藥物組合的協同作用在提高藥物療效或者減輕藥物毒副作用方面非常重要。然而由于藥物之間復雜的作用機制,通過試驗篩選新的藥物組合需要巨大的成本。眾所周知,運用計算模型的虛擬篩選,可以有效降低試驗成本。最近,國外學者運用深度學習模型 DeepSynergy,成功預測了新的藥物組合在癌癥細胞系上的協同作用值。在本研究中,針對 DeepSynergy 采用 two-stage 方法和輸入特征單一的問題,我們提出了一種新的端到端的深度學習模型 MulinputSynergy。該模型通過整合癌癥細胞的基因表達、基因突變、基因拷貝數特征和抗癌藥物化學特征來預測藥物組合的協同作用值。為了解決細胞系某些特征維度數高的問題,我們使用卷積神經網絡對基因特征降維。實驗結果表明,本文提出的模型優于 DeepSynergy 深度學習模型,均方誤差從 197 下降到 176,平均絕對誤差從 9.48 下降到 8.77,決定系數從 0.53 提升到 0.58。研究結果表明本文模型能從多種特征中學習抗癌藥物與細胞系的潛在關系,并且快速準確地定位有效的藥物組合。
引用本文: 陳希, 秦玉芳, 陳明, 張重陽. 基于多輸入神經網絡的藥物組合協同作用預測. 生物醫學工程學雜志, 2020, 37(4): 676-682, 691. doi: 10.7507/1001-5515.201907049 復制
版權信息: ?四川大學華西醫院華西期刊社《生物醫學工程學雜志》版權所有,未經授權不得轉載、改編
引言
基于基因組、蛋白質組的高通量篩選方法和藥物設計雖然取得了巨大的進展,但過去十年批準的藥物數量仍遠小于實際需求。例如,目前全球各國已批準上市的抗癌藥物僅有 130~150 種。因為可用藥物少,耐藥性成為阻礙癌癥化療成功的主要原因之一[1-2],因而組合靶向治療成為解決這個難題的一種重要方法[3]。充分了解藥物組合之間的協同/拮抗機制以及化療的作用機制[4],對于快速方便地尋找針對特定癌癥類型的藥物組合具有十分重要的意義。
隨著高通量篩選和虛擬篩選成為現代藥物研究方法的重要組成部分[5],計算機仿真實驗在生物醫學科學中的應用越來越廣泛[6],利用計算模型尋找潛在的化合物組合成為一種重要的解決方法。例如,He 等[7]提出一種用于高通量篩選癌癥細胞藥物組合效應的實驗計算管道,提升了識別潛在藥物組合的速度。但是因為傳統計算模型通常沒有足夠的模擬能力,所以很難確定有效的藥物組合[8]。機器學習模型經過十幾年的巨大發展,利用其超強的學習能力尋找潛在有效的化合物組合變得至關重要[9]。深度神經網絡模型作為當下最先進的機器學習模型,在圖像處理、語言識別、自動駕駛等領域屢創記錄。由于其處理大規模數據的能力,在藥物設計領域得到了初步的應用。例如,Aliper 等[10]首次提出基于轉錄組數據的深度學習神經網絡,用于識別不同生物系統和不同條件下的多種藥物的藥理特性。Chang 等[11]首次將深度學習模型應用于驗證藥物再利用的可行性。Feng 等[12]設計了一個基于深度神經網絡的框架——蛋白與藥物分子相互作用預測(protein and drug molecular interaction prediction,PADME)來預測化合物與蛋白質之間的相互作用強度。Zong 等[13]提出基于相似性的藥物靶點預測方法,利用深度學習方法 DeepWalk 計算頂點相似性。Preuer 等[14]提出了基于神經網絡的 DeepSynergy 模型,成功預測了抗癌藥物組合。
細胞系基因組數據的突出特點是特征維度高,降維難度大。DeepSynergy 模型選擇的方法是使用第三方工具 Factor Analysis for Robust Microarray Summarization(FARMS)[15]進行降維。該工具是基于貝葉斯極大后驗法的因子分析模型,用于提取從 Affymetrix 芯片獲得的基因表達數據,但是作為無監督學習方法,在提取基因表達的過程中會引入誤差,而且不如神經網絡的學習能力強。此外,DeepSynergy 模型僅考慮了癌癥細胞系的基因表達特征,而忽略了與癌癥密切相關的基因拷貝數變異和基因突變等特征。針對上述問題,本文提出一種多輸入的端到端的深度學習模型——MulinputSynergy。該模型整合抗癌藥物的化學特征和癌癥細胞系的基因表達特征、基因突變特征、基因拷貝數特征,利用卷積層進行降維,最后通過殘差前饋神經網絡進行回歸預測。這種端到端的 one-stage 模型,數據流從輸入到輸出只通過一個模型完成,使得深度學習模型在學習預測的過程中有監督地降維,預測性能得到一定的提高。
1 數據及預處理
1.1 高通量藥物組合篩選數據
高通量藥物組合篩選數據集從 O’Neil 等[16]的論文下載(http://mct.aacrjournals.org/content/molcanther/suppl/2016/03/16/1535-7163.MCT-15-0843.DC1/156849_1_supp_1_w2lrww.xls)。具體來說,該數據集涵蓋了 583 種不同的藥物組合,每種組合都對來自 7 種不同組織類型的 39 個人類癌癥細胞系進行了測試。583 種藥物組合來源于 38 種抗癌藥物(14 個試驗藥物,24 個批準藥物)。按照 O’Neil 等的實驗,這 38 種抗癌藥物中的 22 種藥物(14 個實驗藥物和 8 個批準藥物)兩兩配對構成 231 種藥物組合,剩下的 16 種批準藥物與這 22 種藥物兩兩配對構成 352 種藥物組合。單個藥物有 4 種藥物濃度,2 種藥物形成 4×4 共計 16 個藥物組合劑量方案。每個藥物組合劑量方案重復實驗 4 次,在 96 h 后測量對應于對照組的細胞生長率(0 代表完全抑制,1 代表無抑制作用)。
1.2 藥物組合的協同作用值
藥物組合協同作用值來源于 Preuer 等[14]論文的補充材料(http://www.bioinf.jku.at/software/DeepSynergy/labels.csv)。作者使用 Combenefit[17]批處理程序來計算 Loewe Additivity 模型[18]的值來量化藥物組合協同作用程度。Loewe Additivity 模型使用模型期望的藥物組合反應表面與實際的藥物組合反應表面的偏差來量化藥物組合協同作用程度。按照 O’Neil 等的實驗,每個藥物組合共有 16 個劑量方案即有 16 個細胞生長率,藥物組合反應表面就是 16 個細胞生長率所構成的 4×4 矩陣。經過 Loewe Additivity 模型計算后,每個樣本有 4 個值,即藥物 A、藥物 B、細胞系、協同作用值。
每個樣本(藥物 A、藥物 B、細胞系、協同作用值)的不同藥物順序本質上是同一個樣本。因此,改變每個樣本的藥物組合順序可達到擴充樣本的目的。通過樣本擴充,我們的樣本數是原來的 2 倍。
1.3 化合物描述符和細胞系特征
癌癥細胞系特征有很多,例如基因表達、基因突變、基因拷貝數、microRNA 表達、DNA 甲基化、蛋白質豐度等,目前還沒有一種突出的細胞系基因組學特征能夠代表細胞系。為了使模型的預測性能盡可能較好,可以考慮不同特征的組合,以便計算模型能夠從不同方面學習潛在的聯系。考慮到數據庫之間異質性導致的數據整合困難,我們最終選擇了基因表達、基因突變、基因拷貝數這 3 種特征量化癌癥細胞系。
1.3.1 癌癥細胞系基因組數據
癌癥細胞系的基因表達譜數據來源于 ArrayExpress 數據庫(編號:E-MTAB-3610)[19],該表達譜在 Affymetrix 人類基因組 U219 平臺上測序。癌癥細胞系的基因突變數據來源于 CCLE(Cancer Cell Line Encyclopedia),下載地址為 https://data.broadinstitute.org/ccle/CCLE_DepMap_18q3_maf_20180718.txt。癌癥細胞系的拷貝數變異數據來源于 CCLP(COSMIC Cell Lines Project),下載地址為 https://cancer.sanger.ac.uk/cell_lines/download。
1.3.2 藥物的化學特征
我們使用軟件 alvaDesc[20](https://chm.kode-solutions.net/products_alvadesc.php)來計算化合物的分子描述符和指紋。alvaDesc 是計算各種分子描述符和指紋的新一代工具,共計算了 5 305 個分子描述符,包括功能組、片段計數、藥效團等。
1.4 數據預處理
高通量藥物組合篩選數據集所測試的 39 個癌癥細胞系并非完全包含在 ArrayExpress、CCLE 和 CCLP 中。Preuer 等在 DeepSynergy 中,對于那些缺失的細胞系采用父代或子代細胞系乃至同組織細胞系代替。例如,對于 39 個癌癥細胞系中的 EFM-192B,采用數據庫存在的 EFM-192A 代替。為了實驗的嚴謹性,我們使用藥物組合篩選數據集和 ArrayExpress、CCLE 和 CCLP 數據庫最大交集的 31 個細胞系,剔除了 8 個細胞系(CAOV3、COLO320DM、DLD1、EFM192B、NCIH23、OVCAR3、UWB1289BRCA1、ZR751)。
為了使訓練數據的數據格式符合深度學習模型的規范且利于模型訓練,我們對訓練數據進行預處理。CCLE 的癌癥細胞系基因突變數據是文本格式,對其使用 one-hot 編碼,1 代表這個基因發生突變,0 則代表未突變。同樣,CCLP 的癌癥細胞系基因拷貝數數據也是文本格式,對其進行數值編碼,基因拷貝數作為該基因的特征值。歸一化藥物化學特征和癌癥細胞系的基因表達、基因突變、基因拷貝數特征,使特征縮放在(0,1)區間。最后對照高通量篩選數據集的每條數據項(藥物 A-藥物 B-癌癥細胞系-藥物組合協同作用值),形成藥物化學特征和癌癥細胞系的基因表達特征、基因突變特征、基因拷貝數特征 4 個特征張量。
2 模型
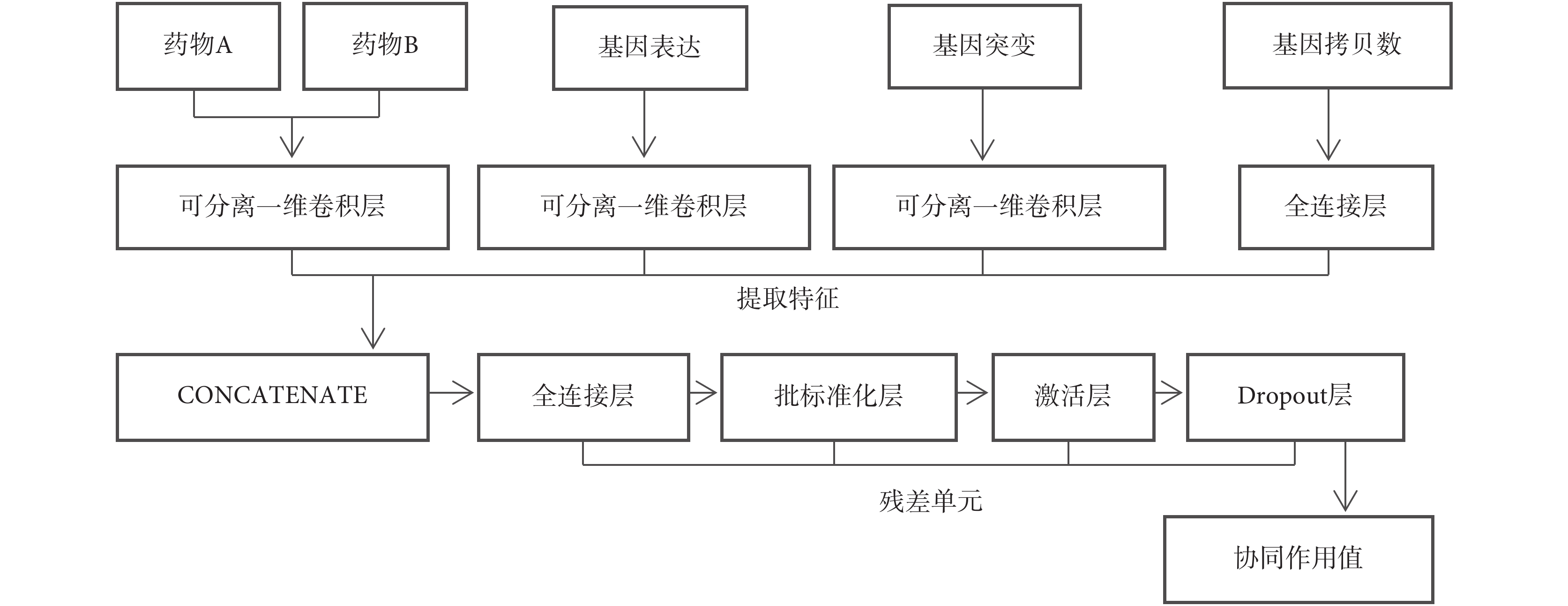
本文提出的深度學習模型以細胞系和藥物組合預處理之后的特征為輸入,以協同作用值為輸出。模型的結構如圖 1 所示。該結構由兩個層次組成,第一個層次用于降維、提取特征,第二個層次用于預測協同值。在第一個層次中,基因表達、基因突變、基因拷貝數和藥物化學特征的特征維度從幾千到幾十萬,我們針對這 4 個特征設計了各自的子模型,用來編碼新的特征向量。所有編碼的特征連接起來形成新的特征向量,構成下一個層次的輸入。第二個層次子模型是一個深度殘差前饋神經網絡模型,比特征編碼網絡具有更多的層,且相鄰兩個殘差單元之間進行殘差連接,最后輸出協同作用值的預測。
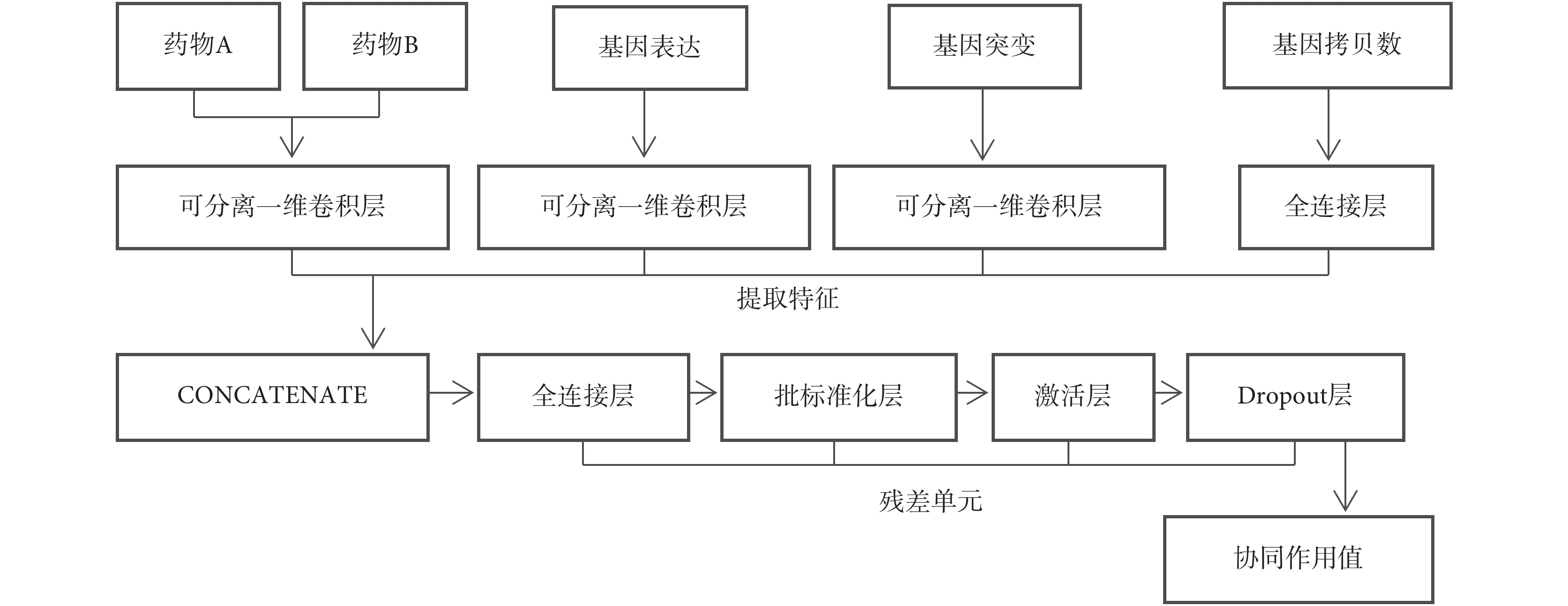 圖1
MulinputSynergy 的模型結構
Figure1.
The architecture of MulinputSynergy
圖1
MulinputSynergy 的模型結構
Figure1.
The architecture of MulinputSynergy
為了構建一個性能優異的端到端的深度學習模型,我們需要對模型進行調參實驗。采用卷積層提取特征時,我們對比一維卷積和二維卷積,發現使用一維卷積效果比二維卷積效果略好。對比圖像領域,我們不使用池化層來減少特征維數,而是減小卷積層通道數來達到提取特征的目的。
2.1 模型建模
2.1.1 藥物特征子模型
在不同的藥物組合中,同一藥物提取的特征應該是相同的。例如,藥物組合(ABT-888,AZD1775)和藥物組合(AZD1775,BEZ-235),其中藥物 AZD1775 提取的特征應該是相同的。所以在神經網絡模型中,訓練 2 個不同的藥物化學特征提取模型權重是不可取的。因此,在 MulinputSynergy 模型中,我們共享藥物 A 特征子模型和藥物 B 特征子模型的神經網絡權重,盡可能減少藥物順序對于實驗結果的影響。
每個藥物的特征向量共有 5 305 維,我們先將其末尾填充 24 個 0 值,然后將其變形為 73*73 的特征矩陣。我們使用卷積神經網絡提取藥物特征,該子模型有 3 層一維卷積層,卷積層的卷積核通道數為[36, 18, 10],每層的卷積核大小為 1,卷積步長也為 1,然后連接一個比率為 0.5 的 dropout 層,最終藥物的特征向量有 730 維。
2.1.2 基因表達特征子模型
我們使用卷積神經網絡提取基因表達特征。來源于 ArrayExpress 數據庫的基因表達譜特征有 553 536 維,將其變形為 744*744 的特征矩陣。該子模型有 4 層一維卷積層,卷積層的卷積核通道數為[186, 46, 11, 3],每層的卷積核大小為 1,卷積步長也為 1,然后連接一個比率為 0.5 的 dropout 層,最終基因表達特征的特征向量有 2 232 維。
2.1.3 基因突變特征子模型
基因突變文本經過 one-hot 編碼,每個細胞系基因突變的特征向量有 13 294 個維度。我們使用卷積神經網絡提取基因突變特征,首先將其末尾填充 46 個 0 值,然后將其變形為 114*115 的特征矩陣。該子模型有 2 層一維卷積層,卷積層的卷積核通道數為[60, 10],每層的卷積核大小為 1,卷積步長也為 1,然后連接一個比率為 0.5 的 dropout 層,最終基因突變特征的特征向量有 1 140 維。
2.1.4 基因拷貝數特征子模型
同樣的,基因拷貝數變異特征經過編碼,每個細胞系基因拷貝數的特征向量有 2 682 個維度。因為特征維度較小,我們使用前饋神經網絡提取基因拷貝數特征。該子模型有 3 層全連接層,全連接層的神經元數為[512, 512, 512],然后連接一個比率為 0.5 的 dropout 層,最終基因拷貝數特征的特征向量有 512 維。
2.1.5 深度殘差神經網絡回歸模型
對上述 4 個輸入模型輸出的 4 個張量進行連接合并,將組合的張量通過深度殘差神經網絡回歸模型進行回歸預測。深度殘差神經網絡回歸模型有 5 個殘差單元,每個殘差單元的神經元數為 2 048,然后連接一個比率為 0.5 的 dropout 層,隱藏層使用 relu 激活函數,輸出層使用 linear 激活函數。優化器使用 adam 函數來最小化均方誤差損失函數。使用回調函數監控測試集上的損失值,保存損失值最小的模型。優化器的學習率設置為 10?5,最終模型輸出協同作用預測值。
2.2 模型比較
我們將本文模型 MulinputSynergy 與 Preuer 等[14]提出的深度學習模型 DeepSynergy 進行比較。為了使 2 種模型具有可比性,DeepSynergy 也在剔除 8 個細胞系的數據集上運行。MulinputSynergy 與 DeepSynergy 都運行 200 輪。
2.3 交叉驗證
對于模型,我們使用 5 折交叉驗證方案來評估模型的性能。為了確保模型的魯棒性,每種藥物組合只出現在 5 折中的一折里。我們使用其中一折的數據作為驗證集來優化模型參數,即驗證模型在未知的藥物組合上的性能。
3 結果
3.1 模型性能評估
我們比較兩種模型對未知藥物組合協同值的預測能力。由于常用的均方誤差(mean squared error,MSE)和平均絕對誤差(mean absolute error,MAE)依賴于數據集預測值的大小,為了進一步描述模型的預測性能,我們還采用了其他回歸性能指標和分類性能指標。
3.1.1 回歸性能指標
我們采用 MSE、MAE、可釋方差值(explained vriance score,EVS)、皮爾遜相關系數(Pearson correlation coefficient,以下簡稱 Pearson)和決定系數(coefficient of determination,R2)來衡量模型的回歸性能。MSE 即預測值與真實值差的平方,這是回歸問題常用的性能指標。MAE 是預測值與真實值之差的絕對值,優點是對異常值有更強的魯棒性。皮爾遜相關系數用于衡量真實值和預測值之間的相關程度,其值范圍在[?1, 1]之間,絕對值越接近 1 相關性越強。R2介于 0~1 之間,越接近 1,回歸擬合效果越好。可釋方差值取值范圍是[0, 1],越接近于 1 說明自變量越能解釋因變量的方差變化,值越小則說明效果越差。表 1 展示了兩種模型在測試集上的回歸性能對比。
對比兩種模型的回歸性能指標,本文模型的均方誤差從 197.14 下降到 176.69,相對提升 10.4%,平均絕對誤差從 9.48 下降到 8.77,相對提升 7.5%。DeepSynergy 的可釋方差值、皮爾遜相關系數、R2決定系數分別為 0.53、0.73、0.53,而 MulinputSynergy 分別為 0.58、0.76、0.57,相對改進了 9.4%、4.1%、7.5%。因此,在回歸性能方面,本文的 MulinputSynergy 模型得到了提升。
3.1.2 分類性能指標
我們采用準確率(accuracy,ACC)、查準率(precision,PREC)、受試者工作特征(receiver operating characteristic,ROC)曲線下的面積(area under curve,AUC)(ROC AUC)、PR(precision-recall)曲線下的面積(PR AUC)、Kappa 系數(Cohen’s Kappa)、F1 分數(F1 score,F1)來衡量模型的分類性能。準確率常用來判斷分類器分類結果的好壞。受試者工作特征曲線下的面積、PR 曲線下的面積常用來衡量分類器的泛化能力。Kappa 系數用于一致性檢驗,也可以用于衡量分類精度。查準率是針對模型預測結果而言的,表示的是預測為正的樣例中真正的正樣例的比率。F1 分數是統計學中用來衡量二分類模型精確度的一種指標,同時兼顧了分類模型的精確率和召回率。由于訓練數據中存在大量的非協同非拮抗藥物組合(協同值在 0 左右),因此,選擇一個適當的閾值來將預測的協同作用值進行二值化是很重要的。我們將二分類閾值設置為 30,協同作用值高于 30 的藥物組合被認為是正類,低于 30 的藥物組合被認為是拮抗的和低協同的,這點與 DeepSynergy 模型相同。表 2 展示了兩種模型在測試集上的分類性能對比。
對比兩種模型的分類性能指標,本文模型的查準率從 0.57 上升到 0.63,相對提升 10.5%,PR 曲線下的面積從 0.53 上升到 0.59,相對提升 10.2%。DeepSynergy 的受試者工作特征曲線下的面積、Kappa 系數、F1 分數分別為 0.93、0.47、0.50,而 MulinputSynergy 分別為 0.94、0.50、0.52,相對改進了 1.1%、6.4%、4.0%。在準確率方面,兩個模型的性能持平。因此,在分類性能方面,本文的 MulinputSynergy 模型亦得到提升。
3.2 細胞系/藥物分組分析
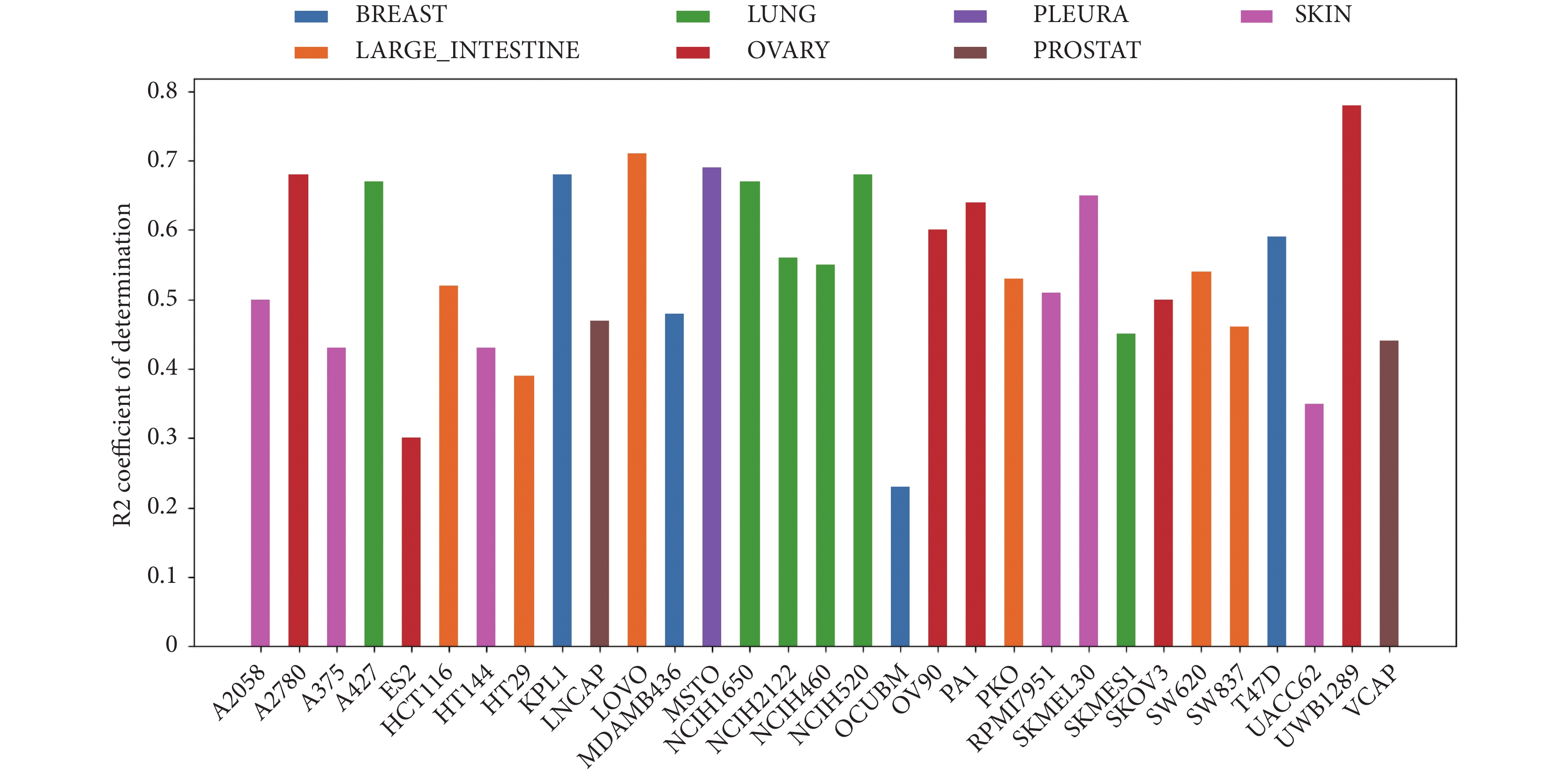
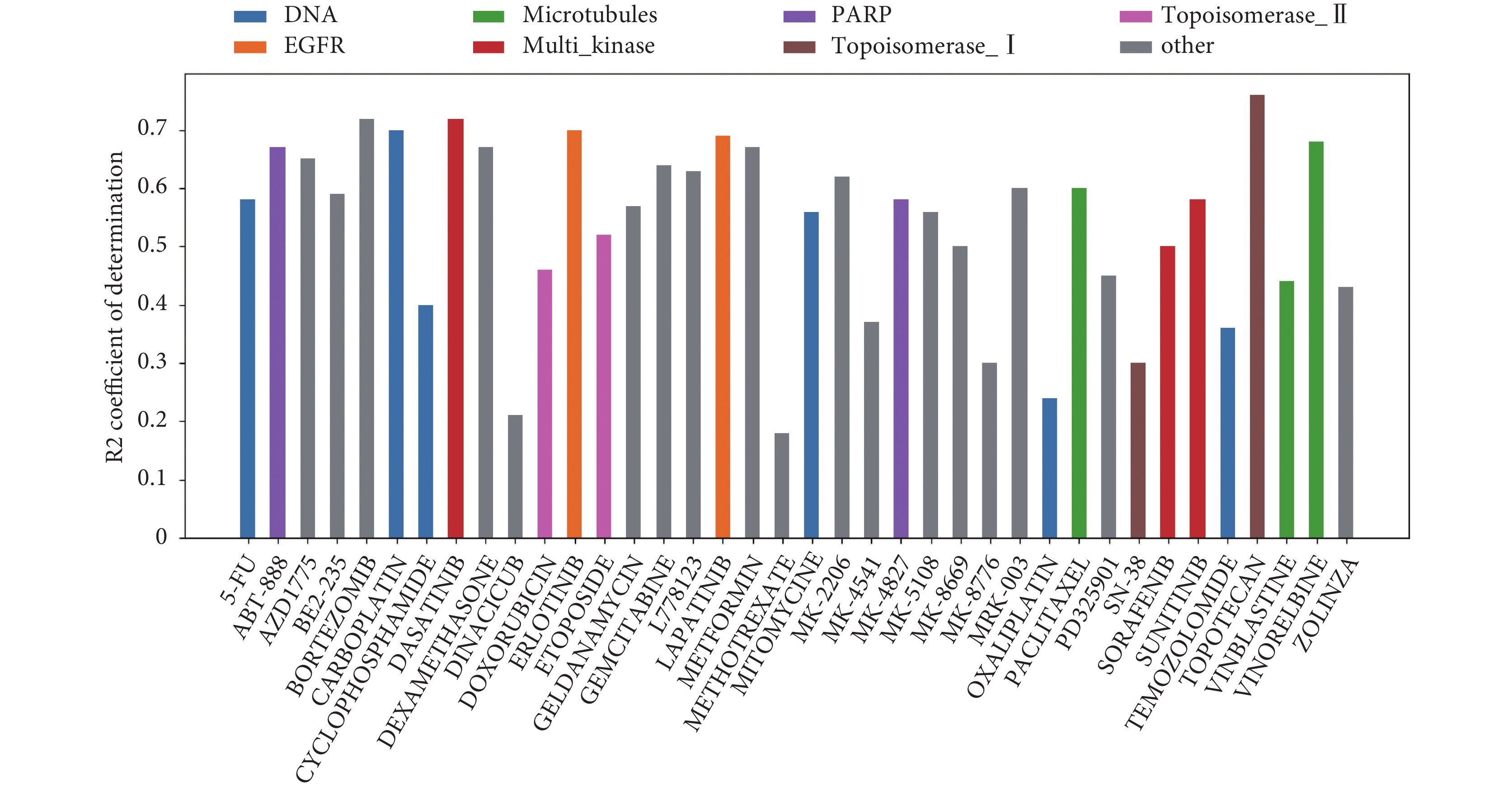
此外,我們通過分析每個細胞系/每個藥物的 R2決定系數來分析它們之間的協同性能。圖 2 和圖 3 分別顯示了每個藥物和每個細胞系的結果。圖 2 中,在 x 軸上顯示細胞系的名稱,y 軸上顯示 R2 決定系數大小,條形圖的顏色與細胞系來源組織相對應。圖 3 中,在 x 軸上顯示藥物的名稱,y 軸上顯示 R2 決定系數大小,條形圖的顏色與藥物靶向目標相對應。
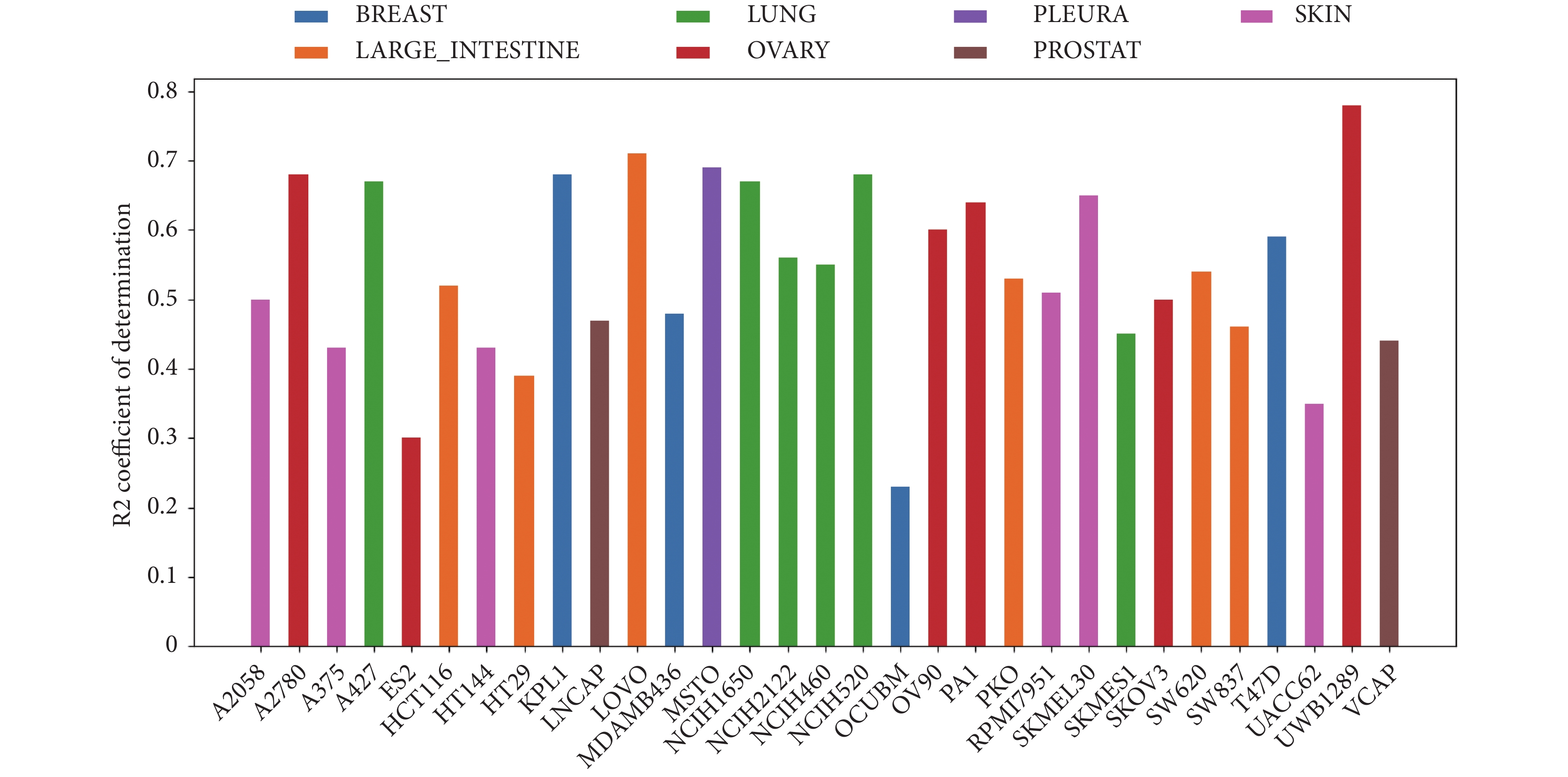 圖2
每個細胞系預測協同值的決定系數
Figure2.
Coefficient of determination of predicted synergy scores for each cell line
圖2
每個細胞系預測協同值的決定系數
Figure2.
Coefficient of determination of predicted synergy scores for each cell line
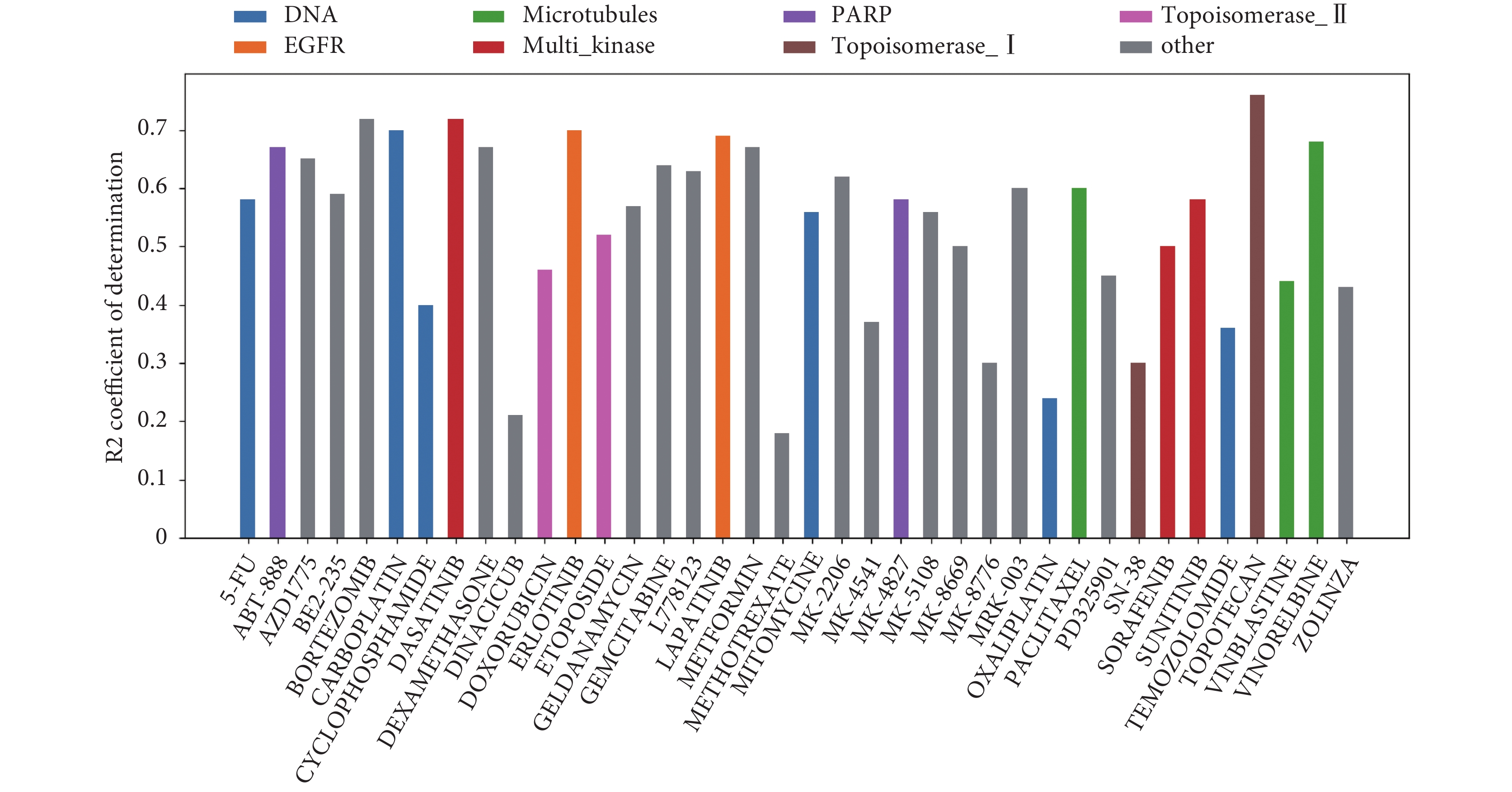 圖3
每個藥物預測協同值的決定系數
Figure3.
Coefficient of determination of predicted synergy scores for each drug
圖3
每個藥物預測協同值的決定系數
Figure3.
Coefficient of determination of predicted synergy scores for each drug
如圖 2 所示,對于每個細胞系,R2決定系數的變化范圍從 0.23 到 0.78,有 11 個細胞系的 R2決定系數小于 0.5,大約 65% 的細胞系的系數大于 0.5,有 11 個細胞系的系數大于 0.6,其中 LOVO 大腸癌和 UWB1289 卵巢癌細胞系的 R2決定系數大于 0.7,表現出較好的性能。從圖中可以看出,不同細胞系之間的預測精度差異較大,這可能是由于不同細胞系的生物學機制不同造成的。
如圖 3 所示,對于每種抗癌藥物,R2決定系數的變化范圍從 0.18 到 0.76,有 12 個藥物的 R2決定系數小于 0.5,約 68% 的藥物的系數大于 0.5,約 42% 的藥物的系數大于 0.6,有 5 個藥物(BORTEZOMIB、CARBOPLATIN、DASATINIB、ERLOTINIB、TOPOTECAN)的系數大于 0.7。在圖 3 中,沒有觀察到藥物靶向目標和 R2決定系數相關性之間的明顯聯系。因此,靶向目標特定的機制并不能解釋性能差異。從圖中可以看出,不同藥物之間的預測精度差異較大,這可能是由于不同的藥物組合對特定細胞系的協同/拮抗作用機制不同造成的。
3.3 虛擬篩選分析
計算模型的一個重要應用是高通量虛擬篩選,一個有效的計算模型可以幫助降低篩選成本。首先,在測試集上,我們按照協同值從大到小的順序對每個細胞系上測試的藥物組合進行有效性排序。然后定義了 TopN 指標來描述模型性能。TopN 定義如下:預測的前 N 個有效藥物組合中包含真實的最有效藥物組合的細胞系數/細胞系總數,N 取大于 1 的自然數且不大于待測藥物組合數。TopN 指標介于 0~1 之間,且越大越好。我們分別取 N 為 1、2、3、4、5、20 和 40,計算它們在 MulinputSynergy 和 DeepSynergy 模型下的值,然后進行對比,如表 3 所示。
我們統計出測試集上共有 116 個藥物組合。依據 MulinputSynergy 的 Top20 指標 0.95,對于每個細胞系有 95% 的概率,只要試驗模型預測的排名前 20 的藥物組合就能得出真實的最佳藥物組合,對比原本需要試驗 116 個藥物組合,減少 82.8% 的待測藥物組合。對比兩種模型的 TopN 指標,本文模型的 Top2 從 0.41 上升到 0.49,相對提升 19.5%,Top3 從 0.50 上升到 0.58,相對提升 16.0%,以此類推,Top4 相對提升 14.6%,Top5 相對提升 19.0%,Top20 相對提升 8.0%,Top40 相對提升 2.1%。
3.4 誤差分析
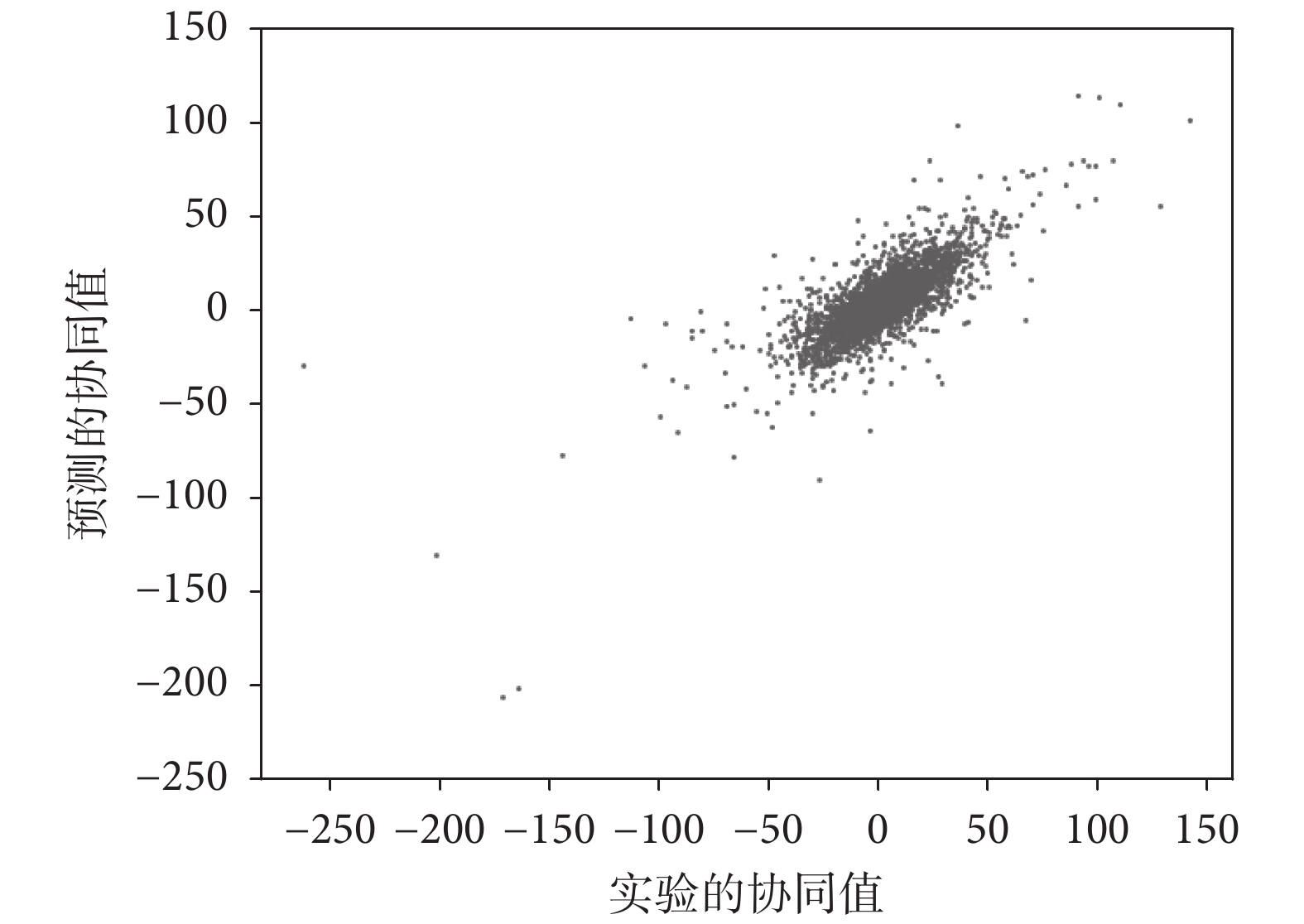
圖 4 給出了協同值的真實值和預測值之間的散點圖。我們計算每個樣本點協同值的實驗值與預測值的絕對誤差,發現約 97% 的樣本點預測值與真實值之差小于 30,約 70% 的樣本點小于 10,約 42% 的樣本點小于 5,約 27% 的樣本點小于 3。總體來說,大部分的預測效果是較好的。
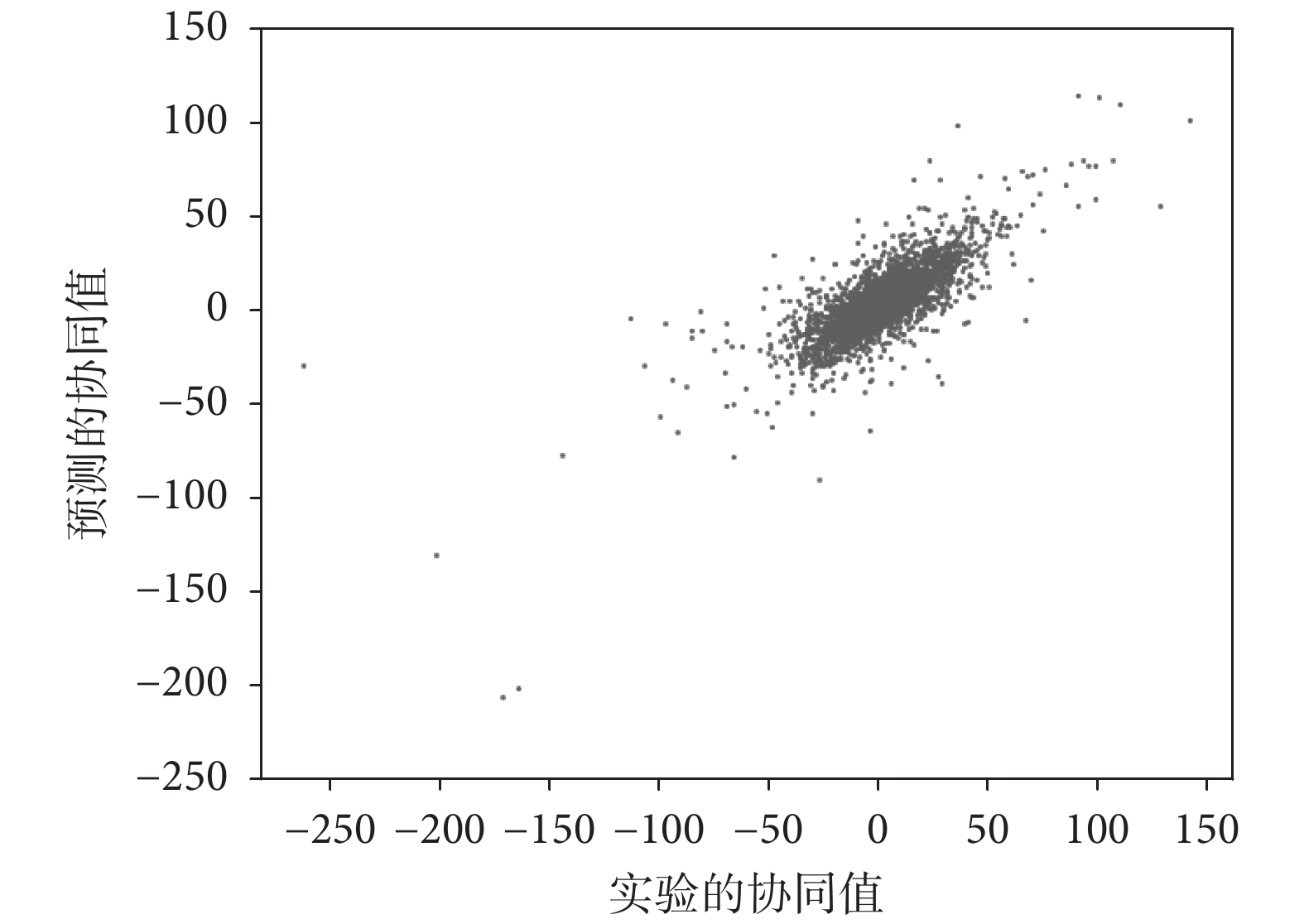 圖4
預測的和實驗的藥物組合協同值散點圖
Figure4.
Scatter plots of predicted and experimental synergistic values of drug combinations
圖4
預測的和實驗的藥物組合協同值散點圖
Figure4.
Scatter plots of predicted and experimental synergistic values of drug combinations
4 結論
本文提出一種基于多輸入神經網絡的抗癌藥物組合協同作用的預測方法,該方法利用卷積神經網絡一維卷積層降維和殘差前饋神經網絡構建回歸預測模型,能更加方便、快速降低數據的維度,減少模型訓練的復雜度。并且該方法能夠從多維度特征學習抗癌藥物的潛在協同關系,其預測模型的預測精度高,可以幫助臨床實驗減少時間和金錢成本,快速定位有效的藥物組合。
利益沖突聲明:本文全體作者均聲明不存在利益沖突。
引言
基于基因組、蛋白質組的高通量篩選方法和藥物設計雖然取得了巨大的進展,但過去十年批準的藥物數量仍遠小于實際需求。例如,目前全球各國已批準上市的抗癌藥物僅有 130~150 種。因為可用藥物少,耐藥性成為阻礙癌癥化療成功的主要原因之一[1-2],因而組合靶向治療成為解決這個難題的一種重要方法[3]。充分了解藥物組合之間的協同/拮抗機制以及化療的作用機制[4],對于快速方便地尋找針對特定癌癥類型的藥物組合具有十分重要的意義。
隨著高通量篩選和虛擬篩選成為現代藥物研究方法的重要組成部分[5],計算機仿真實驗在生物醫學科學中的應用越來越廣泛[6],利用計算模型尋找潛在的化合物組合成為一種重要的解決方法。例如,He 等[7]提出一種用于高通量篩選癌癥細胞藥物組合效應的實驗計算管道,提升了識別潛在藥物組合的速度。但是因為傳統計算模型通常沒有足夠的模擬能力,所以很難確定有效的藥物組合[8]。機器學習模型經過十幾年的巨大發展,利用其超強的學習能力尋找潛在有效的化合物組合變得至關重要[9]。深度神經網絡模型作為當下最先進的機器學習模型,在圖像處理、語言識別、自動駕駛等領域屢創記錄。由于其處理大規模數據的能力,在藥物設計領域得到了初步的應用。例如,Aliper 等[10]首次提出基于轉錄組數據的深度學習神經網絡,用于識別不同生物系統和不同條件下的多種藥物的藥理特性。Chang 等[11]首次將深度學習模型應用于驗證藥物再利用的可行性。Feng 等[12]設計了一個基于深度神經網絡的框架——蛋白與藥物分子相互作用預測(protein and drug molecular interaction prediction,PADME)來預測化合物與蛋白質之間的相互作用強度。Zong 等[13]提出基于相似性的藥物靶點預測方法,利用深度學習方法 DeepWalk 計算頂點相似性。Preuer 等[14]提出了基于神經網絡的 DeepSynergy 模型,成功預測了抗癌藥物組合。
細胞系基因組數據的突出特點是特征維度高,降維難度大。DeepSynergy 模型選擇的方法是使用第三方工具 Factor Analysis for Robust Microarray Summarization(FARMS)[15]進行降維。該工具是基于貝葉斯極大后驗法的因子分析模型,用于提取從 Affymetrix 芯片獲得的基因表達數據,但是作為無監督學習方法,在提取基因表達的過程中會引入誤差,而且不如神經網絡的學習能力強。此外,DeepSynergy 模型僅考慮了癌癥細胞系的基因表達特征,而忽略了與癌癥密切相關的基因拷貝數變異和基因突變等特征。針對上述問題,本文提出一種多輸入的端到端的深度學習模型——MulinputSynergy。該模型整合抗癌藥物的化學特征和癌癥細胞系的基因表達特征、基因突變特征、基因拷貝數特征,利用卷積層進行降維,最后通過殘差前饋神經網絡進行回歸預測。這種端到端的 one-stage 模型,數據流從輸入到輸出只通過一個模型完成,使得深度學習模型在學習預測的過程中有監督地降維,預測性能得到一定的提高。
1 數據及預處理
1.1 高通量藥物組合篩選數據
高通量藥物組合篩選數據集從 O’Neil 等[16]的論文下載(http://mct.aacrjournals.org/content/molcanther/suppl/2016/03/16/1535-7163.MCT-15-0843.DC1/156849_1_supp_1_w2lrww.xls)。具體來說,該數據集涵蓋了 583 種不同的藥物組合,每種組合都對來自 7 種不同組織類型的 39 個人類癌癥細胞系進行了測試。583 種藥物組合來源于 38 種抗癌藥物(14 個試驗藥物,24 個批準藥物)。按照 O’Neil 等的實驗,這 38 種抗癌藥物中的 22 種藥物(14 個實驗藥物和 8 個批準藥物)兩兩配對構成 231 種藥物組合,剩下的 16 種批準藥物與這 22 種藥物兩兩配對構成 352 種藥物組合。單個藥物有 4 種藥物濃度,2 種藥物形成 4×4 共計 16 個藥物組合劑量方案。每個藥物組合劑量方案重復實驗 4 次,在 96 h 后測量對應于對照組的細胞生長率(0 代表完全抑制,1 代表無抑制作用)。
1.2 藥物組合的協同作用值
藥物組合協同作用值來源于 Preuer 等[14]論文的補充材料(http://www.bioinf.jku.at/software/DeepSynergy/labels.csv)。作者使用 Combenefit[17]批處理程序來計算 Loewe Additivity 模型[18]的值來量化藥物組合協同作用程度。Loewe Additivity 模型使用模型期望的藥物組合反應表面與實際的藥物組合反應表面的偏差來量化藥物組合協同作用程度。按照 O’Neil 等的實驗,每個藥物組合共有 16 個劑量方案即有 16 個細胞生長率,藥物組合反應表面就是 16 個細胞生長率所構成的 4×4 矩陣。經過 Loewe Additivity 模型計算后,每個樣本有 4 個值,即藥物 A、藥物 B、細胞系、協同作用值。
每個樣本(藥物 A、藥物 B、細胞系、協同作用值)的不同藥物順序本質上是同一個樣本。因此,改變每個樣本的藥物組合順序可達到擴充樣本的目的。通過樣本擴充,我們的樣本數是原來的 2 倍。
1.3 化合物描述符和細胞系特征
癌癥細胞系特征有很多,例如基因表達、基因突變、基因拷貝數、microRNA 表達、DNA 甲基化、蛋白質豐度等,目前還沒有一種突出的細胞系基因組學特征能夠代表細胞系。為了使模型的預測性能盡可能較好,可以考慮不同特征的組合,以便計算模型能夠從不同方面學習潛在的聯系。考慮到數據庫之間異質性導致的數據整合困難,我們最終選擇了基因表達、基因突變、基因拷貝數這 3 種特征量化癌癥細胞系。
1.3.1 癌癥細胞系基因組數據
癌癥細胞系的基因表達譜數據來源于 ArrayExpress 數據庫(編號:E-MTAB-3610)[19],該表達譜在 Affymetrix 人類基因組 U219 平臺上測序。癌癥細胞系的基因突變數據來源于 CCLE(Cancer Cell Line Encyclopedia),下載地址為 https://data.broadinstitute.org/ccle/CCLE_DepMap_18q3_maf_20180718.txt。癌癥細胞系的拷貝數變異數據來源于 CCLP(COSMIC Cell Lines Project),下載地址為 https://cancer.sanger.ac.uk/cell_lines/download。
1.3.2 藥物的化學特征
我們使用軟件 alvaDesc[20](https://chm.kode-solutions.net/products_alvadesc.php)來計算化合物的分子描述符和指紋。alvaDesc 是計算各種分子描述符和指紋的新一代工具,共計算了 5 305 個分子描述符,包括功能組、片段計數、藥效團等。
1.4 數據預處理
高通量藥物組合篩選數據集所測試的 39 個癌癥細胞系并非完全包含在 ArrayExpress、CCLE 和 CCLP 中。Preuer 等在 DeepSynergy 中,對于那些缺失的細胞系采用父代或子代細胞系乃至同組織細胞系代替。例如,對于 39 個癌癥細胞系中的 EFM-192B,采用數據庫存在的 EFM-192A 代替。為了實驗的嚴謹性,我們使用藥物組合篩選數據集和 ArrayExpress、CCLE 和 CCLP 數據庫最大交集的 31 個細胞系,剔除了 8 個細胞系(CAOV3、COLO320DM、DLD1、EFM192B、NCIH23、OVCAR3、UWB1289BRCA1、ZR751)。
為了使訓練數據的數據格式符合深度學習模型的規范且利于模型訓練,我們對訓練數據進行預處理。CCLE 的癌癥細胞系基因突變數據是文本格式,對其使用 one-hot 編碼,1 代表這個基因發生突變,0 則代表未突變。同樣,CCLP 的癌癥細胞系基因拷貝數數據也是文本格式,對其進行數值編碼,基因拷貝數作為該基因的特征值。歸一化藥物化學特征和癌癥細胞系的基因表達、基因突變、基因拷貝數特征,使特征縮放在(0,1)區間。最后對照高通量篩選數據集的每條數據項(藥物 A-藥物 B-癌癥細胞系-藥物組合協同作用值),形成藥物化學特征和癌癥細胞系的基因表達特征、基因突變特征、基因拷貝數特征 4 個特征張量。
2 模型
本文提出的深度學習模型以細胞系和藥物組合預處理之后的特征為輸入,以協同作用值為輸出。模型的結構如圖 1 所示。該結構由兩個層次組成,第一個層次用于降維、提取特征,第二個層次用于預測協同值。在第一個層次中,基因表達、基因突變、基因拷貝數和藥物化學特征的特征維度從幾千到幾十萬,我們針對這 4 個特征設計了各自的子模型,用來編碼新的特征向量。所有編碼的特征連接起來形成新的特征向量,構成下一個層次的輸入。第二個層次子模型是一個深度殘差前饋神經網絡模型,比特征編碼網絡具有更多的層,且相鄰兩個殘差單元之間進行殘差連接,最后輸出協同作用值的預測。
圖1
MulinputSynergy 的模型結構
Figure1.
The architecture of MulinputSynergy
為了構建一個性能優異的端到端的深度學習模型,我們需要對模型進行調參實驗。采用卷積層提取特征時,我們對比一維卷積和二維卷積,發現使用一維卷積效果比二維卷積效果略好。對比圖像領域,我們不使用池化層來減少特征維數,而是減小卷積層通道數來達到提取特征的目的。
2.1 模型建模
2.1.1 藥物特征子模型
在不同的藥物組合中,同一藥物提取的特征應該是相同的。例如,藥物組合(ABT-888,AZD1775)和藥物組合(AZD1775,BEZ-235),其中藥物 AZD1775 提取的特征應該是相同的。所以在神經網絡模型中,訓練 2 個不同的藥物化學特征提取模型權重是不可取的。因此,在 MulinputSynergy 模型中,我們共享藥物 A 特征子模型和藥物 B 特征子模型的神經網絡權重,盡可能減少藥物順序對于實驗結果的影響。
每個藥物的特征向量共有 5 305 維,我們先將其末尾填充 24 個 0 值,然后將其變形為 73*73 的特征矩陣。我們使用卷積神經網絡提取藥物特征,該子模型有 3 層一維卷積層,卷積層的卷積核通道數為[36, 18, 10],每層的卷積核大小為 1,卷積步長也為 1,然后連接一個比率為 0.5 的 dropout 層,最終藥物的特征向量有 730 維。
2.1.2 基因表達特征子模型
我們使用卷積神經網絡提取基因表達特征。來源于 ArrayExpress 數據庫的基因表達譜特征有 553 536 維,將其變形為 744*744 的特征矩陣。該子模型有 4 層一維卷積層,卷積層的卷積核通道數為[186, 46, 11, 3],每層的卷積核大小為 1,卷積步長也為 1,然后連接一個比率為 0.5 的 dropout 層,最終基因表達特征的特征向量有 2 232 維。
2.1.3 基因突變特征子模型
基因突變文本經過 one-hot 編碼,每個細胞系基因突變的特征向量有 13 294 個維度。我們使用卷積神經網絡提取基因突變特征,首先將其末尾填充 46 個 0 值,然后將其變形為 114*115 的特征矩陣。該子模型有 2 層一維卷積層,卷積層的卷積核通道數為[60, 10],每層的卷積核大小為 1,卷積步長也為 1,然后連接一個比率為 0.5 的 dropout 層,最終基因突變特征的特征向量有 1 140 維。
2.1.4 基因拷貝數特征子模型
同樣的,基因拷貝數變異特征經過編碼,每個細胞系基因拷貝數的特征向量有 2 682 個維度。因為特征維度較小,我們使用前饋神經網絡提取基因拷貝數特征。該子模型有 3 層全連接層,全連接層的神經元數為[512, 512, 512],然后連接一個比率為 0.5 的 dropout 層,最終基因拷貝數特征的特征向量有 512 維。
2.1.5 深度殘差神經網絡回歸模型
對上述 4 個輸入模型輸出的 4 個張量進行連接合并,將組合的張量通過深度殘差神經網絡回歸模型進行回歸預測。深度殘差神經網絡回歸模型有 5 個殘差單元,每個殘差單元的神經元數為 2 048,然后連接一個比率為 0.5 的 dropout 層,隱藏層使用 relu 激活函數,輸出層使用 linear 激活函數。優化器使用 adam 函數來最小化均方誤差損失函數。使用回調函數監控測試集上的損失值,保存損失值最小的模型。優化器的學習率設置為 10?5,最終模型輸出協同作用預測值。
2.2 模型比較
我們將本文模型 MulinputSynergy 與 Preuer 等[14]提出的深度學習模型 DeepSynergy 進行比較。為了使 2 種模型具有可比性,DeepSynergy 也在剔除 8 個細胞系的數據集上運行。MulinputSynergy 與 DeepSynergy 都運行 200 輪。
2.3 交叉驗證
對于模型,我們使用 5 折交叉驗證方案來評估模型的性能。為了確保模型的魯棒性,每種藥物組合只出現在 5 折中的一折里。我們使用其中一折的數據作為驗證集來優化模型參數,即驗證模型在未知的藥物組合上的性能。
3 結果
3.1 模型性能評估
我們比較兩種模型對未知藥物組合協同值的預測能力。由于常用的均方誤差(mean squared error,MSE)和平均絕對誤差(mean absolute error,MAE)依賴于數據集預測值的大小,為了進一步描述模型的預測性能,我們還采用了其他回歸性能指標和分類性能指標。
3.1.1 回歸性能指標
我們采用 MSE、MAE、可釋方差值(explained vriance score,EVS)、皮爾遜相關系數(Pearson correlation coefficient,以下簡稱 Pearson)和決定系數(coefficient of determination,R2)來衡量模型的回歸性能。MSE 即預測值與真實值差的平方,這是回歸問題常用的性能指標。MAE 是預測值與真實值之差的絕對值,優點是對異常值有更強的魯棒性。皮爾遜相關系數用于衡量真實值和預測值之間的相關程度,其值范圍在[?1, 1]之間,絕對值越接近 1 相關性越強。R2介于 0~1 之間,越接近 1,回歸擬合效果越好。可釋方差值取值范圍是[0, 1],越接近于 1 說明自變量越能解釋因變量的方差變化,值越小則說明效果越差。表 1 展示了兩種模型在測試集上的回歸性能對比。
對比兩種模型的回歸性能指標,本文模型的均方誤差從 197.14 下降到 176.69,相對提升 10.4%,平均絕對誤差從 9.48 下降到 8.77,相對提升 7.5%。DeepSynergy 的可釋方差值、皮爾遜相關系數、R2決定系數分別為 0.53、0.73、0.53,而 MulinputSynergy 分別為 0.58、0.76、0.57,相對改進了 9.4%、4.1%、7.5%。因此,在回歸性能方面,本文的 MulinputSynergy 模型得到了提升。
3.1.2 分類性能指標
我們采用準確率(accuracy,ACC)、查準率(precision,PREC)、受試者工作特征(receiver operating characteristic,ROC)曲線下的面積(area under curve,AUC)(ROC AUC)、PR(precision-recall)曲線下的面積(PR AUC)、Kappa 系數(Cohen’s Kappa)、F1 分數(F1 score,F1)來衡量模型的分類性能。準確率常用來判斷分類器分類結果的好壞。受試者工作特征曲線下的面積、PR 曲線下的面積常用來衡量分類器的泛化能力。Kappa 系數用于一致性檢驗,也可以用于衡量分類精度。查準率是針對模型預測結果而言的,表示的是預測為正的樣例中真正的正樣例的比率。F1 分數是統計學中用來衡量二分類模型精確度的一種指標,同時兼顧了分類模型的精確率和召回率。由于訓練數據中存在大量的非協同非拮抗藥物組合(協同值在 0 左右),因此,選擇一個適當的閾值來將預測的協同作用值進行二值化是很重要的。我們將二分類閾值設置為 30,協同作用值高于 30 的藥物組合被認為是正類,低于 30 的藥物組合被認為是拮抗的和低協同的,這點與 DeepSynergy 模型相同。表 2 展示了兩種模型在測試集上的分類性能對比。
對比兩種模型的分類性能指標,本文模型的查準率從 0.57 上升到 0.63,相對提升 10.5%,PR 曲線下的面積從 0.53 上升到 0.59,相對提升 10.2%。DeepSynergy 的受試者工作特征曲線下的面積、Kappa 系數、F1 分數分別為 0.93、0.47、0.50,而 MulinputSynergy 分別為 0.94、0.50、0.52,相對改進了 1.1%、6.4%、4.0%。在準確率方面,兩個模型的性能持平。因此,在分類性能方面,本文的 MulinputSynergy 模型亦得到提升。
3.2 細胞系/藥物分組分析
此外,我們通過分析每個細胞系/每個藥物的 R2決定系數來分析它們之間的協同性能。圖 2 和圖 3 分別顯示了每個藥物和每個細胞系的結果。圖 2 中,在 x 軸上顯示細胞系的名稱,y 軸上顯示 R2 決定系數大小,條形圖的顏色與細胞系來源組織相對應。圖 3 中,在 x 軸上顯示藥物的名稱,y 軸上顯示 R2 決定系數大小,條形圖的顏色與藥物靶向目標相對應。
圖2
每個細胞系預測協同值的決定系數
Figure2.
Coefficient of determination of predicted synergy scores for each cell line
圖3
每個藥物預測協同值的決定系數
Figure3.
Coefficient of determination of predicted synergy scores for each drug
如圖 2 所示,對于每個細胞系,R2決定系數的變化范圍從 0.23 到 0.78,有 11 個細胞系的 R2決定系數小于 0.5,大約 65% 的細胞系的系數大于 0.5,有 11 個細胞系的系數大于 0.6,其中 LOVO 大腸癌和 UWB1289 卵巢癌細胞系的 R2決定系數大于 0.7,表現出較好的性能。從圖中可以看出,不同細胞系之間的預測精度差異較大,這可能是由于不同細胞系的生物學機制不同造成的。
如圖 3 所示,對于每種抗癌藥物,R2決定系數的變化范圍從 0.18 到 0.76,有 12 個藥物的 R2決定系數小于 0.5,約 68% 的藥物的系數大于 0.5,約 42% 的藥物的系數大于 0.6,有 5 個藥物(BORTEZOMIB、CARBOPLATIN、DASATINIB、ERLOTINIB、TOPOTECAN)的系數大于 0.7。在圖 3 中,沒有觀察到藥物靶向目標和 R2決定系數相關性之間的明顯聯系。因此,靶向目標特定的機制并不能解釋性能差異。從圖中可以看出,不同藥物之間的預測精度差異較大,這可能是由于不同的藥物組合對特定細胞系的協同/拮抗作用機制不同造成的。
3.3 虛擬篩選分析
計算模型的一個重要應用是高通量虛擬篩選,一個有效的計算模型可以幫助降低篩選成本。首先,在測試集上,我們按照協同值從大到小的順序對每個細胞系上測試的藥物組合進行有效性排序。然后定義了 TopN 指標來描述模型性能。TopN 定義如下:預測的前 N 個有效藥物組合中包含真實的最有效藥物組合的細胞系數/細胞系總數,N 取大于 1 的自然數且不大于待測藥物組合數。TopN 指標介于 0~1 之間,且越大越好。我們分別取 N 為 1、2、3、4、5、20 和 40,計算它們在 MulinputSynergy 和 DeepSynergy 模型下的值,然后進行對比,如表 3 所示。
我們統計出測試集上共有 116 個藥物組合。依據 MulinputSynergy 的 Top20 指標 0.95,對于每個細胞系有 95% 的概率,只要試驗模型預測的排名前 20 的藥物組合就能得出真實的最佳藥物組合,對比原本需要試驗 116 個藥物組合,減少 82.8% 的待測藥物組合。對比兩種模型的 TopN 指標,本文模型的 Top2 從 0.41 上升到 0.49,相對提升 19.5%,Top3 從 0.50 上升到 0.58,相對提升 16.0%,以此類推,Top4 相對提升 14.6%,Top5 相對提升 19.0%,Top20 相對提升 8.0%,Top40 相對提升 2.1%。
3.4 誤差分析
圖 4 給出了協同值的真實值和預測值之間的散點圖。我們計算每個樣本點協同值的實驗值與預測值的絕對誤差,發現約 97% 的樣本點預測值與真實值之差小于 30,約 70% 的樣本點小于 10,約 42% 的樣本點小于 5,約 27% 的樣本點小于 3。總體來說,大部分的預測效果是較好的。
圖4
預測的和實驗的藥物組合協同值散點圖
Figure4.
Scatter plots of predicted and experimental synergistic values of drug combinations
4 結論
本文提出一種基于多輸入神經網絡的抗癌藥物組合協同作用的預測方法,該方法利用卷積神經網絡一維卷積層降維和殘差前饋神經網絡構建回歸預測模型,能更加方便、快速降低數據的維度,減少模型訓練的復雜度。并且該方法能夠從多維度特征學習抗癌藥物的潛在協同關系,其預測模型的預測精度高,可以幫助臨床實驗減少時間和金錢成本,快速定位有效的藥物組合。
利益沖突聲明:本文全體作者均聲明不存在利益沖突。