引用本文: 史麗英, 盧躍兵, 孫爽, 徐利輝, 劉婷, 白大勇, 孫先桃. KIF11基因變異致小頭畸形伴或不伴脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征眼部臨床特征分析. 中華眼底病雜志, 2024, 40(11): 825-832. doi: 10.3760/cma.j.cn511434-20240122-00043 復制
版權信息: ?四川大學華西醫院華西期刊社《中華眼底病雜志》版權所有,未經授權不得轉載、改編
小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征(MCLMR)是一種罕見的常染色體顯性遺傳疾病。其涉及一系列中樞神經系統和眼部發育異常的臨床表現,特征多樣且相互重疊。小頭畸形根據其嚴重程度可分為輕度至重度,常伴隨發育遲緩。此外,該病癥還通常表現為一系列典型的面部特征,包括瞼裂上斜、寬鼻梁及圓而尖的鼻尖、較長的人中區域、較薄的上唇、突出的下頜以及外翻的耳朵。脈絡膜視網膜病變是其最常見的眼部異常,視網膜褶皺、小眼畸形、近視和遠視散光也有報道,有些患者無明顯眼表型。淋巴水腫一般為先天性,常見于足背部。在GeneCard網站搜索到已知的與MCLMR相關基因包括KIF11、FZD4、CTNNB1、LRP5、PRSS23、LOC126806659、TSPAN12、CCBE1、TUBGCP4、TUBGCP6等。目前國內關于MCLMR研究較少,且缺乏詳細的眼部臨床特征描述。本研究對KIF11基因變異所致MCLMR一家系的眼部臨床表現和致病基因進行分析。現將結果報道如下。
1 對象和方法
回顧性臨床研究。本研究經河南省兒童醫院倫理委員會審批(批文號:2024-K-010);嚴格遵循《赫爾辛基宣言》原則;所有參與者及未成年受試者的監護人均獲知情并簽署書面知情同意書。
2023年9月于河南省兒童醫院臨床檢查并經基因檢測確診的MCLMR一家系先證者及3名家系成員(圖1)納入本研究。詳細詢問病史、家族史,并行最佳矯正視力(BCVA)、驗光、裂隙燈顯微鏡、眼底彩色照相、熒光素眼底血管造影(FFA)、光相干斷層掃描(OCT)、閃光視覺誘發電位(F-VEP)、全視野視網膜電圖(ERG)檢查以及眼軸長度(AL)測量。同時行頭顱+眼眶MRI檢查和基因檢測。先證者符合MCLMR臨床診斷標準[1-2]。
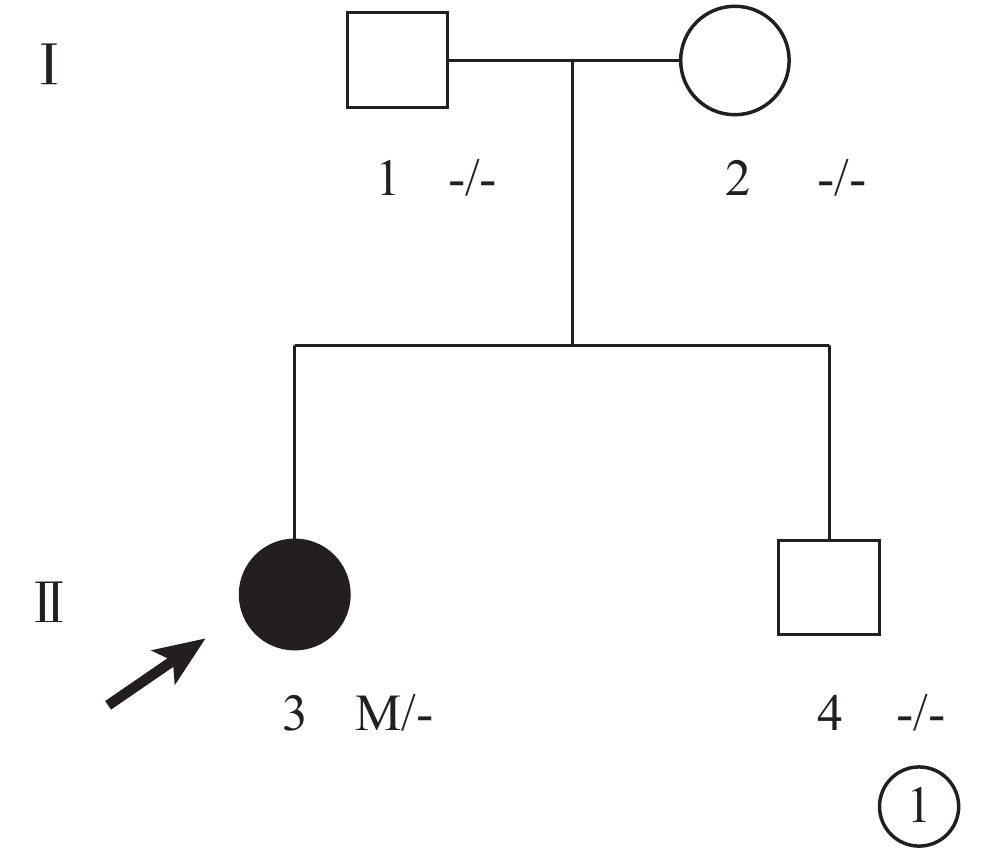
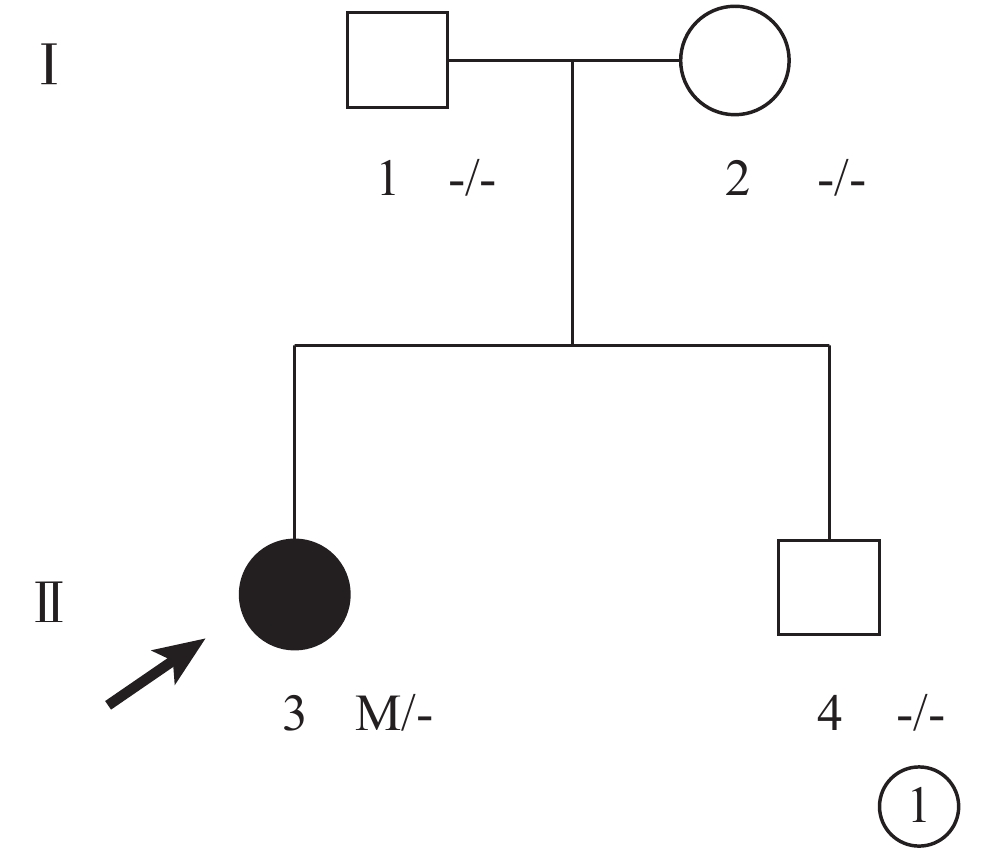 圖1
MCLMR家系圖 □:正常男性;○:正常女性;●:女性患者;↗:先證者;M:c.895A>G(p.Ile299Val); MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征
圖1
MCLMR家系圖 □:正常男性;○:正常女性;●:女性患者;↗:先證者;M:c.895A>G(p.Ile299Val); MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征
采集先證者及其父母、胞弟外周靜脈血樣3 ml,乙二胺四乙酸抗凝。行全外顯子組測序、染色體拷貝數變異檢測、染色體核型分析及線粒體全基因檢測(武漢良培醫學檢驗實驗室)。測序儀下機reads利用BWA(版本:0.7.16a)軟件與人類參考基因組GRCh38比對,生成的bam文件采用GATK軟件(版本:4.0.8.1)進行變異檢測分析,包括標記聚合酶鏈反應(PCR)重復序列、SNV和插入缺失標記(InDel)變異。采用Annova軟件(r版本:20200608)對變異文件vcf進行注釋。注釋數據庫為外顯子組聚集聯盟(ExAC)數據庫(http://exac.broadinstitute.org/)、dbSNP數據庫(https://www.ncbi.nlm.nih.gov/snp/)、千人基因組計劃數據庫(1000Genome,http://www.1000genomes.org/)、人群基因頻率數據庫(gnomAD,https://gnomad.broadinstitute.org/)、InterPro數據庫(https://www.ebi.ac.uk/interpro/)、ClinVar數據庫(https://www.ncbi.nlm.nih.gov/clinvar/)、人類孟德爾遺傳在線數據庫(OMIM,https://omim.org/)、人類基因變異數據庫(HGMD,http://www.hgmd.cf.ac.uk/ac/index.php)等。
變異位點篩選原則:(1)篩選出外顯子區域變異、剪切區域變異、非同義變異位點;(2)ExAC_EAS、ExAC_ALL、1000Genome、gnomAD等人群頻率數據庫中正常人變異攜帶率<1%;(3)使用SIFT、Polyphen2、LRT、MutationTaster等預測軟件對基因位點變異對其蛋白功能影響進行預測;(4)參考OMIM、HGMD、ClinVar等數據庫對致病變異位點進行評估。對檢測出的可疑致病突變,均采用Sanger測序進行驗證。根據美國醫學遺傳學與基因組學學會(ACMG)2015年發布的《序列變異解讀標準和指南》進行變異解析,并評估其臨床意義[3]。
計算機檢索美國國立醫學圖書館醫學數據庫PubMed、萬方數據庫相關文獻。中文檢索詞為“KIF11”、“小頭畸形”、“脈絡膜視網膜病變”;英文檢索詞為“KIF11”、“microcephaly”、“chorioretinopathy”。檢索時間為2012年1月1日至2023年12月31日。共檢索到相關文獻20篇,其中英文文獻、中文文獻分別為19篇[1, 2, 4-20]、1篇[21]。
2 結果
先證者(Ⅱ-1),女,8歲5個月。監護人述患兒自幼視力差、眼球不自主轉動。1年前當地醫院檢查發現屈光不正、眼底病變、眼球震顫;給予戴鏡治療、弱視訓練,視力無明顯提高。智力發育輕度遲緩,輕度學習困難,語言發育及生長發育基本正常。既往身體健康,無外傷及手術史;足月生產,生后無窒息、缺氧病史;無全身淋巴水腫病史,腦部及心臟無明顯異常。其父親(Ⅰ-1)輕度眼球震顫、斜視;母親(Ⅰ-2)、胞弟(Ⅱ-2)眼部表型未見明顯異常。
先證者(Ⅱ-1)表現為頭顱偏小、下頜較小、額部狹窄、雙耳較大且向外突出、鼻頭寬且圓、瞼裂輕度上斜以及人中較長(圖2)。其右眼、左眼祼眼視力分別為0.04、0.05,BCVA分別為 0.08(+2.25 DS/ -2.25 DC×10°)、0.1(+2.25 DS/ -2.25 DC×180°)。雙眼眼球震顫呈水平鐘擺型,無明顯中間帶及異常頭位。遠近角膜映光基本正位,間斷出現內斜視+10°,交替遮蓋內→正,雙眼及單眼眼球運動各方向到位。眼前節未見明顯異常。右眼、左眼AL分別為20.95、21.25 mm(參考值23.19~23.37 mm)。右眼角膜第1、2個主子午線(K1、K2)曲率值分別為43.50、45.77 D;左眼K1、K2曲率值分別為43.51、43.51 D。右眼、左眼角膜散光值分別為2.26、2.03 D。雙眼視盤邊界清楚,顏色淡,杯盤比約2/3;視網膜血管纖細,視網膜呈“豹紋狀”改變,黃斑中心凹反光消失,黃斑區及下方大片脈絡膜視網膜黃白色病灶,大片狀融合,呈萎縮樣改變,小片狀向周邊擴展,色素不均勻(圖3)。FFA檢查,雙眼動靜脈充盈時間正常,黃斑區及下方視網膜斑駁樣熒光著染;晚期無明顯熒光素滲漏(圖4)。OCT檢查,因患兒眼球震顫,注視功能差,可掃描范圍內黃斑中心凹形態淺,橢圓體帶缺失,層次欠清楚,視網膜全層變薄,黃斑下方脈絡膜視網膜全層明顯萎縮(圖5)。F-VEP檢查,雙眼未引出P100波波形(角膜電極,鎮靜狀態下)(圖6)。全視野ERG檢查,視桿反應:雙眼未引出a、b波波形;最大混合反應:雙眼a、b波振幅重度降低;振蕩電位:雙眼未引出波形;視錐反應:30 Hz閃爍光刺激條件下,雙眼a、b波振幅重度降低(角膜電極,鎮靜狀態下)(圖7)。頭顱+眼眶MRI檢查,左側顳部蛛網膜下腔增寬,腦實質內未見明顯異常信號,腦室系統大小形態正常,腦溝、腦回未見異常,中線結構居中,幕下小腦及腦干結構未見明顯異常;雙側眼眶未見明顯異常。
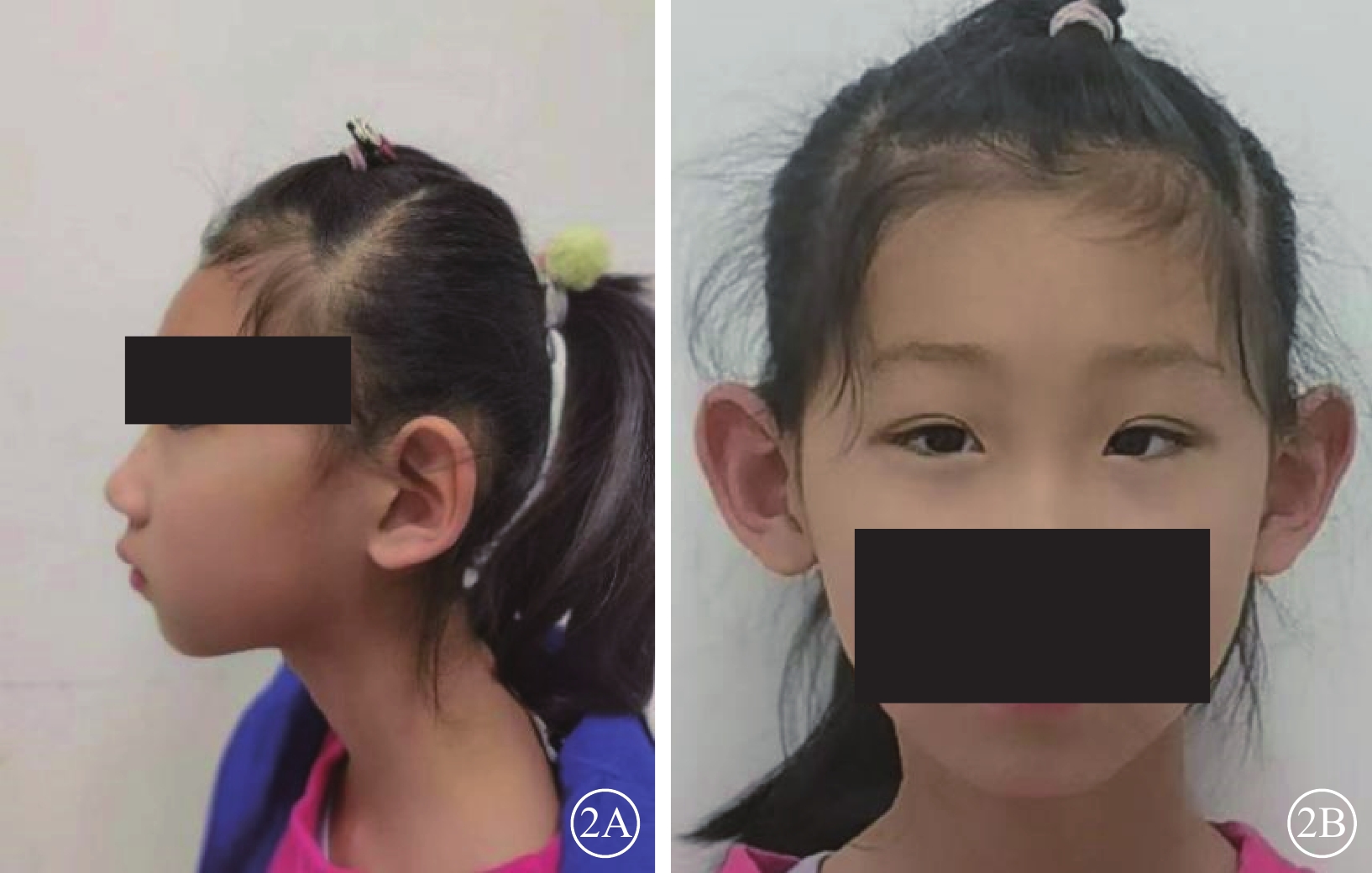
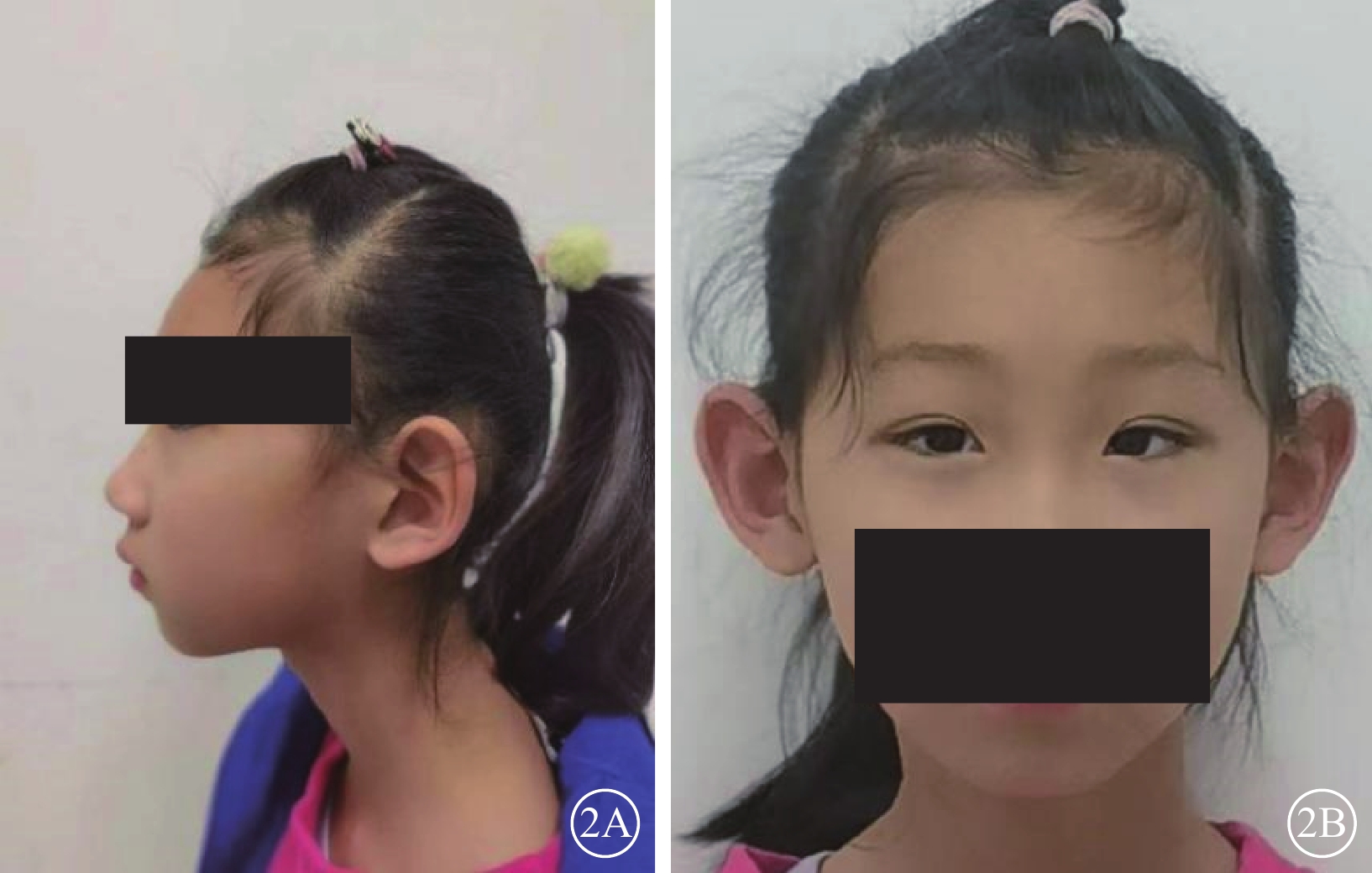 圖2
MCLMR家系先證者(Ⅱ-1)面部外觀像 2A、2B分別示側位、正位,頭顱小,下頜較小,額部窄小,雙側耳朵大并突出,鼻頭寬鼻尖圓,人中較長 MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征
圖2
MCLMR家系先證者(Ⅱ-1)面部外觀像 2A、2B分別示側位、正位,頭顱小,下頜較小,額部窄小,雙側耳朵大并突出,鼻頭寬鼻尖圓,人中較長 MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征
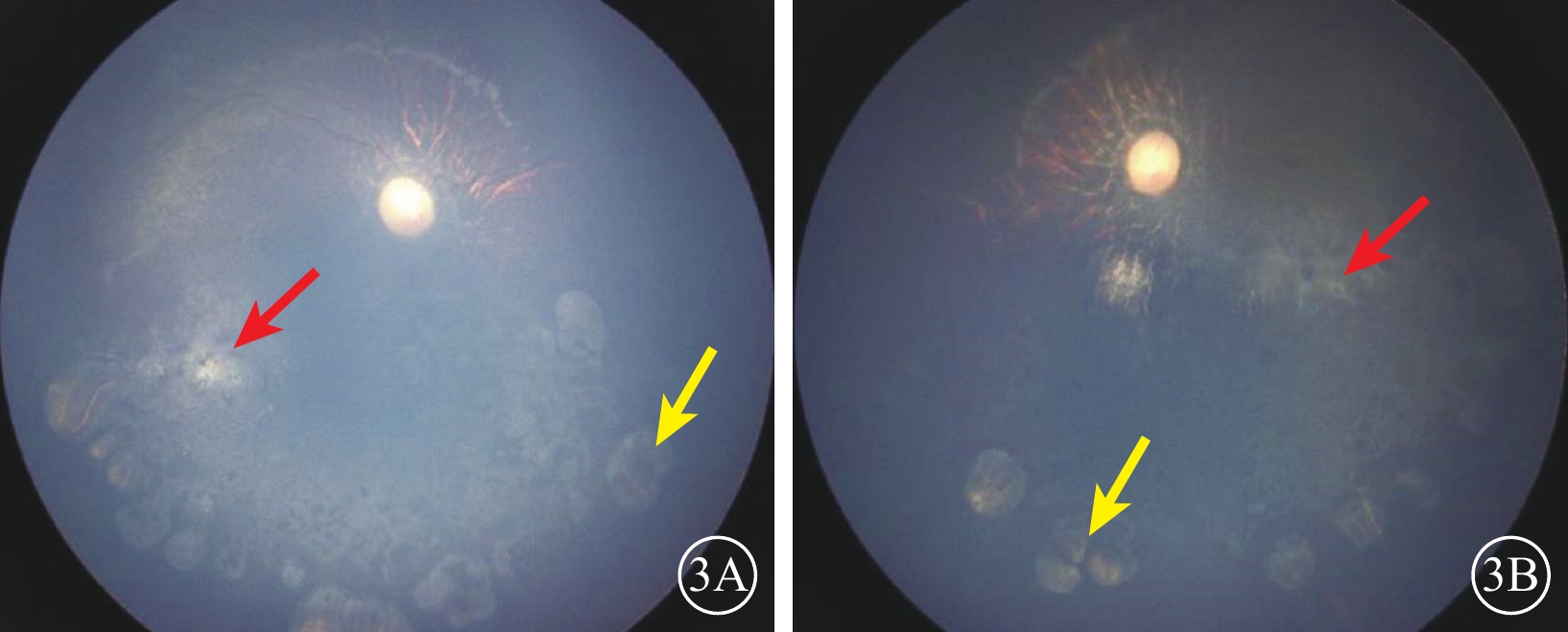
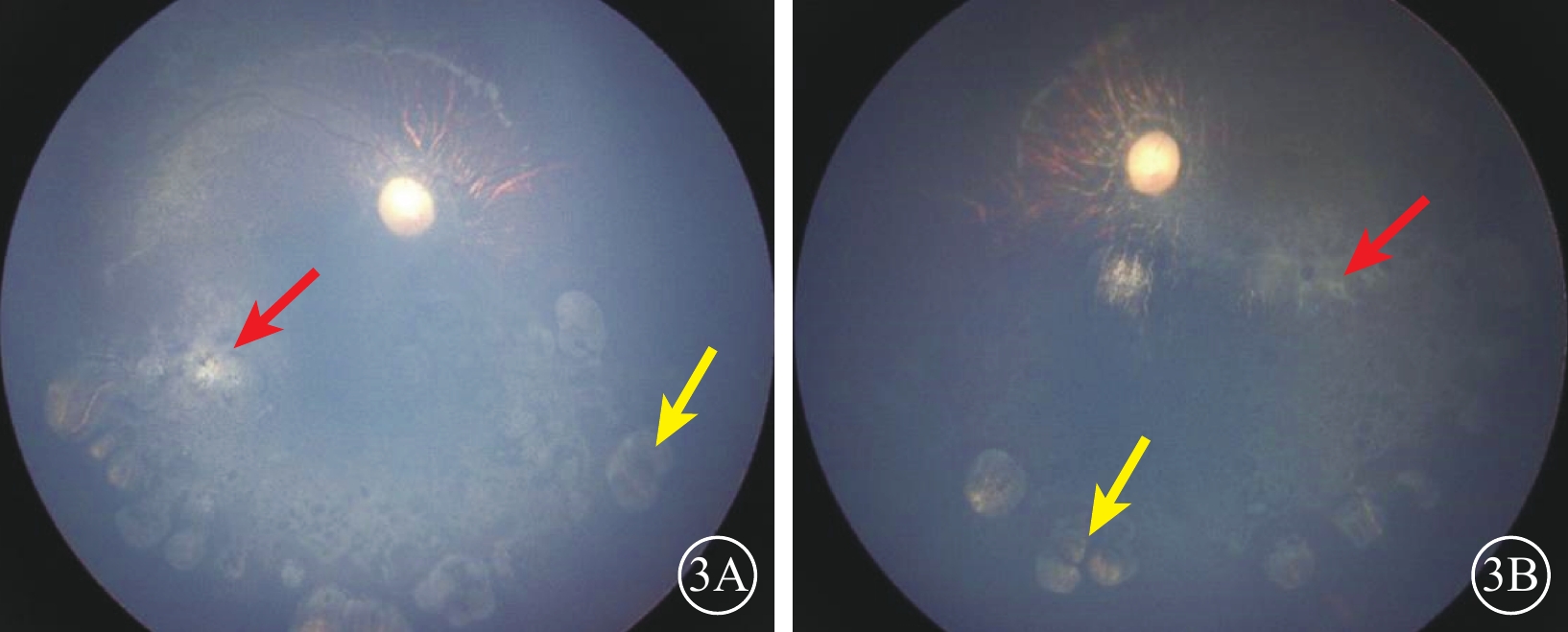 圖3
MCLMR家系先證者(Ⅱ-1)雙眼彩色眼底像 3A、3B分別示右眼、左眼,雙眼視盤顏色淡,視網膜血管纖細,視網膜“豹紋狀”改變;雙眼對稱性黃斑區及下方大片視網膜脈絡膜黃白色萎縮性病灶(紅箭),小片狀病灶向周邊擴展(黃箭) MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征
圖3
MCLMR家系先證者(Ⅱ-1)雙眼彩色眼底像 3A、3B分別示右眼、左眼,雙眼視盤顏色淡,視網膜血管纖細,視網膜“豹紋狀”改變;雙眼對稱性黃斑區及下方大片視網膜脈絡膜黃白色萎縮性病灶(紅箭),小片狀病灶向周邊擴展(黃箭) MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征
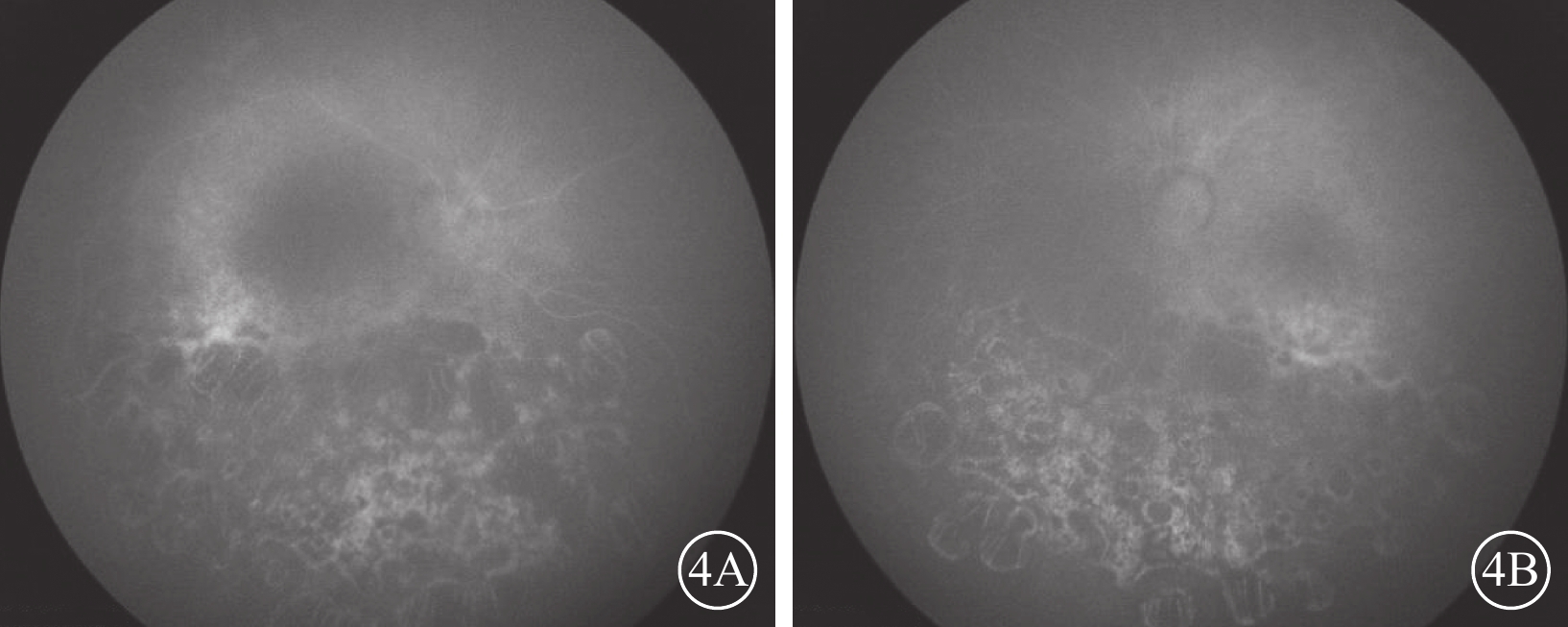
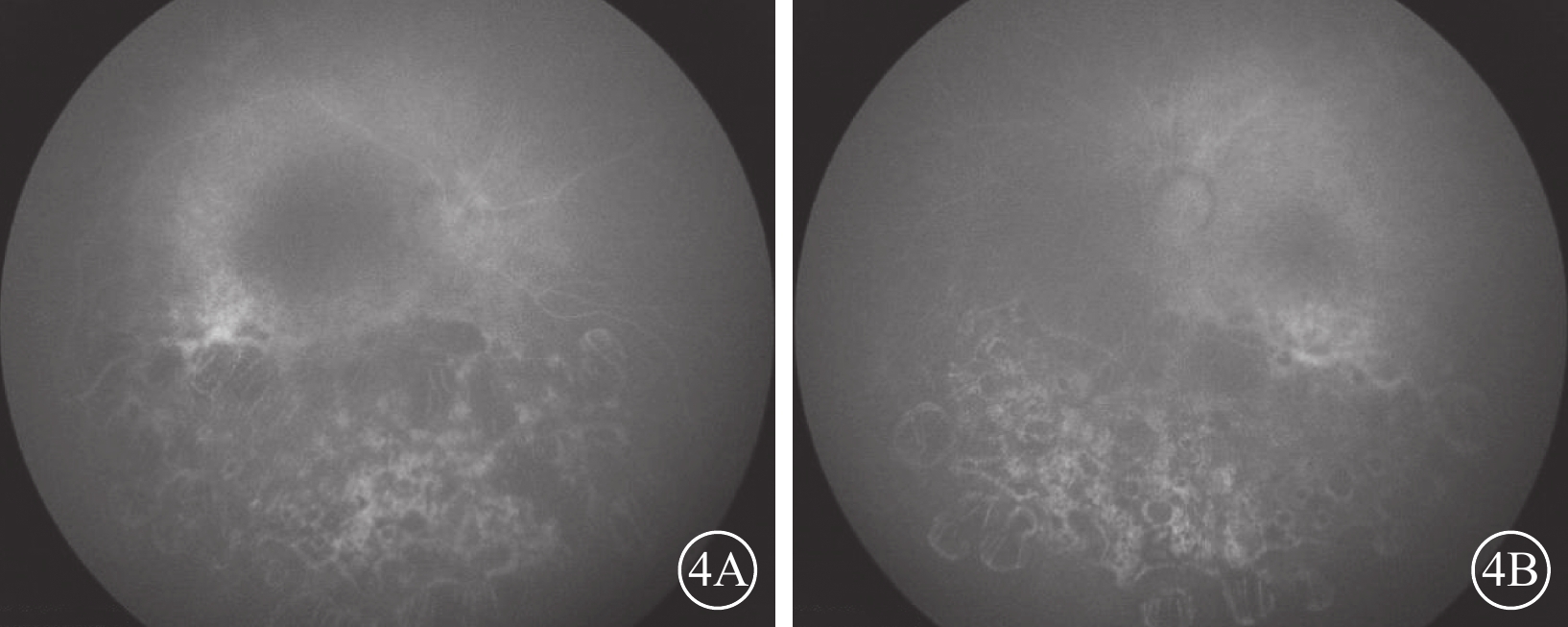 圖4
MCLMR家系先證者(Ⅱ-1)雙眼FFA晚期像 4A、4B分別示右眼、左眼,雙眼與眼底病變范圍一致的視網膜斑駁樣熒光著染,無明顯熒光素滲漏 MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征;FFA:熒光素眼底血管造影
圖4
MCLMR家系先證者(Ⅱ-1)雙眼FFA晚期像 4A、4B分別示右眼、左眼,雙眼與眼底病變范圍一致的視網膜斑駁樣熒光著染,無明顯熒光素滲漏 MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征;FFA:熒光素眼底血管造影
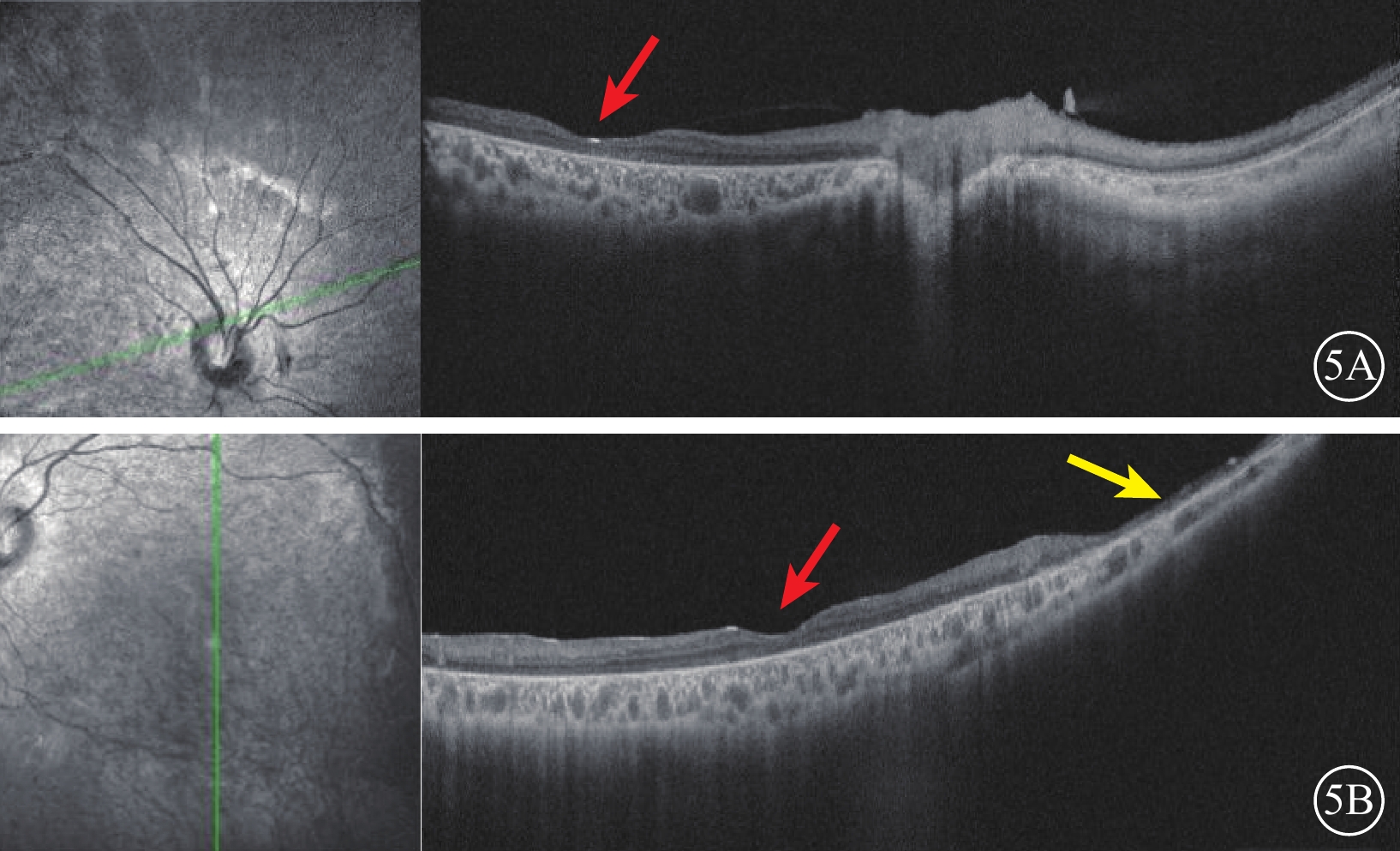
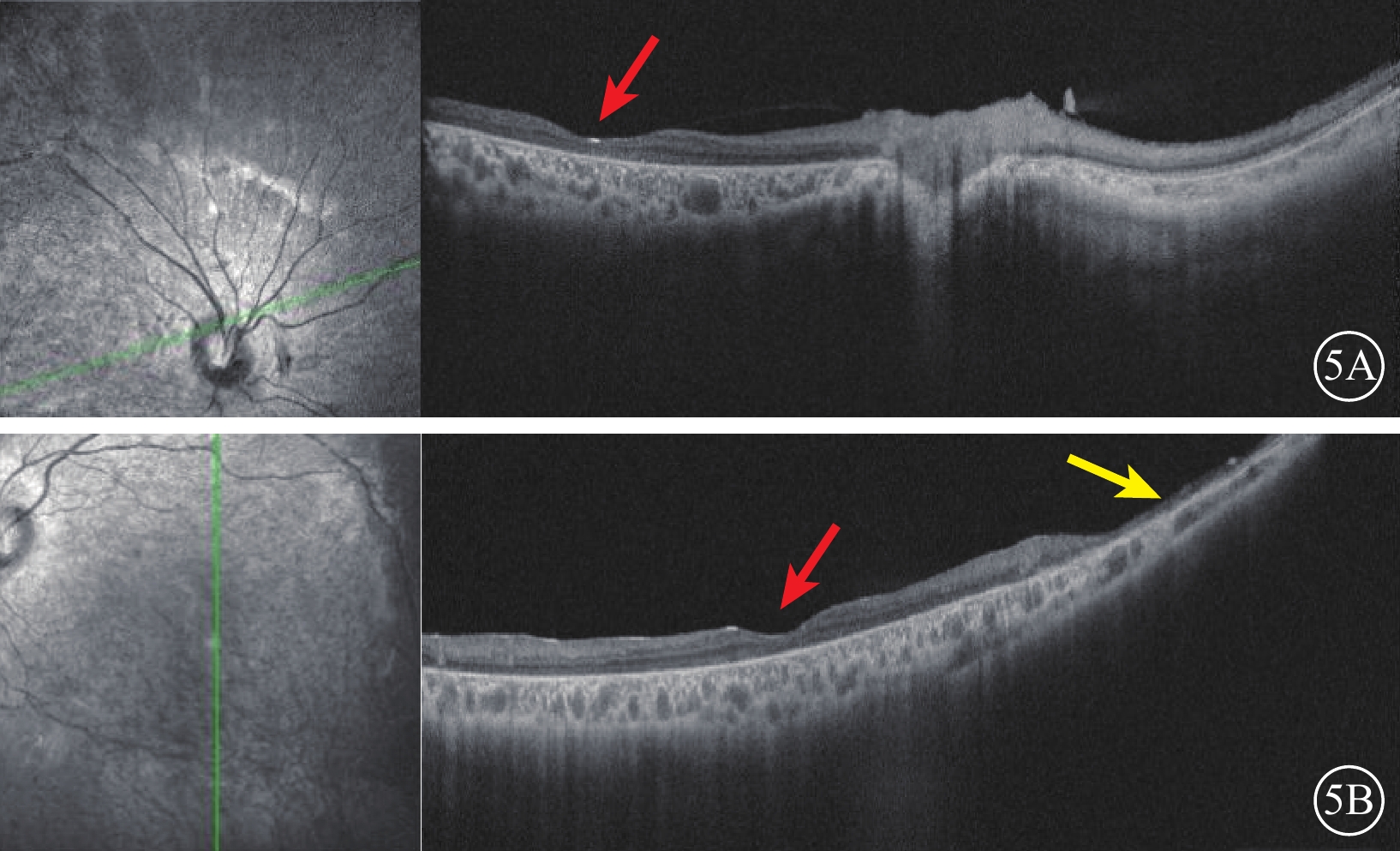 圖5
MCLMR家系先證者(Ⅱ-1)雙眼OCT像 5A、5B分別示右眼、左眼,左圖為掃描方向和部位,右圖為檢查結果。黃斑中心凹形態變淺(紅箭),全層視網膜變薄,黃斑下方脈絡膜視網膜萎縮極薄(黃箭) MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征;OCT:光相干斷層掃描
圖5
MCLMR家系先證者(Ⅱ-1)雙眼OCT像 5A、5B分別示右眼、左眼,左圖為掃描方向和部位,右圖為檢查結果。黃斑中心凹形態變淺(紅箭),全層視網膜變薄,黃斑下方脈絡膜視網膜萎縮極薄(黃箭) MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征;OCT:光相干斷層掃描
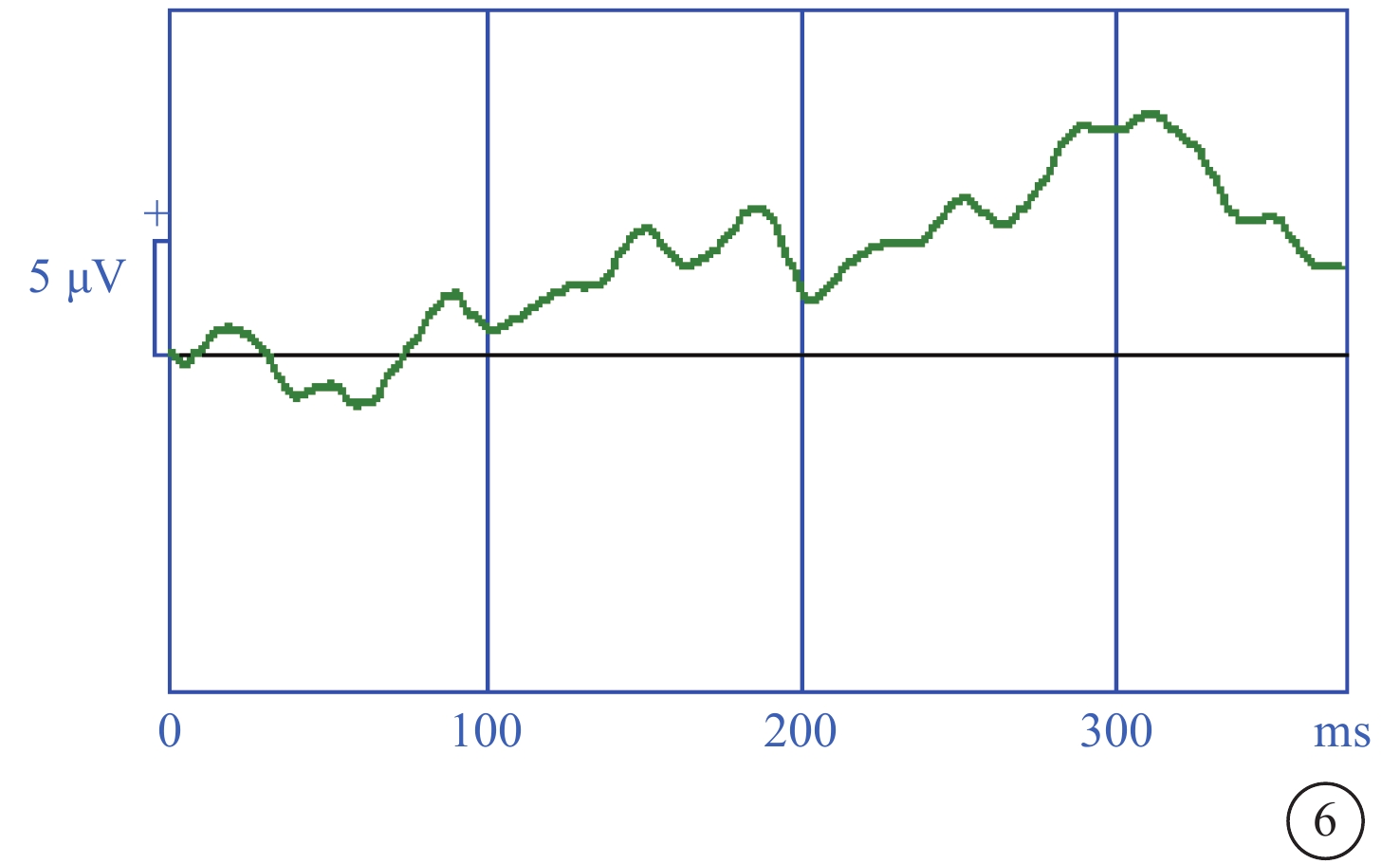
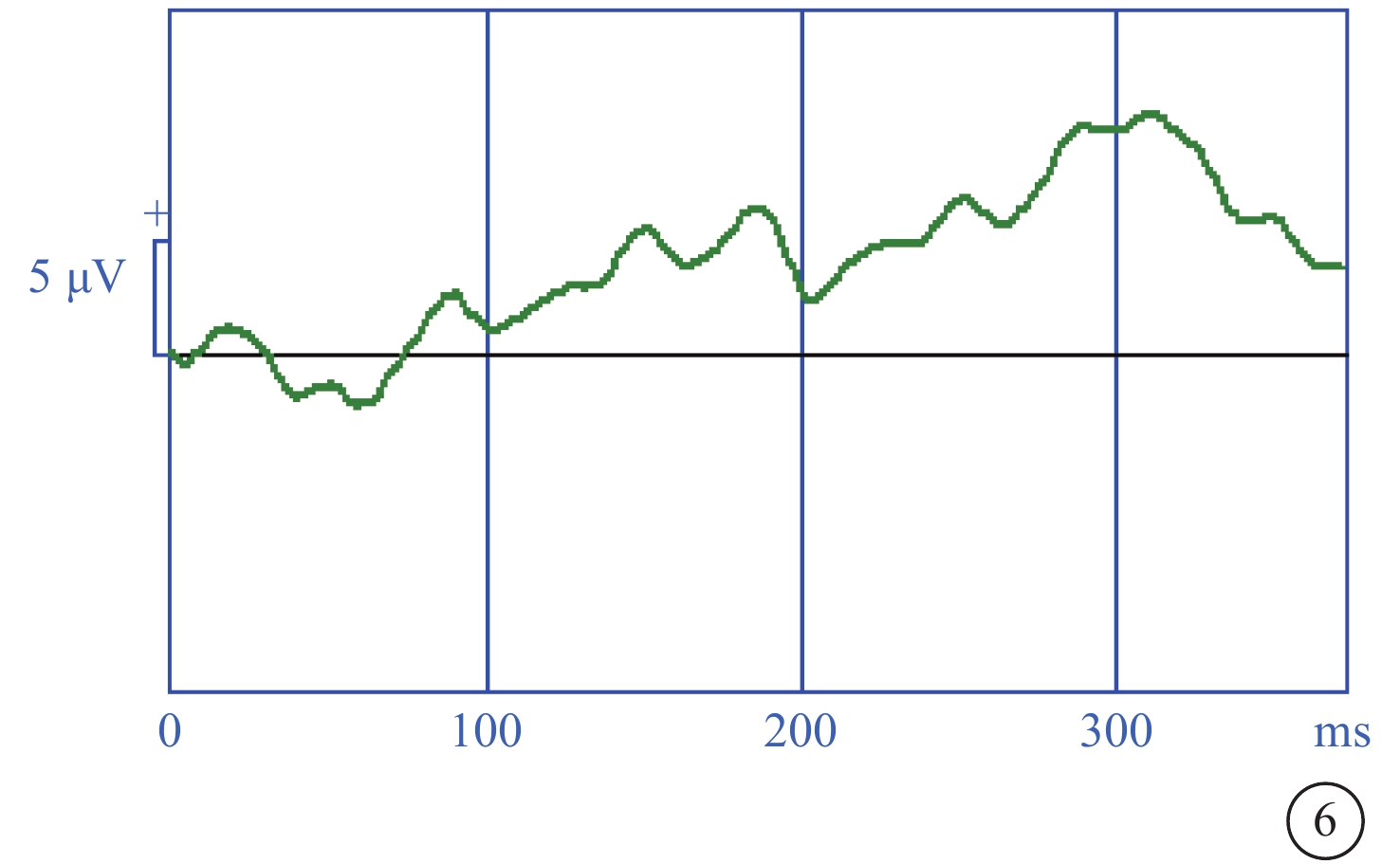 圖6
MCLMR家系先證者(Ⅱ-1)右眼視覺誘發電位圖 未引出可分析的P100波波形 MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙縮合征
圖6
MCLMR家系先證者(Ⅱ-1)右眼視覺誘發電位圖 未引出可分析的P100波波形 MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙縮合征
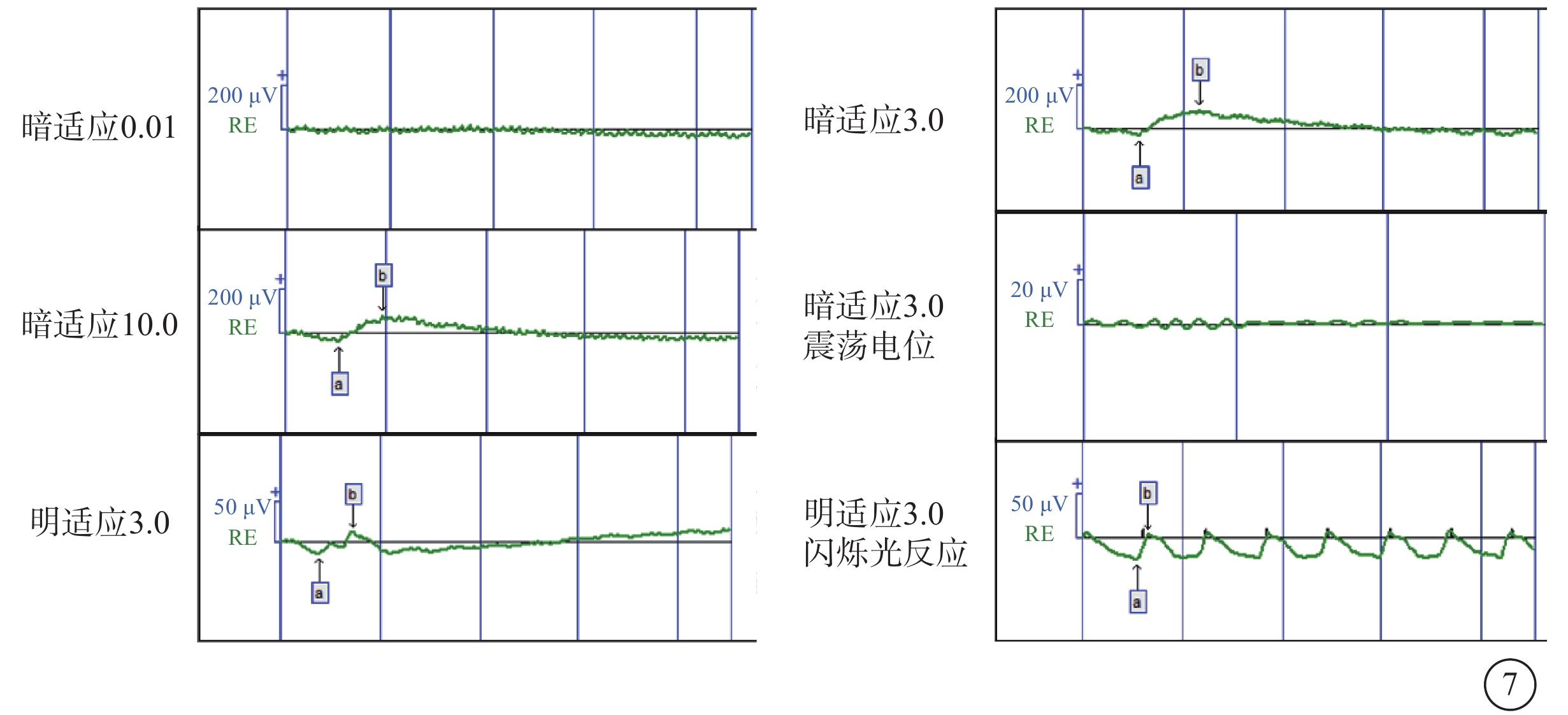
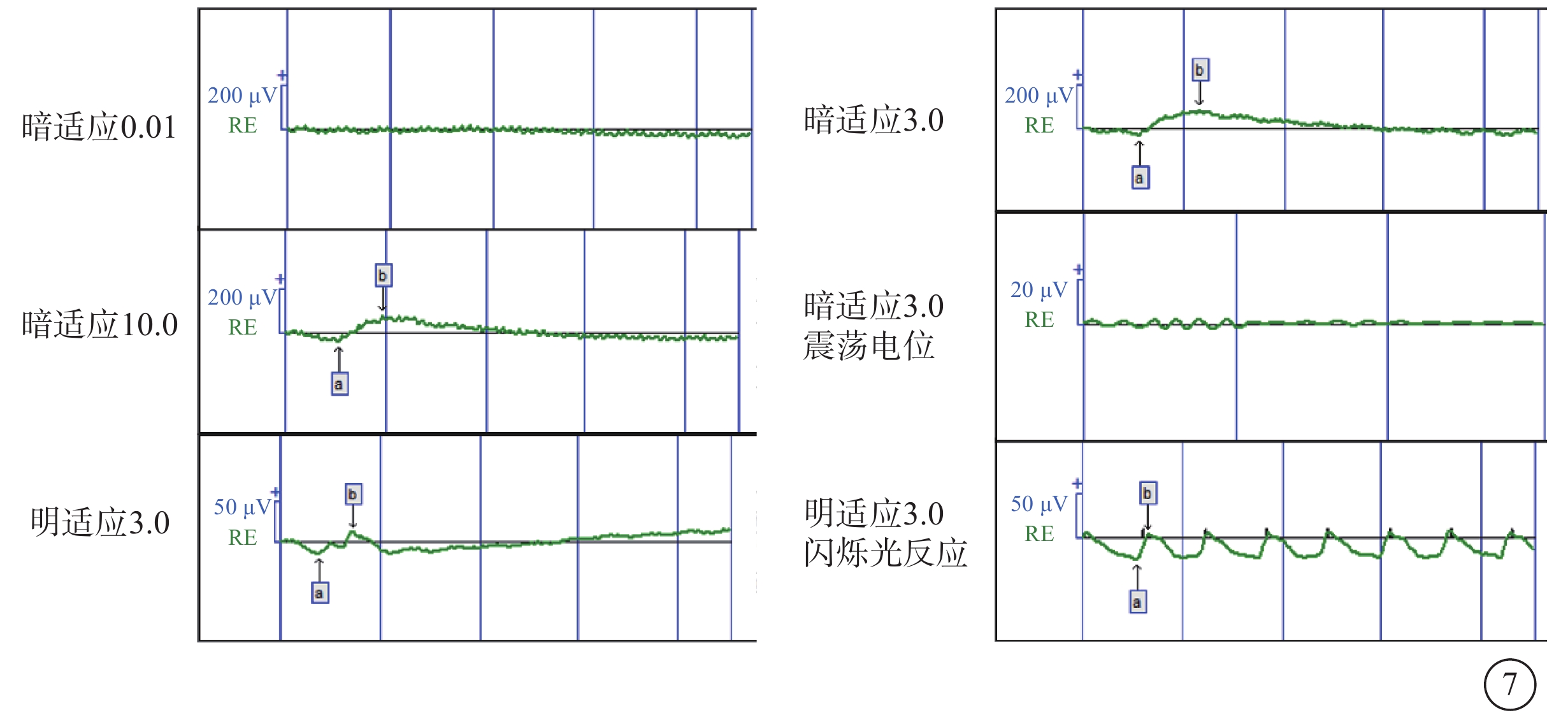 圖7
MCLMR家系先證者(Ⅱ-1)右眼全視野視網膜電圖像 視桿反應、振蕩電位,未引出波形;最大混合反應及30 Hz閃爍光刺激條件下的視錐反應a、b波振幅重度降低 MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征
圖7
MCLMR家系先證者(Ⅱ-1)右眼全視野視網膜電圖像 視桿反應、振蕩電位,未引出波形;最大混合反應及30 Hz閃爍光刺激條件下的視錐反應a、b波振幅重度降低 MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征
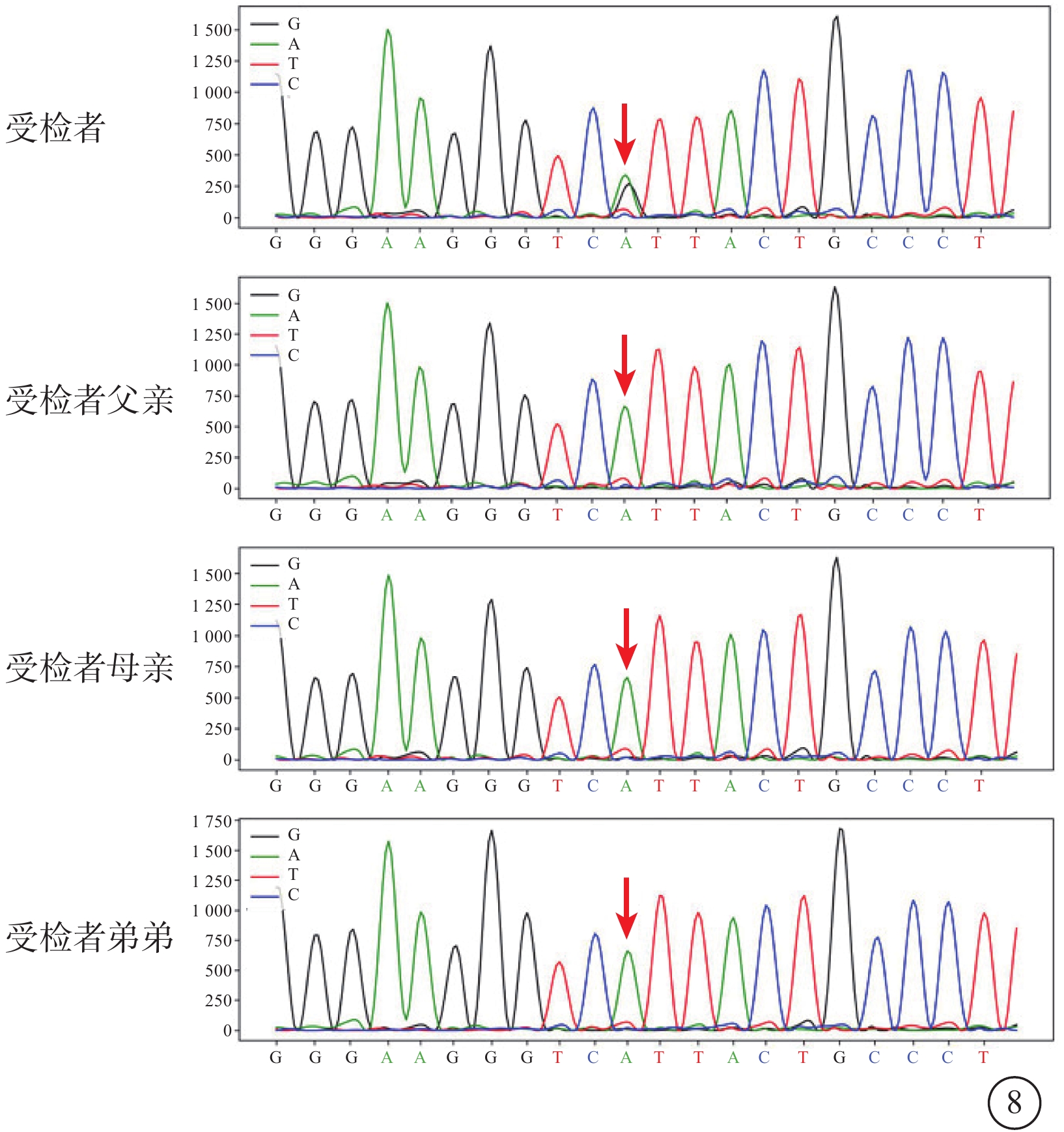
全外顯子組測序結果顯示,先證者(Ⅱ-1)KIF11基因第8號外顯子存在c.895A>G(p.Ile299Val)雜合錯義變異;其父親(Ⅰ-1)、母親(Ⅰ-2)、胞弟(Ⅱ-2)均為野生型(圖8)。ClinVar已有該變異位點致病性分析,結果為可能致病。根據ACMG指南,該變異評級為可能致病性變異(PS2+PM2_ supporting+PP3)。證據如下:該變異為經雙親驗證的新發(de novo)變異,且無家族史(PS2);在ExAC、gnomAD、1000Genome、dbSNP等數據庫中均未見收錄,屬于低頻變異(PM2_ supporting);SIFT、Polyphen2、REVEL等生物信息學預測軟件均預測有害(PP3)。
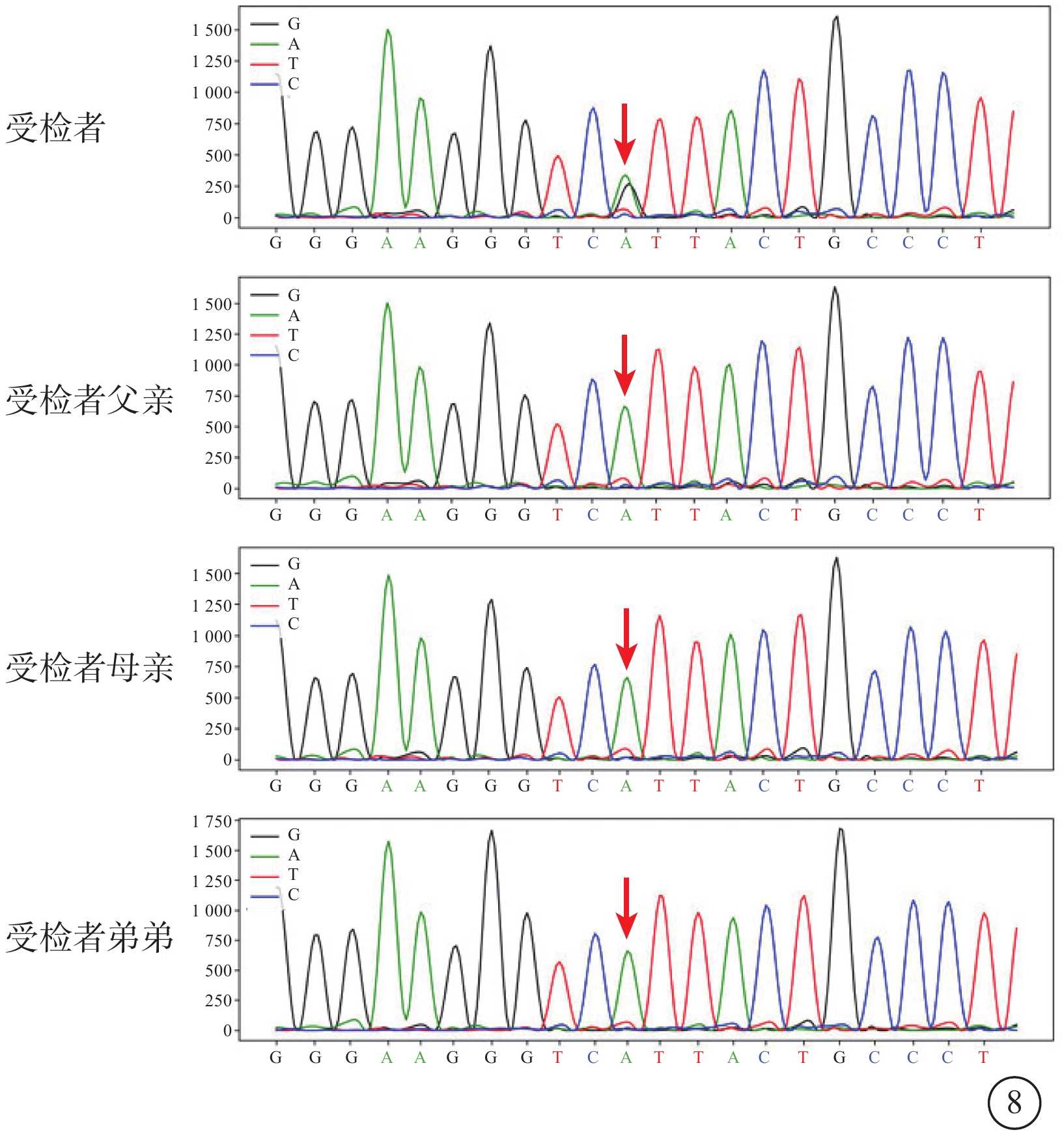 圖8
MCLMR家系基因測序圖 先證者(Ⅱ-1)KIF11基因第8號外顯子c.895A>G雜合錯義變異(紅箭);先證者父親(Ⅰ-1)、母親(Ⅰ-2)、胞弟(Ⅱ-2)均為野生型(紅箭) MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征
圖8
MCLMR家系基因測序圖 先證者(Ⅱ-1)KIF11基因第8號外顯子c.895A>G雜合錯義變異(紅箭);先證者父親(Ⅰ-1)、母親(Ⅰ-2)、胞弟(Ⅱ-2)均為野生型(紅箭) MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征
共檢索到相關文獻20篇,包含患者109例。其中,男性55例(50.5%,55/109),女性54例(49.5%,54/109)。有、無家族史分別為61、48例。小頭畸形98例(89.9%,98/109);小頜畸形4例(3.7%,4/109);顱縫早閉2例(1.8%,2/109)。淋巴水腫50例(45.9%,50/109)。其中,先天性雙側下肢淋巴水腫49例,嬰兒期逐漸減輕至痊愈;成人輕度創傷后淋巴水腫1例,成年期發現淋巴水腫并呈間歇性1例。智力發育障礙83例(76.1%,83/109),其中輕中度、重度分別為70、13例。行頭顱MRI檢查11例。其中,頭顱偏小伴腦回發育簡單化、腦體積減少、腦溝減少、腦回寬大、額葉縮短、胼胝體發育不全或變薄、小腦蚓部變小、枕大池增大分別為11、3、2、4、1、4、2、2例。
眼部異常92例(84.4%,92/109)。其中,脈絡膜視網膜病變69例(63.3%,69/109);視網膜皺襞20例(18.3%,20/109);視網膜脫離17例(15.6%,17/109);眼球震顫10例(9.2%,10/109);散光20例(18.3%,20/109);近視10例(9.2%,10/109);近視/近視散光5例(4.6%,5/109);遠視10例(9.2%,10/109);遠視/遠視散光9例(8.3%,9/109)。眼部其他臨床表現,小眼畸形、小角膜、斜視、虹膜角膜發育不良、永存原始玻璃體增生癥、雙側視神經發育不良伴萎縮性改變分別為6(5.5%,6/109)、5(4.6%,5/109)、6(5.5%,6/109)、2(1.8%,2/109)、3(2.8%,3/109)、1(0.9%,1/109)例;視網膜營養不良8例(7.3%,8/109);閉角型青光眼、白內障分別為3(2.8%,3/109)、6(5.5%,6/109)例;皮質核白內障、假性視神經缺損、持續性透明動脈1例(0.9%,1/109);黃斑區色素斑點和中心凹反光消失、牛眼樣黃斑病變、糖尿病視網膜病變分別為1(0.9%,1/109)、2(1.8%,2/109)、1(0.9%,1/109)例。黃斑區弱自發熒光3例(2.8%,3/109)。行OCT檢查4例,表現為不同程度的視網膜變薄。行ERG檢查7例,各反應視錐視桿細胞輕中度功能障礙、黃斑區視錐細胞功能嚴重障礙。
其他全身系統臨床表現,包括發育遲緩13例;癲癇6例;多動癥8例,其中需藥物控制2例;神經性耳聾5例;學習困難30例(27.5%);骨骼發育異常5例;肌張力異常4例;舌中線裂1例;胃食管反流1例;心臟異常6例;不明原因輕度雙側腎鈣質沉著癥1例;囊性纖維化1例;臍疝和尿道下裂2例;乳糜胸1例;睡眠困難2例。
3 討論
KIF11基因(OMIM:* 148760)位于染色體10q23.33區域,共22個外顯子,編碼1 056個氨基酸。在淋巴結(RPKM 9.9)、骨髓(RPKM 8.6)和其他18個組織中廣泛表達。該基因編碼屬于驅動蛋白樣蛋白家族的驅動蛋白KIF11。該基因產物在染色體定位、中心體分離和在細胞有絲分裂期間形成紡錘體等過程中起重要作用[22]。KIF11基因變異多種多樣,包括錯義變異、無義變異、移碼變異和剪接位點變異,并均勻分布在整個KIF11基因中,常見于18、8、6、3號外顯子,常見突變包括Val264Argfs*26、Tyr211Ilefs*4、Glu669Lysfs*7、Thr71Argfs*8等[11]。Birtel等[23]發現,除KIF11基因相關視網膜病變中眼部表型的顯著變異性外,還發現進行性視網膜變性,表明KIF11基因在眼部發育和維持視網膜形態和功能方面發揮重要作用。Guo等[15]發現,KIF11基因的一種新變異c.2922G>T通過影響RNA剪接而與小頭畸形產生關聯。
MCLMR是一種罕見的常染色體顯性遺傳性疾病。小頭畸形與脈絡膜視網膜病變、進行性視桿視錐細胞營養不良、視網膜皺襞等3種不同視網膜表型有關[16]。既往文獻將MCLMR分為2種不同類型,即小頭畸形、淋巴水腫和脈絡膜視網膜發育不良綜合征 (MLCRD) 和脈絡膜視網膜發育不良、小頭畸形和智力低下綜合征(CDMMR)[11]。Alzial等[24]于1980年首次描述了MLCRD;其后Feingold和Bartoshesky[25]報告了2例無血緣關系MCLMR患者。MCLMR常伴隨瞼裂上傾、耳朵外突、鼻寬且鼻頭圓潤、人中長、上唇薄、小頜畸形以及前額突出(罕見)、瞼裂下斜(部分患者)、內眥贅皮(部分患者)、上顎較窄等特殊面容[9]。小頭畸形從輕度至重度不等,顱腦MRI可見腦部較小,但通常結構正常[26-27]。智力發育障礙可從正常發育至輕度、中度、重度發育障礙。淋巴水腫在該綜合征3個核心特征中變異最大,通常為先天性雙側發病,累及下肢,足背部水腫最常見,通常發生于新生兒期,多數可自發消退[4]。但也有成人發病和間歇性淋巴水腫的報道[9]。本例患兒年齡較大,嬰兒時期有無淋巴水腫其監護人已描述不清。Jones等[9]發現,MCLMR患兒有輕度至中度學習困難。
MCLMR患者常伴發眼部病變,最典型且常見的視網膜異常為雙眼脈絡膜視網膜萎縮性病變[4, 6]。Jones等[9]研究中患者主要眼部異常為脈絡膜視網膜病變、遠視、近視、雙側視網膜皺襞和小眼癥。雙側脈絡膜視網膜病變伴黃斑區病變是該綜合征最常見的眼部表現,表現為邊界清晰的脈絡膜視網膜發育不良或萎縮、視網膜血管變細和視盤蒼白,OCT檢查可見局灶性脈絡膜視網膜萎縮[1, 6]。本家系先證者OCT表現與此一致。而Chang等[17]發現,KIF11基因相關視網膜病變主要視網膜特征是視網膜皺襞、牽拉性視網膜脫離和脈絡膜視網膜發育不良。伴脈絡膜視網膜病變患者視力下降明顯且可降至光感,嚴重者致盲。其他眼部臨床表現包括眼球震顫、近視、高度遠視、散光、角膜混濁、小角膜、白內障、晶狀體后纖維化腫塊、部分或完全性視網膜脫離、視盤異常、視網膜皺褶、視網膜色素異常、視神經萎縮、視神經發育不全,其嚴重程度在不同個體間差異較大[4, 9]。KIF11基因變異患者表現出具有不同表達性和家族內變異性的特定眼表型,既往報道中黃斑萎縮和功能障礙無一致性病變,眼底病變表現為非進行性,這些發現有助于提供準確診斷,從而改善MCLMR患者的管理和隨訪[16]。脈絡膜視網膜病變從輕度至重度不等,常為雙側發病,也有部分單眼發病患者。Shurygina等[6]發現,脈絡膜視網膜病變常見進展方式包括脈絡膜視網膜萎縮面積擴大和玻璃體視網膜牽拉增強導致視網膜皺襞或脫離。Balikova等[16]報道的一組MCLMR患者其最小分辨角對數BCVA為0.12~3.00,多數患者為遠視伴散光,本家系先證者與此一致。
由于MCLMR基因變異位置、性質不同導致臨床異質性及不完全外顯性,因此MCLMR患者脈絡膜視網膜病變初期需與急性后極部多灶性鱗狀色素上皮病變(APMPPE)、匍行性脈絡膜炎、家族性滲出性玻璃體視網膜病變(FEVR)等相鑒別。APMPPE主要發生于視網膜色素上皮(RPE)層和脈絡膜毛細血管層,典型表現為視力突然下降,眼底后極部出現多發性、黃白色、扁平“魚鱗狀”病變,急性階段以RPE和脈絡膜淺層多灶性損害為特征,病變局限于后極部,可彼此融合,病灶吸收后呈淺淡的邊界清晰的色素脫失斑。本家系先證者眼底后極部可見大片萎縮樣改變,病灶周圍可見小片狀病變,似乎視網膜病變也是從局灶性病變逐漸融合成大片病變。APMPPE與MCLMR鑒別要點是發病年齡及病變進展。匍行性脈絡膜炎是一種罕見的雙眼慢性進行性脈絡膜炎癥,主要累及脈絡膜毛細血管層和RPE,常見于中青年,可能與自身免疫、感染因素、血管病變有關。鑒別要點是MCLMR患兒頭顱小,伴或不伴有智力發育低、淋巴水腫等。基因檢測有助于明確診斷。FEVR是一種視網膜血管發育異常疾病,可導致牽拉性視網膜皺襞和脫離。早期兩者極其相似,容易誤診[21]。既往研究發現,部分攜帶KIF11基因變異的FEVR患者具有綜合征特征,如小頭畸形、智力低下、脈絡膜視網膜病變、外周視網膜血管發育不全和異常環狀血管,表明MCLMR與FEVR之間存在復雜的表型重疊[7, 16, 18-19, 28-29]。Wang等[30]在44.2%(31/70)的KIF11基因相關視網膜病變患者中檢測到脈絡膜視網膜發育不良,而攜帶其他基因變異的FEVR患者,這一比例僅為1.3%(1/70)。脈絡膜視網膜發育不良是KIF11基因相關視網膜病變的主要表型[30]。
本研究在PubMed數據庫和萬方數據庫檢索到KIF11基因變異導致脈絡膜視網膜病變共20篇文獻,加上本研究病例,共總結了109例KIF11基因變異導致的MCLMR患者的臨床特征。該病起病隱匿,臨床上易被漏診或誤診。每個臨床癥狀并非完全外顯,嚴重程度各異,導致MCLMR患者之間表型差異顯著。不同家系及個體在各項臨床特征上的表現具有不同的外顯率。小頭畸形的嚴重程度不一,目前所報道的MCLMR病例中尚未出現極其嚴重的小頭畸形情況。在這些病例中,約90%的患者表現出小頭畸形的癥狀,但也有部分患兒并未明顯表現出這一特征。回顧病例發現,幾乎所有患兒脈絡膜視網膜萎縮性病變均位于視網膜下方,而FEVR病變多起源于顳側視網膜。這種發病特點的具體原因有待通過增加更多病例來進行深入研究。患兒智力發育障礙可能與小頭畸形引起的頭顱腦回發育簡單化、視力低下等相關。視網膜脫離患兒眼底病變較重,需手術治療,考慮與視網膜皺襞引起的視網膜牽拉有關,表明這部分患兒的病情具有進展性。此外,部分患兒還存在學習困難、睡眠困難、多動癥等其他系統癥狀,這些癥狀可能與患兒智力低下、視力低下有關。
本研究結果表明,對于兒童不明原因脈絡膜視網膜萎縮性病變伴眼球震顫、視力低下者,應注意排查有無遺傳代謝性疾病,行基因檢測明確是否為致病基因變異引發的全身綜合征。本研究存在的局限性和不足:(1)鑒于MCLMR的發病率較低,本研究僅收集到1例患兒的完整臨床資料。由于患兒伴有其他系統性的合并癥,在治療過程中可能會集中于處理較為嚴重的合并癥,從而忽略其他癥狀,這容易導致漏診。(2)眼部并發癥主要表現為脈絡膜視網膜萎縮性病變,但對于這一眼底病變是先天形成還是后天逐漸發展而成,目前尚不明確。不同文獻對此的報道結果也不一致,并且由于目前缺乏有效的治療方法,導致MCLMR患兒在眼科治療中的依從性較差,這也限制了對該病眼部病變進行更深入研究的可能性。
小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征(MCLMR)是一種罕見的常染色體顯性遺傳疾病。其涉及一系列中樞神經系統和眼部發育異常的臨床表現,特征多樣且相互重疊。小頭畸形根據其嚴重程度可分為輕度至重度,常伴隨發育遲緩。此外,該病癥還通常表現為一系列典型的面部特征,包括瞼裂上斜、寬鼻梁及圓而尖的鼻尖、較長的人中區域、較薄的上唇、突出的下頜以及外翻的耳朵。脈絡膜視網膜病變是其最常見的眼部異常,視網膜褶皺、小眼畸形、近視和遠視散光也有報道,有些患者無明顯眼表型。淋巴水腫一般為先天性,常見于足背部。在GeneCard網站搜索到已知的與MCLMR相關基因包括KIF11、FZD4、CTNNB1、LRP5、PRSS23、LOC126806659、TSPAN12、CCBE1、TUBGCP4、TUBGCP6等。目前國內關于MCLMR研究較少,且缺乏詳細的眼部臨床特征描述。本研究對KIF11基因變異所致MCLMR一家系的眼部臨床表現和致病基因進行分析。現將結果報道如下。
1 對象和方法
回顧性臨床研究。本研究經河南省兒童醫院倫理委員會審批(批文號:2024-K-010);嚴格遵循《赫爾辛基宣言》原則;所有參與者及未成年受試者的監護人均獲知情并簽署書面知情同意書。
2023年9月于河南省兒童醫院臨床檢查并經基因檢測確診的MCLMR一家系先證者及3名家系成員(圖1)納入本研究。詳細詢問病史、家族史,并行最佳矯正視力(BCVA)、驗光、裂隙燈顯微鏡、眼底彩色照相、熒光素眼底血管造影(FFA)、光相干斷層掃描(OCT)、閃光視覺誘發電位(F-VEP)、全視野視網膜電圖(ERG)檢查以及眼軸長度(AL)測量。同時行頭顱+眼眶MRI檢查和基因檢測。先證者符合MCLMR臨床診斷標準[1-2]。
圖1
MCLMR家系圖 □:正常男性;○:正常女性;●:女性患者;↗:先證者;M:c.895A>G(p.Ile299Val); MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征
采集先證者及其父母、胞弟外周靜脈血樣3 ml,乙二胺四乙酸抗凝。行全外顯子組測序、染色體拷貝數變異檢測、染色體核型分析及線粒體全基因檢測(武漢良培醫學檢驗實驗室)。測序儀下機reads利用BWA(版本:0.7.16a)軟件與人類參考基因組GRCh38比對,生成的bam文件采用GATK軟件(版本:4.0.8.1)進行變異檢測分析,包括標記聚合酶鏈反應(PCR)重復序列、SNV和插入缺失標記(InDel)變異。采用Annova軟件(r版本:20200608)對變異文件vcf進行注釋。注釋數據庫為外顯子組聚集聯盟(ExAC)數據庫(http://exac.broadinstitute.org/)、dbSNP數據庫(https://www.ncbi.nlm.nih.gov/snp/)、千人基因組計劃數據庫(1000Genome,http://www.1000genomes.org/)、人群基因頻率數據庫(gnomAD,https://gnomad.broadinstitute.org/)、InterPro數據庫(https://www.ebi.ac.uk/interpro/)、ClinVar數據庫(https://www.ncbi.nlm.nih.gov/clinvar/)、人類孟德爾遺傳在線數據庫(OMIM,https://omim.org/)、人類基因變異數據庫(HGMD,http://www.hgmd.cf.ac.uk/ac/index.php)等。
變異位點篩選原則:(1)篩選出外顯子區域變異、剪切區域變異、非同義變異位點;(2)ExAC_EAS、ExAC_ALL、1000Genome、gnomAD等人群頻率數據庫中正常人變異攜帶率<1%;(3)使用SIFT、Polyphen2、LRT、MutationTaster等預測軟件對基因位點變異對其蛋白功能影響進行預測;(4)參考OMIM、HGMD、ClinVar等數據庫對致病變異位點進行評估。對檢測出的可疑致病突變,均采用Sanger測序進行驗證。根據美國醫學遺傳學與基因組學學會(ACMG)2015年發布的《序列變異解讀標準和指南》進行變異解析,并評估其臨床意義[3]。
計算機檢索美國國立醫學圖書館醫學數據庫PubMed、萬方數據庫相關文獻。中文檢索詞為“KIF11”、“小頭畸形”、“脈絡膜視網膜病變”;英文檢索詞為“KIF11”、“microcephaly”、“chorioretinopathy”。檢索時間為2012年1月1日至2023年12月31日。共檢索到相關文獻20篇,其中英文文獻、中文文獻分別為19篇[1, 2, 4-20]、1篇[21]。
2 結果
先證者(Ⅱ-1),女,8歲5個月。監護人述患兒自幼視力差、眼球不自主轉動。1年前當地醫院檢查發現屈光不正、眼底病變、眼球震顫;給予戴鏡治療、弱視訓練,視力無明顯提高。智力發育輕度遲緩,輕度學習困難,語言發育及生長發育基本正常。既往身體健康,無外傷及手術史;足月生產,生后無窒息、缺氧病史;無全身淋巴水腫病史,腦部及心臟無明顯異常。其父親(Ⅰ-1)輕度眼球震顫、斜視;母親(Ⅰ-2)、胞弟(Ⅱ-2)眼部表型未見明顯異常。
先證者(Ⅱ-1)表現為頭顱偏小、下頜較小、額部狹窄、雙耳較大且向外突出、鼻頭寬且圓、瞼裂輕度上斜以及人中較長(圖2)。其右眼、左眼祼眼視力分別為0.04、0.05,BCVA分別為 0.08(+2.25 DS/ -2.25 DC×10°)、0.1(+2.25 DS/ -2.25 DC×180°)。雙眼眼球震顫呈水平鐘擺型,無明顯中間帶及異常頭位。遠近角膜映光基本正位,間斷出現內斜視+10°,交替遮蓋內→正,雙眼及單眼眼球運動各方向到位。眼前節未見明顯異常。右眼、左眼AL分別為20.95、21.25 mm(參考值23.19~23.37 mm)。右眼角膜第1、2個主子午線(K1、K2)曲率值分別為43.50、45.77 D;左眼K1、K2曲率值分別為43.51、43.51 D。右眼、左眼角膜散光值分別為2.26、2.03 D。雙眼視盤邊界清楚,顏色淡,杯盤比約2/3;視網膜血管纖細,視網膜呈“豹紋狀”改變,黃斑中心凹反光消失,黃斑區及下方大片脈絡膜視網膜黃白色病灶,大片狀融合,呈萎縮樣改變,小片狀向周邊擴展,色素不均勻(圖3)。FFA檢查,雙眼動靜脈充盈時間正常,黃斑區及下方視網膜斑駁樣熒光著染;晚期無明顯熒光素滲漏(圖4)。OCT檢查,因患兒眼球震顫,注視功能差,可掃描范圍內黃斑中心凹形態淺,橢圓體帶缺失,層次欠清楚,視網膜全層變薄,黃斑下方脈絡膜視網膜全層明顯萎縮(圖5)。F-VEP檢查,雙眼未引出P100波波形(角膜電極,鎮靜狀態下)(圖6)。全視野ERG檢查,視桿反應:雙眼未引出a、b波波形;最大混合反應:雙眼a、b波振幅重度降低;振蕩電位:雙眼未引出波形;視錐反應:30 Hz閃爍光刺激條件下,雙眼a、b波振幅重度降低(角膜電極,鎮靜狀態下)(圖7)。頭顱+眼眶MRI檢查,左側顳部蛛網膜下腔增寬,腦實質內未見明顯異常信號,腦室系統大小形態正常,腦溝、腦回未見異常,中線結構居中,幕下小腦及腦干結構未見明顯異常;雙側眼眶未見明顯異常。
圖2
MCLMR家系先證者(Ⅱ-1)面部外觀像 2A、2B分別示側位、正位,頭顱小,下頜較小,額部窄小,雙側耳朵大并突出,鼻頭寬鼻尖圓,人中較長 MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征
圖3
MCLMR家系先證者(Ⅱ-1)雙眼彩色眼底像 3A、3B分別示右眼、左眼,雙眼視盤顏色淡,視網膜血管纖細,視網膜“豹紋狀”改變;雙眼對稱性黃斑區及下方大片視網膜脈絡膜黃白色萎縮性病灶(紅箭),小片狀病灶向周邊擴展(黃箭) MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征
圖4
MCLMR家系先證者(Ⅱ-1)雙眼FFA晚期像 4A、4B分別示右眼、左眼,雙眼與眼底病變范圍一致的視網膜斑駁樣熒光著染,無明顯熒光素滲漏 MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征;FFA:熒光素眼底血管造影
圖5
MCLMR家系先證者(Ⅱ-1)雙眼OCT像 5A、5B分別示右眼、左眼,左圖為掃描方向和部位,右圖為檢查結果。黃斑中心凹形態變淺(紅箭),全層視網膜變薄,黃斑下方脈絡膜視網膜萎縮極薄(黃箭) MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征;OCT:光相干斷層掃描
圖6
MCLMR家系先證者(Ⅱ-1)右眼視覺誘發電位圖 未引出可分析的P100波波形 MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙縮合征
圖7
MCLMR家系先證者(Ⅱ-1)右眼全視野視網膜電圖像 視桿反應、振蕩電位,未引出波形;最大混合反應及30 Hz閃爍光刺激條件下的視錐反應a、b波振幅重度降低 MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征
全外顯子組測序結果顯示,先證者(Ⅱ-1)KIF11基因第8號外顯子存在c.895A>G(p.Ile299Val)雜合錯義變異;其父親(Ⅰ-1)、母親(Ⅰ-2)、胞弟(Ⅱ-2)均為野生型(圖8)。ClinVar已有該變異位點致病性分析,結果為可能致病。根據ACMG指南,該變異評級為可能致病性變異(PS2+PM2_ supporting+PP3)。證據如下:該變異為經雙親驗證的新發(de novo)變異,且無家族史(PS2);在ExAC、gnomAD、1000Genome、dbSNP等數據庫中均未見收錄,屬于低頻變異(PM2_ supporting);SIFT、Polyphen2、REVEL等生物信息學預測軟件均預測有害(PP3)。
圖8
MCLMR家系基因測序圖 先證者(Ⅱ-1)KIF11基因第8號外顯子c.895A>G雜合錯義變異(紅箭);先證者父親(Ⅰ-1)、母親(Ⅰ-2)、胞弟(Ⅱ-2)均為野生型(紅箭) MCLMR:小頭畸形伴或不伴有脈絡膜視網膜病變、淋巴水腫或智力障礙綜合征
共檢索到相關文獻20篇,包含患者109例。其中,男性55例(50.5%,55/109),女性54例(49.5%,54/109)。有、無家族史分別為61、48例。小頭畸形98例(89.9%,98/109);小頜畸形4例(3.7%,4/109);顱縫早閉2例(1.8%,2/109)。淋巴水腫50例(45.9%,50/109)。其中,先天性雙側下肢淋巴水腫49例,嬰兒期逐漸減輕至痊愈;成人輕度創傷后淋巴水腫1例,成年期發現淋巴水腫并呈間歇性1例。智力發育障礙83例(76.1%,83/109),其中輕中度、重度分別為70、13例。行頭顱MRI檢查11例。其中,頭顱偏小伴腦回發育簡單化、腦體積減少、腦溝減少、腦回寬大、額葉縮短、胼胝體發育不全或變薄、小腦蚓部變小、枕大池增大分別為11、3、2、4、1、4、2、2例。
眼部異常92例(84.4%,92/109)。其中,脈絡膜視網膜病變69例(63.3%,69/109);視網膜皺襞20例(18.3%,20/109);視網膜脫離17例(15.6%,17/109);眼球震顫10例(9.2%,10/109);散光20例(18.3%,20/109);近視10例(9.2%,10/109);近視/近視散光5例(4.6%,5/109);遠視10例(9.2%,10/109);遠視/遠視散光9例(8.3%,9/109)。眼部其他臨床表現,小眼畸形、小角膜、斜視、虹膜角膜發育不良、永存原始玻璃體增生癥、雙側視神經發育不良伴萎縮性改變分別為6(5.5%,6/109)、5(4.6%,5/109)、6(5.5%,6/109)、2(1.8%,2/109)、3(2.8%,3/109)、1(0.9%,1/109)例;視網膜營養不良8例(7.3%,8/109);閉角型青光眼、白內障分別為3(2.8%,3/109)、6(5.5%,6/109)例;皮質核白內障、假性視神經缺損、持續性透明動脈1例(0.9%,1/109);黃斑區色素斑點和中心凹反光消失、牛眼樣黃斑病變、糖尿病視網膜病變分別為1(0.9%,1/109)、2(1.8%,2/109)、1(0.9%,1/109)例。黃斑區弱自發熒光3例(2.8%,3/109)。行OCT檢查4例,表現為不同程度的視網膜變薄。行ERG檢查7例,各反應視錐視桿細胞輕中度功能障礙、黃斑區視錐細胞功能嚴重障礙。
其他全身系統臨床表現,包括發育遲緩13例;癲癇6例;多動癥8例,其中需藥物控制2例;神經性耳聾5例;學習困難30例(27.5%);骨骼發育異常5例;肌張力異常4例;舌中線裂1例;胃食管反流1例;心臟異常6例;不明原因輕度雙側腎鈣質沉著癥1例;囊性纖維化1例;臍疝和尿道下裂2例;乳糜胸1例;睡眠困難2例。
3 討論
KIF11基因(OMIM:* 148760)位于染色體10q23.33區域,共22個外顯子,編碼1 056個氨基酸。在淋巴結(RPKM 9.9)、骨髓(RPKM 8.6)和其他18個組織中廣泛表達。該基因編碼屬于驅動蛋白樣蛋白家族的驅動蛋白KIF11。該基因產物在染色體定位、中心體分離和在細胞有絲分裂期間形成紡錘體等過程中起重要作用[22]。KIF11基因變異多種多樣,包括錯義變異、無義變異、移碼變異和剪接位點變異,并均勻分布在整個KIF11基因中,常見于18、8、6、3號外顯子,常見突變包括Val264Argfs*26、Tyr211Ilefs*4、Glu669Lysfs*7、Thr71Argfs*8等[11]。Birtel等[23]發現,除KIF11基因相關視網膜病變中眼部表型的顯著變異性外,還發現進行性視網膜變性,表明KIF11基因在眼部發育和維持視網膜形態和功能方面發揮重要作用。Guo等[15]發現,KIF11基因的一種新變異c.2922G>T通過影響RNA剪接而與小頭畸形產生關聯。
MCLMR是一種罕見的常染色體顯性遺傳性疾病。小頭畸形與脈絡膜視網膜病變、進行性視桿視錐細胞營養不良、視網膜皺襞等3種不同視網膜表型有關[16]。既往文獻將MCLMR分為2種不同類型,即小頭畸形、淋巴水腫和脈絡膜視網膜發育不良綜合征 (MLCRD) 和脈絡膜視網膜發育不良、小頭畸形和智力低下綜合征(CDMMR)[11]。Alzial等[24]于1980年首次描述了MLCRD;其后Feingold和Bartoshesky[25]報告了2例無血緣關系MCLMR患者。MCLMR常伴隨瞼裂上傾、耳朵外突、鼻寬且鼻頭圓潤、人中長、上唇薄、小頜畸形以及前額突出(罕見)、瞼裂下斜(部分患者)、內眥贅皮(部分患者)、上顎較窄等特殊面容[9]。小頭畸形從輕度至重度不等,顱腦MRI可見腦部較小,但通常結構正常[26-27]。智力發育障礙可從正常發育至輕度、中度、重度發育障礙。淋巴水腫在該綜合征3個核心特征中變異最大,通常為先天性雙側發病,累及下肢,足背部水腫最常見,通常發生于新生兒期,多數可自發消退[4]。但也有成人發病和間歇性淋巴水腫的報道[9]。本例患兒年齡較大,嬰兒時期有無淋巴水腫其監護人已描述不清。Jones等[9]發現,MCLMR患兒有輕度至中度學習困難。
MCLMR患者常伴發眼部病變,最典型且常見的視網膜異常為雙眼脈絡膜視網膜萎縮性病變[4, 6]。Jones等[9]研究中患者主要眼部異常為脈絡膜視網膜病變、遠視、近視、雙側視網膜皺襞和小眼癥。雙側脈絡膜視網膜病變伴黃斑區病變是該綜合征最常見的眼部表現,表現為邊界清晰的脈絡膜視網膜發育不良或萎縮、視網膜血管變細和視盤蒼白,OCT檢查可見局灶性脈絡膜視網膜萎縮[1, 6]。本家系先證者OCT表現與此一致。而Chang等[17]發現,KIF11基因相關視網膜病變主要視網膜特征是視網膜皺襞、牽拉性視網膜脫離和脈絡膜視網膜發育不良。伴脈絡膜視網膜病變患者視力下降明顯且可降至光感,嚴重者致盲。其他眼部臨床表現包括眼球震顫、近視、高度遠視、散光、角膜混濁、小角膜、白內障、晶狀體后纖維化腫塊、部分或完全性視網膜脫離、視盤異常、視網膜皺褶、視網膜色素異常、視神經萎縮、視神經發育不全,其嚴重程度在不同個體間差異較大[4, 9]。KIF11基因變異患者表現出具有不同表達性和家族內變異性的特定眼表型,既往報道中黃斑萎縮和功能障礙無一致性病變,眼底病變表現為非進行性,這些發現有助于提供準確診斷,從而改善MCLMR患者的管理和隨訪[16]。脈絡膜視網膜病變從輕度至重度不等,常為雙側發病,也有部分單眼發病患者。Shurygina等[6]發現,脈絡膜視網膜病變常見進展方式包括脈絡膜視網膜萎縮面積擴大和玻璃體視網膜牽拉增強導致視網膜皺襞或脫離。Balikova等[16]報道的一組MCLMR患者其最小分辨角對數BCVA為0.12~3.00,多數患者為遠視伴散光,本家系先證者與此一致。
由于MCLMR基因變異位置、性質不同導致臨床異質性及不完全外顯性,因此MCLMR患者脈絡膜視網膜病變初期需與急性后極部多灶性鱗狀色素上皮病變(APMPPE)、匍行性脈絡膜炎、家族性滲出性玻璃體視網膜病變(FEVR)等相鑒別。APMPPE主要發生于視網膜色素上皮(RPE)層和脈絡膜毛細血管層,典型表現為視力突然下降,眼底后極部出現多發性、黃白色、扁平“魚鱗狀”病變,急性階段以RPE和脈絡膜淺層多灶性損害為特征,病變局限于后極部,可彼此融合,病灶吸收后呈淺淡的邊界清晰的色素脫失斑。本家系先證者眼底后極部可見大片萎縮樣改變,病灶周圍可見小片狀病變,似乎視網膜病變也是從局灶性病變逐漸融合成大片病變。APMPPE與MCLMR鑒別要點是發病年齡及病變進展。匍行性脈絡膜炎是一種罕見的雙眼慢性進行性脈絡膜炎癥,主要累及脈絡膜毛細血管層和RPE,常見于中青年,可能與自身免疫、感染因素、血管病變有關。鑒別要點是MCLMR患兒頭顱小,伴或不伴有智力發育低、淋巴水腫等。基因檢測有助于明確診斷。FEVR是一種視網膜血管發育異常疾病,可導致牽拉性視網膜皺襞和脫離。早期兩者極其相似,容易誤診[21]。既往研究發現,部分攜帶KIF11基因變異的FEVR患者具有綜合征特征,如小頭畸形、智力低下、脈絡膜視網膜病變、外周視網膜血管發育不全和異常環狀血管,表明MCLMR與FEVR之間存在復雜的表型重疊[7, 16, 18-19, 28-29]。Wang等[30]在44.2%(31/70)的KIF11基因相關視網膜病變患者中檢測到脈絡膜視網膜發育不良,而攜帶其他基因變異的FEVR患者,這一比例僅為1.3%(1/70)。脈絡膜視網膜發育不良是KIF11基因相關視網膜病變的主要表型[30]。
本研究在PubMed數據庫和萬方數據庫檢索到KIF11基因變異導致脈絡膜視網膜病變共20篇文獻,加上本研究病例,共總結了109例KIF11基因變異導致的MCLMR患者的臨床特征。該病起病隱匿,臨床上易被漏診或誤診。每個臨床癥狀并非完全外顯,嚴重程度各異,導致MCLMR患者之間表型差異顯著。不同家系及個體在各項臨床特征上的表現具有不同的外顯率。小頭畸形的嚴重程度不一,目前所報道的MCLMR病例中尚未出現極其嚴重的小頭畸形情況。在這些病例中,約90%的患者表現出小頭畸形的癥狀,但也有部分患兒并未明顯表現出這一特征。回顧病例發現,幾乎所有患兒脈絡膜視網膜萎縮性病變均位于視網膜下方,而FEVR病變多起源于顳側視網膜。這種發病特點的具體原因有待通過增加更多病例來進行深入研究。患兒智力發育障礙可能與小頭畸形引起的頭顱腦回發育簡單化、視力低下等相關。視網膜脫離患兒眼底病變較重,需手術治療,考慮與視網膜皺襞引起的視網膜牽拉有關,表明這部分患兒的病情具有進展性。此外,部分患兒還存在學習困難、睡眠困難、多動癥等其他系統癥狀,這些癥狀可能與患兒智力低下、視力低下有關。
本研究結果表明,對于兒童不明原因脈絡膜視網膜萎縮性病變伴眼球震顫、視力低下者,應注意排查有無遺傳代謝性疾病,行基因檢測明確是否為致病基因變異引發的全身綜合征。本研究存在的局限性和不足:(1)鑒于MCLMR的發病率較低,本研究僅收集到1例患兒的完整臨床資料。由于患兒伴有其他系統性的合并癥,在治療過程中可能會集中于處理較為嚴重的合并癥,從而忽略其他癥狀,這容易導致漏診。(2)眼部并發癥主要表現為脈絡膜視網膜萎縮性病變,但對于這一眼底病變是先天形成還是后天逐漸發展而成,目前尚不明確。不同文獻對此的報道結果也不一致,并且由于目前缺乏有效的治療方法,導致MCLMR患兒在眼科治療中的依從性較差,這也限制了對該病眼部病變進行更深入研究的可能性。