:黃芪甲苷;GFAP:神經膠質纖維酸性蛋白
3 討論
光感受器細胞不可逆的進行性死亡是導致光感受器退行性病變的核心病理機制之一。因此,尋找能夠拮抗光感受器死亡的藥物是當前領域內的重點研究方向之一[1]。本課題組前期研究發現,傳統明目中藥黃芪中的主要活性成分AS-A能夠抑制光氧化應激和DNA損傷介導的光感受器細胞死亡[2]。本研究在此基礎上,進一步研究了AS-A在NaIO3誘導的光感受器退行性病變模型中的藥理學作用和機制。通過免疫組織化學法分析RPE細胞膜上的主要結構蛋白ZO-1的表達情況,評估AS-A對RPE細胞結構完整性的保護作用。此外,采用TUNEL法評估了AS-A對NaIO3誘導的光感受器細胞死亡的影響。結果表明,AS-A可顯著抑制NaIO3誘導的RPE細胞損傷,抑制繼發的光感受器細胞死亡,為AS-A的光感受器保護作用提供了新的認識。
由于視網膜退行性病變過程中伴隨的炎癥反應,壞死性凋亡逐漸被視為是光感受器死亡的重要機制[3-4]。在壞死性凋亡過程中,RIPK1與主要壞死性凋亡調節因子RIPK3結合形成促壞死復合體。壞死性RIPK1/RIPK3復合物激活MLKL,通過引起膜破裂和細胞溶解來執行壞死性細胞死亡[5]。本課題組前期研究已證明,AS-A可以抑制光氧化應激以及DNA損傷誘導介導的視網膜壞死性凋亡和炎癥相關基因的上調,提示AS-A在抑制壞死性凋亡和炎癥方面的潛在藥理學作用[2]。有研究表明,NaIO3可以誘導RPE細胞發生壞死性凋亡[6-7]。RIPK3-綠色熒光蛋白轉基因小鼠在注射NaIO3后,RPE細胞中RIPK3出現聚集,而注射RIPK1抑制劑Nec-1可以顯著抑制RPE細胞的壞死性凋亡[6]。本研究發現,AS-A同樣可以顯著抑制NaIO3誘導的壞死性凋亡基因Mlkl、Ripk3以及炎癥相關基因Ccl2、Il-1B和Tnf的表達顯著上調,從而減輕光感受器細胞死亡。
壞死性凋亡釋放的細胞內容物還會激活視網膜中的常駐免疫細胞小膠質細胞,引發炎癥反應[8-9]。在受到嚴格調控下,適度激活的小膠質細胞有助于視網膜損傷的修復。然而,持續和過度激活的小膠質細胞會分泌大量促炎細胞因子,導致嚴重的神經炎癥反應,進一步加劇光感受器退行性病變[10-11]。本研究結果顯示,NaIO3組小鼠視網膜下間隙和ONL層可見大量激活呈阿米巴樣的小膠質細胞,而AS-A可以顯著抑制小膠質細胞的激活和遷移。
此外,本研究還發現AS-A具有抑制Müller細胞反應性膠質增生的作用。Müller細胞是視網膜特有的神經膠質細胞,為視網膜神經元的結構、代謝和功能提供支持[12-14]。在光感受器退行性病變過程中,Müller細胞率先感知視網膜損傷,其中間絲蛋白,如GFAP的表達上調,導致反應性膠質增生,形成致密而堅硬的膠質瘢痕,包裹受損的部位。然而,持續過度的反應性膠質增生會影響剩余健康的視網膜神經元細胞,加重光感受器退行性病變[15-16]。本研究結果顯示,AS-A可以顯著抑制NaIO3誘導的GFAP上調,抑制減輕Müller細胞反應性膠質增生。
本研究結果表明,AS-A對NaIO3誘導的RPE損傷及光感受器細胞死亡具有一定的拮抗作用。這一作用在一定程度上涉及對壞死性凋亡和神經炎癥反應相關表型的抑制。此外,AS-A可顯著減輕NaIO3誘導的視網膜小膠質細胞激活和Müller細胞反應性膠質增生,進一步佐證了其對光感受器退行性改變相關視網膜穩態失衡的有效干預作用。這些研究結果為今后AS-A在預防和治療光感受器退行性改變相關疾病中的應用提供了新的藥理學證據。盡管如此,本研究仍存在一些不足之處,需要在未來的研究中進一步探討。首先,本研究未能揭示AS-A是否能夠直接調控壞死性凋亡途徑。未來的研究需要通過分子和細胞生物學的方法,進一步揭示AS-A在調控壞死性凋亡中的具體作用和機制。其次,盡管已有研究表明,AS-A在體外具有直接抑制小膠質細胞激活的作用[17],但其在體內對光感受器變性相關的小膠質細胞活化的直接抑制作用尚未得到證實。因此,未來的研究需要明確AS-A是否能夠通過直接作用于小膠質細胞來發揮協同干預光感受器退行性病變的效應。此外,Müller細胞在光感受器退行性病變中的功能異常及其與AS-A干預效應之間的關系也尚未得到充分研究。在光感受器退行性病變過程中,Müller細胞的谷氨酰胺合成酶表達下調可能導致神經遞質谷氨酸轉化障礙,從而加劇神經元的興奮性中毒和光感受器細胞的死亡[18],但AS-A是否能夠直接干預Müller細胞的病理生理變化,目前還不清楚。綜上所述,未來的研究應當集中在以下幾個方面:一是深入研究AS-A的分子作用機制,特別是其對壞死性凋亡途徑的調控;二是在體內模型中探究AS-A對小膠質細胞活化的直接抑制作用及其對光感受器退行性病變的協同干預效應;三是研究AS-A對Müller細胞病理生理變化的影響,以及其在光感受器退行性病變中的潛在保護作用。這些研究將有助于我們更全面地理解AS-A的藥理學特性,為其在光感受器退行性病變相關疾病的臨床治療中的應用提供更加堅實的科學依據。
視網膜光感受器細胞是負責啟動視覺的一級神經元,其不可逆死亡是導致年齡相關性黃斑變性(AMD)和視網膜色素變性等光感受器退行性疾病患者視力損害的核心原因。但目前尚無有效干預光感受器細胞死亡的措施[1]。本課題組前期研究發現,傳統明目中藥黃芪的主要活性成分黃芪甲苷(AS-A)具有光感受器細胞保護作用[2]。AS-A能夠顯著抑制光誘導的視網膜氧化應激和光感受器細胞死亡,減輕光感受器細胞DNA損傷、多聚ADP核糖聚合酶活化和核蛋白高遷移率族蛋白B1的釋放。在分子水平,AS-A干預可拮抗光誘導的視網膜全基因表達譜改變,特別是抑制了光損傷條件下視網膜壞死性凋亡和炎癥反應相關基因表達的上調。同時,AS-A對光損傷視網膜小膠質細胞活化具有顯著的抑制作用。此外,AS-A可減輕DNA烷化劑甲磺酸甲酯(MMS)誘導的DNA損傷介導的光感受器細胞退行性改變,抑制MMS誘導的視網膜壞死性凋亡和促炎基因表達上調以及小膠質細胞的活化[2]。碘酸鈉(NaIO3)可選擇性誘導視網膜色素上皮(RPE)氧化損傷,繼而導致光感受器退行性改變的發生。因此,為更全面理解AS-A在干預光感受器退行性病變的藥理學作用,本研究進一步在NaIO3誘導RPE損傷介導的光感受器細胞退行性病變模型中探究其作用。現將結果報道如下。
1 材料和方法
本研究經上海中醫藥大學岳陽中西醫結合醫院動物保護與使用委員會審核通過(動物倫理批號:YYLAC-2019-021-2)。
1.1 實驗動物及主要材料
健康雄性C57BL/6J小鼠60只,6~8周齡,體重(24±2)g,無特定病原體級,購自上海斯萊克實驗動物有限公司。飼養于光照12 h/12 h晝夜明暗交替,溫度(20±2)℃,濕度35%~55%條件下,自由進食和飲水。
AS-A(B20564,上海源葉生物科技有限公司);NaIO3(71702,美國Sigma公司);緊密連接蛋白(ZO-1)抗體(ab221547,美國Abcam公司);離子鈣接頭蛋白1(Iba1)抗體(019-19741,日本FUJIFILM Wako Pure Chemical公司);Müller細胞特異性標志物神經膠質纖維酸性蛋白(GFAP)抗體(Z0334,美國DAKO公司);綿羊抗兔Cy3二抗(C2306,美國Sigma公司);原位末端標記(TUNEL)試劑盒(美國Promega公司);TRIzol(美國Invitrogen公司);PrimeScript RT Master Mix(日本TaKaRa公司);LightCycler 480 SYBR Green I Master(德國Roche公司);光相干斷層掃描(OCT)儀(美國Phoenix Research Labs公司);光學顯微鏡、熒光顯微鏡(德國Leica公司);實時熒光定量聚合酶鏈反應(qPCR)系統(德國Roche公司)。
1.2 方法
分組、建模及給藥。采用隨機數字表法將小鼠分為正常對照組(NC組)、NaIO3模型組(NaIO3組)、AS-A組,每組20只。AS-A、NaIO3分別溶于30%二甲基亞砜(DMSO)溶液、生理鹽水中。建模前30 min,AS-A組小鼠參照課題組前期研究結果按100 mg/kg的劑量腹腔注射100 μl AS-A;NC組、NaIO3組小鼠腹腔注射等體積30% DMSO作為溶劑對照。30 min后,NaIO3組、AS-A組小鼠按30 mg/kg的劑量腹腔注射100 μl NaIO3溶液;NC組給予生理鹽水作為溶劑對照。其后,AS-A組小鼠每天早晚間隔12 h給藥2次,直到實驗結束。
眼底彩色照相、OCT檢查。建模后4 d,各組小鼠腹腔注射鹽酸氯胺酮(82.5 mg/kg)和塞拉嗪(8.25 mg/kg)混合溶液麻醉,1%托吡卡胺散瞳,行眼底彩色照相和OCT檢查,采集眼底圖像和視網膜橫截面圖像并測量顳側、鼻側視網膜外核層(ONL)厚度。
蘇木精-伊紅(HE)染色觀察各組小鼠視網膜形態結構。建模后4 d,小鼠過量麻醉處死,摘除眼球,置于4%多聚甲醛中固定24 h,常規脫水、浸蠟后包埋,按4 μm厚度切片。制備好的石蠟切片脫蠟,水化后,浸于HE染液中染色,重新脫水,透明,中性樹脂封片。光學顯微鏡下觀察拍照。
ZO-1染色觀察RPE細胞結構。建模后1 d,小鼠過量麻醉處死,摘除眼球并去除前房、晶狀體、神經視網膜,100%甲醇室溫固定30 min,含10%綿羊血清抗原封閉液室溫孵育30 min,ZO-1抗體(1∶500)4℃孵育過夜,綿羊抗兔Cy3二抗(1:1 000)37 ℃孵育30 min,鋪片,甘油明膠封片。
免疫組織化學染色觀察視網膜中Iba1、GFAP表達;TUNEL法檢測光感受器細胞死亡情況。建模后3 d,小鼠過量麻醉處死,摘除眼球,常規制作石蠟切片。石蠟切片檸檬酸鈉溶液加熱修復抗原30 min,封閉非特異性抗原,一抗GFAP(1:500)4 ℃孵育過夜,綿羊抗兔Cy3二抗(1:1 000)37 ℃孵育30 min;眼球去除角膜、晶狀體制成視杯,4%多聚甲醛中固定2 h,蔗糖梯度脫水,OCT包埋劑包埋,按12 μm厚度制備冰凍切片,一抗Iba1(1:200)和綿羊抗兔Cy3二抗(1:1 000)染色,4′, 6-二氨基-2-苯基吲哚(DAPI)復染,甘油明膠封片。石蠟切片按說明書進行原位TUNEL染色。
qPCR檢測各組小鼠視網膜趨化因子配體2(Ccl2)、白細胞介素-1β(IL-1β)、混合譜系激酶結構域樣蛋白(Mlkl)、受體相互作用蛋白激酶3(Ripk3)、腫瘤壞死因子(Tnf)等基因的mRNA相對表達量。建模后1 d,過量麻醉處死小鼠,摘除眼球剝離視網膜,Trizol提取總RNA,反轉錄試劑逆轉錄成cDNA,按相應基因配置SYBR Green qPCR擴增反應體系進行qPCR檢測。應用Express 3.0軟件設計引物序列(表1)。以18S rRNA作為內參照,根據2?[Ct(目標)?Ct(18S rRNA)]計算基因表達的倍數變化。
1.3 統計學方法
采用GraphPad Prism 10軟件行統計學分析。經正態性檢驗和方差齊性檢驗后,符合正態分布的計量資料以均數±標準差(x±s)表示。兩組間比較采用獨立樣本t檢驗;多組間比較采用單因素方差分析;事后多重比較采用Tukey檢驗。P<0.05為差異有統計學意義。
2 結果
2.1 AS-A抑制NaIO3誘導的視網膜結構損傷
建模后4 d,NC組、AS-A組小鼠眼底未見明顯異常;NaIO3組小鼠眼底可見大量散在黃白色玻璃膜疣樣結構(圖1A~1C)。與NC組比較,NaIO3組小鼠顳側和鼻側RPE層出現大量強反射區;與NaIO3組比較,AS-A組小鼠顳側和鼻側RPE層中強反射區數量減少(圖1D~1I)。與NC組、AS-A組比較,NaIO3組小鼠視網膜顳側、鼻側ONL厚度均顯著變薄,差異有統計學意義(F=123.00、41.37,P<0.001)(圖2)。
圖1
AS-A抑制NaIO3誘導的小鼠視網膜結構損傷
1A~1C分別示NC組、NaIO3組、AS-A組小鼠彩色眼底像,建模后4 d,與NC組、AS-A組比較,NaIO3組小鼠眼底可見大量散在黃白色玻璃膜疣樣結構(紅箭)。1D、1E分別示NC組小鼠顳側、鼻側視網膜OCT像;1F、1G示NaIO3組小鼠顳側、鼻側視網膜OCT像;1H、1I示AS-A組小鼠顳側、鼻側視網膜OCT像。與NC組比較,NaIO3組小鼠顳側和鼻側RPE層出現大量強反射區(白箭);NaIO3組、AS-A組 與NaIO3組比較,AS-A組小鼠顳側和鼻側RPE層中強反射區數量減少(白箭) NC:正常對照;NaIO3:碘酸鈉;AS-A:黃芪甲苷;OCT:光相干斷層掃描;RPE:視網膜色素上皮;ELM:外界膜(紅線所在處);INL:內核層(黃線以下弱反射區);ONL:外核層(紅、黃線之間白線所示弱反射區);OPL:外叢狀層(黃線所在處)
圖2
各組小鼠視網膜外核層厚度比較(n=5),aP<0.001
NC:正常對照;NaIO3:碘酸鈉;AS-A:黃芪甲苷;ONL:外核層
2.2 AS-A保護RPE細胞形態結構
建模后1 d,NC組、AS-A組小鼠RPE細胞呈六邊形結構,ZO-1均勻分布于其膜上。NaIO3組小鼠視網膜中央、中間、周邊RPE細胞膜上ZO-1大量丟失,RPE細胞明顯萎縮(圖3)。
圖3
AS-A抑制NaIO3誘導RPE細胞結構損傷
3A~3C、3D~3F、3G~3I分別示NC組、NaIO3組、AS-A組小鼠視網膜中央、中間、周邊RPE細胞熒光顯微鏡像(標尺:50 μm),ZO-1呈綠色熒光表達。NC組(3A~3C)、AS-A組(3G~3I),RPE細胞呈六邊形結構,ZO-1均勻分布于RPE細胞膜上;NaIO3組建模后1 d,小鼠視網膜中央、中間、周邊ZO-1大量丟失,RPE細胞明顯萎縮(3D~3F) NC:正常對照;NaIO3:碘酸鈉;AS-A:黃芪甲苷;RPE:視網膜色素上皮;ZO-1:緊密連接蛋白-1
2.3 AS-A抑制NaIO3誘導光感受器細胞丟失
NC組小鼠視網膜各層形態結構完整(圖4A,4B);AS-A組小鼠RPE層色素整齊,光感受器內外節結構完整,ONL細胞核層數顯著增加(圖4C,4D);NaIO3組小鼠RPE層可見圓形黑色沉積物,光感受器內外節結構嚴重受損,ONL細胞核層數顯著減少(圖4E,4F)。與NC組、AS-A組比較,NaIO3組小鼠視網膜顳側、鼻側ONL細胞核層數顯著降低,差異有統計學意義(F=38.82、65.55,P<0.001)(圖4G)。
圖4
AS-A抑制NaIO3誘導的光感受器細胞丟失
4A、4B,4C、4D、4E、4F分別示NC組、NaIO3組、AS-A組建模后4 d小鼠視網膜光學顯微鏡像(蘇木精-伊紅染色,標尺:50 μm),NC組小鼠視網膜各層形態結構完整;NaIO3組RPE層可見圓形黑色沉積物(紅箭),IS、OS結構嚴重受損,ONL細胞核層數顯著減少;AS-A組小鼠RPE層色素整齊,IS、OS結構完整,ONL細胞核層數顯著增加。4G示NC組、AS-A組、NaIO3組小鼠視網膜ONL細胞核層數比較(
2.4 AS-A抑制NaIO3誘導的光感受器細胞死亡
建模后3 d,與NC組比較,NaIO3組小鼠視網膜ONL可見大量TUNEL陽性細胞核;與NaIO3組比較,AS-A組小鼠視網膜ONL中TUNEL陽性細胞核數量顯著減少,差異有統計學意義(t=2.657,P<0.05)(圖5)。
圖5
AS-A抑制NaIO3誘導的光感受器細胞死亡
5A~5C分別示NC組、NaIO3組、AS-A組熒光顯微鏡像(標尺:50 μm),DAPI標記的細胞核呈藍色熒光;TUNEL染色的光感受器死亡細胞呈綠色熒光。5D示NaIO3組、AS-A組陽性細胞核相對熒光面積比較(
2.5 AS-A抑制NaIO3誘導的視網膜壞死性凋亡和炎癥相關基因的表達上調
建模后1 d,與NC組、AS-A組比較,NaIO3組視網膜中Mlkl、Ripk3、Ccl2、Il-1β、Tnf mRNA相對表達量顯著升高,差異有統計學意義(F=39.18、10.66、53.51、41.40、24.13,P<0.001)(圖6)。
圖6
NC組、NaIO3組、AS-A組小鼠視網膜中Mlkl、Ripk3、Ccl2、Il-1β、Tnf mRNA相對表達量比較(n=5),aP<0.001
NC:正常對照;NaIO3:碘酸鈉;AS-A:黃芪甲苷;
2.6 AS-A抑制NaIO3誘導的小膠質細胞激活
建模后3 d,NC組小鼠僅內層視網膜可見少量Iba1陽性表達;NaIO3組小鼠ONL和視網膜下間隙處見大量Iba1陽性表達;AS-A組小鼠ONL和視網膜下間隙處可見少量Iba1陽性表達(圖7A~7C)。與NaIO3組比較,AS-A組小鼠ONL和視網膜下間隙處Iba1陽性表達顯著降低,差異有統計學意義(t=3.34,P<0.05)(圖7D)。
圖7
AS-A抑制NaIO3誘導的小膠質細胞激活
7A~7C分別示NC組、NaIO3組、AS-A組小鼠視網膜免疫熒光顯微鏡像(標尺:50 μm),DAPI標記細胞核呈藍色熒光,Iba1標記的陽性細胞呈紅色熒光;7D示NaIO3組、AS-A組Iba1相對熒光面積比較(
2.7 AS-A抑制NaIO3誘導的Müller細胞反應性膠質增生
建模后3 d,NC組小鼠視網膜外從狀層、神經纖維層可見少量GFAP陽性表達(圖8A~8C);NaIO3組小鼠中央、中間、周邊視網膜GFAP陽性表達顯著增加(圖8D~8F);AS-A組小鼠視網膜GFAP陽性表達顯著減少,僅局限于外從狀層和神經纖維層(圖8G~8I)。與NC組、AS-A組比較,NaIO3組小鼠視網膜GFAP陽性表達顯著升高,差異有統計學意義(F=9.62,P<0.05)(圖9)。
圖8
AS-A抑制NaIO3誘導Müller細胞反應性膠質增生
8A~8C、8D~8F、8G~8I分別示NC組、NaIO3組、AS-A組小鼠視網膜中央、中間、周邊熒光顯微鏡像(標尺:50 μm),DAPI標記的細胞核呈藍色熒光,GFAP標記的陽性細胞呈紅色熒光 NC:正常對照;NaIO3:碘酸鈉;AS-A:黃芪甲苷;INL:內核層;ONL:外核層;OPL:外從狀層;NFL:神經纖維層;DAPI:4',6-二脒基-2-苯基吲哚;GFAP:神經膠質纖維酸性蛋白
圖9
NC組、NaIO3組、AS-A組GFAP相對熒光面積比較(n=5),aP<0.001,bP<0.05
NC:正常對照;NaIO3:碘酸鈉;AS-A:黃芪甲苷;GFAP:神經膠質纖維酸性蛋白
3 討論
光感受器細胞不可逆的進行性死亡是導致光感受器退行性病變的核心病理機制之一。因此,尋找能夠拮抗光感受器死亡的藥物是當前領域內的重點研究方向之一[1]。本課題組前期研究發現,傳統明目中藥黃芪中的主要活性成分AS-A能夠抑制光氧化應激和DNA損傷介導的光感受器細胞死亡[2]。本研究在此基礎上,進一步研究了AS-A在NaIO3誘導的光感受器退行性病變模型中的藥理學作用和機制。通過免疫組織化學法分析RPE細胞膜上的主要結構蛋白ZO-1的表達情況,評估AS-A對RPE細胞結構完整性的保護作用。此外,采用TUNEL法評估了AS-A對NaIO3誘導的光感受器細胞死亡的影響。結果表明,AS-A可顯著抑制NaIO3誘導的RPE細胞損傷,抑制繼發的光感受器細胞死亡,為AS-A的光感受器保護作用提供了新的認識。
由于視網膜退行性病變過程中伴隨的炎癥反應,壞死性凋亡逐漸被視為是光感受器死亡的重要機制[3-4]。在壞死性凋亡過程中,RIPK1與主要壞死性凋亡調節因子RIPK3結合形成促壞死復合體。壞死性RIPK1/RIPK3復合物激活MLKL,通過引起膜破裂和細胞溶解來執行壞死性細胞死亡[5]。本課題組前期研究已證明,AS-A可以抑制光氧化應激以及DNA損傷誘導介導的視網膜壞死性凋亡和炎癥相關基因的上調,提示AS-A在抑制壞死性凋亡和炎癥方面的潛在藥理學作用[2]。有研究表明,NaIO3可以誘導RPE細胞發生壞死性凋亡[6-7]。RIPK3-綠色熒光蛋白轉基因小鼠在注射NaIO3后,RPE細胞中RIPK3出現聚集,而注射RIPK1抑制劑Nec-1可以顯著抑制RPE細胞的壞死性凋亡[6]。本研究發現,AS-A同樣可以顯著抑制NaIO3誘導的壞死性凋亡基因Mlkl、Ripk3以及炎癥相關基因Ccl2、Il-1B和Tnf的表達顯著上調,從而減輕光感受器細胞死亡。
壞死性凋亡釋放的細胞內容物還會激活視網膜中的常駐免疫細胞小膠質細胞,引發炎癥反應[8-9]。在受到嚴格調控下,適度激活的小膠質細胞有助于視網膜損傷的修復。然而,持續和過度激活的小膠質細胞會分泌大量促炎細胞因子,導致嚴重的神經炎癥反應,進一步加劇光感受器退行性病變[10-11]。本研究結果顯示,NaIO3組小鼠視網膜下間隙和ONL層可見大量激活呈阿米巴樣的小膠質細胞,而AS-A可以顯著抑制小膠質細胞的激活和遷移。
此外,本研究還發現AS-A具有抑制Müller細胞反應性膠質增生的作用。Müller細胞是視網膜特有的神經膠質細胞,為視網膜神經元的結構、代謝和功能提供支持[12-14]。在光感受器退行性病變過程中,Müller細胞率先感知視網膜損傷,其中間絲蛋白,如GFAP的表達上調,導致反應性膠質增生,形成致密而堅硬的膠質瘢痕,包裹受損的部位。然而,持續過度的反應性膠質增生會影響剩余健康的視網膜神經元細胞,加重光感受器退行性病變[15-16]。本研究結果顯示,AS-A可以顯著抑制NaIO3誘導的GFAP上調,抑制減輕Müller細胞反應性膠質增生。
本研究結果表明,AS-A對NaIO3誘導的RPE損傷及光感受器細胞死亡具有一定的拮抗作用。這一作用在一定程度上涉及對壞死性凋亡和神經炎癥反應相關表型的抑制。此外,AS-A可顯著減輕NaIO3誘導的視網膜小膠質細胞激活和Müller細胞反應性膠質增生,進一步佐證了其對光感受器退行性改變相關視網膜穩態失衡的有效干預作用。這些研究結果為今后AS-A在預防和治療光感受器退行性改變相關疾病中的應用提供了新的藥理學證據。盡管如此,本研究仍存在一些不足之處,需要在未來的研究中進一步探討。首先,本研究未能揭示AS-A是否能夠直接調控壞死性凋亡途徑。未來的研究需要通過分子和細胞生物學的方法,進一步揭示AS-A在調控壞死性凋亡中的具體作用和機制。其次,盡管已有研究表明,AS-A在體外具有直接抑制小膠質細胞激活的作用[17],但其在體內對光感受器變性相關的小膠質細胞活化的直接抑制作用尚未得到證實。因此,未來的研究需要明確AS-A是否能夠通過直接作用于小膠質細胞來發揮協同干預光感受器退行性病變的效應。此外,Müller細胞在光感受器退行性病變中的功能異常及其與AS-A干預效應之間的關系也尚未得到充分研究。在光感受器退行性病變過程中,Müller細胞的谷氨酰胺合成酶表達下調可能導致神經遞質谷氨酸轉化障礙,從而加劇神經元的興奮性中毒和光感受器細胞的死亡[18],但AS-A是否能夠直接干預Müller細胞的病理生理變化,目前還不清楚。綜上所述,未來的研究應當集中在以下幾個方面:一是深入研究AS-A的分子作用機制,特別是其對壞死性凋亡途徑的調控;二是在體內模型中探究AS-A對小膠質細胞活化的直接抑制作用及其對光感受器退行性病變的協同干預效應;三是研究AS-A對Müller細胞病理生理變化的影響,以及其在光感受器退行性病變中的潛在保護作用。這些研究將有助于我們更全面地理解AS-A的藥理學特性,為其在光感受器退行性病變相關疾病的臨床治療中的應用提供更加堅實的科學依據。