人類染色體核型分析是診斷遺傳疾病的重要手段,染色體圖像類型識別是分析過程的關鍵步驟,準確高效地識別對自動核型分析具有重大意義。本文提出了一種分段重標定的稠密卷積神經網絡模型(SR-DenseNet),模型各階段先利用稠密連接的網絡層自動提取染色體不同抽象層次的特征,再用壓縮激活(SE)結構對匯集了局部所有特征的層進行特征重標定,對不同特征的重要性顯式地構建可學習的結構;提出了一種模型融合方法,構建了染色體識別模型專家組。在國際公開的哥本哈根染色體識別數據集(G 顯帶)上進行了實驗驗證,該模型的識別錯誤率僅為 1.60%;采用模型融合方法后,識別錯誤率進一步降低到 0.99%。在意大利帕多瓦大學的數據集(Q 顯帶)上,識別錯誤率為 6.67%;模型融合后,進一步降低到 5.98%。實驗結果表明本文所提方法是有效的,具備實現染色體類型識別自動化的潛力。
引用本文: 李建明, 陳斌, 孫曉飛, 馮濤, 張躍飛. 基于分段重標定的稠密卷積神經網絡的分帶染色體圖像類型識別. 生物醫學工程學雜志, 2021, 38(1): 122-130. doi: 10.7507/1001-5515.201912029 復制
版權信息: ?四川大學華西醫院華西期刊社《生物醫學工程學雜志》版權所有,未經授權不得轉載、改編
引言
染色體是人類遺傳物質的重要載體。正常情況下,體細胞包含 23 對(46 條)無結構改變的染色體。染色體在細胞分裂過程中出現染色體結構或數目異常的疾病,稱為染色體病。如果這種情況出現在胚胎發育階段,嚴重者會停止發育并流產,少數存活者可能出現機體畸變、智力低下、發育遲緩等情況[1-2]。目前這種疾病無有效的治療方法,因此對染色體病的遺傳分析和產前診斷是重要的預防手段。
染色體核型分析,是臨床上診斷染色體是否存在異常的常用方法。該技術以細胞分裂中期的染色體為研究對象,并借助顯帶技術對染色體進行數字成像。成像后由專業的遺傳學醫師,人工從圖像中分割染色體與其他雜質。再根據圖像中各染色體如長短、染色體帶型、著絲粒位置、有無隨體、長短臂比例特點對染色體半自動分類配對。其中,把分割得到的單條染色體的圖像分為 24 類(0~22 號常染色體和 X/Y 性染色體)之一的工作即為染色體圖像類型識別(以下簡稱染色體識別)。得到配對的染色體圖像后,按國際人類細胞遺傳命名系統(international system for human cytogenetic nomenclature,ISCN)組織排列,再計數、分析以確定是否存在數目和結構異常。
傳統的染色體核型分析主要依靠專業醫師對采集到的染色體圖像進行預處理、識別配對和異常分析[3]。在分割、識別、計數任務中,以識別配對在臨床操作中最為耗時;且受染色體圖像質量、醫師技術經驗和操作時精力集中程度等一系列因素的影響,識別任務是染色體核型分析效率較低、容易出錯的環節。因而準確高效的染色體識別對自動核型分析具有重大意義。
隨著計算機圖像識別技術的發展,染色體核型自動分析技術受到了國內外眾多研究者的關注[4-8]。2012 年以前的技術大都采用人工設計的特征或淺層人工神經網絡提取特征再進行分類識別的方式。郭宏宇等[4]將模糊理論運用到染色體自動識別系統,并與神經網絡相結合,提出了一種模糊神經網絡模型。蔣欣[5]基于中點法提取染色體中軸,基于幾何特征和灰度分布定位著絲粒,采用了平均灰度投影曲線、灰度梯度投影曲線和形狀投影曲線提取帶紋,再用加權的密度分布(weighted density distribution,WDD)[5]計算和表示帶紋特征;然后采用兩層分類器進行染色體識別。
深度學習技術的出現,促進了計算機圖像研究領域長足的進步,并在大規模自然圖像識別競賽中取得了優異的成績[9-14]。很多學者把相關技術應用在染色體識別任務中,自動提取圖像特征并進行識別,取得了較好的結果[15-20]。Sharma 等[15]提出了結合眾包、預處理和深度學習技術的方法,分割并識別染色體。該研究使用眾包的方法分割出單條染色體后,對彎曲的染色體進行了預處理,但其預處理方法為填充的部分引入了非真實的像素。Qin 等[16]提出了變焦網絡(Varifocal-Net)對染色體進行識別。該網絡是一個二階段的網絡結構,包含全局尺度的網絡(global-scale network,G-Net)和局部尺度的網絡(local-scale network,L-Net)。G-Net 提取全局特征并檢測可用于提取精細特征的區域,L-Net 變焦到 G-Net 定位的區域并進一步提取精細的局部特征。譚凱[19]提出了單條染色體圖像的伸直處理算法,對彎曲染色體圖像進行預處理,以提升后續網絡對染色體的識別效果。Lin 等[20]為染色體識別任務設計了改進的開端(Inception)網絡,結合提出的數據增強方法,取得了較好的識別效果。
本文充分結合壓縮激活網絡(squeeze-and-excitation networks,SE-Net)[13]和稠密卷積網絡(dense convolutional network,DenseNet)[14]的優點,設計了分段重標定(segmental recalibration,SR)的 DenseNet(SR-DenseNet)模型。該網絡利用單條染色體圖像和對應的類別標注信息,自動提取染色體特征,并完成染色體圖像識別任務。然后,本文選擇國際上可公開使用的兩個數據集進行實驗,分別是:哥本哈根染色體數據集(Copenhagen chromosome dataset,CCD)(網址:ftp://ftp.igmm.ed.ac.uk/pub/CromData/gbands/CPR.data/)和意大利帕多瓦大學染色體分類數據集(Padova chromosome dataset for classification,PCDC)(網址:http://bioimlab.dei.unipd.it/Chromosome%20Data%20Set%204Class.htm)[21]。在這兩個數據集上,本文與先進的通用卷積神經網絡[9-14]以及 Sharma 等[15]、Qin 等[16]設計的染色體識別網絡進行了對比實驗。最后,提出了一種模型融合算法,在兩個數據集上均進行了實驗,進一步降低了染色體識別任務的錯誤率。實驗結果表明,本文所提方法是有效的,具備實現染色體識別自動化的潛力,為進一步實現染色體核型分析的自動化打下基礎。
1 SR-DenseNet 網絡模型
1.1 SE-Net 的壓縮激活結構
傳統的卷積神經網絡,在局部感受野上同時對不同通道進行卷積運算(convolution,conv),以提取空間和通道之間的信息。SE-Net 的壓縮激活結構顯式地對通道之間的相關性進行建模,重新標定了各通道之間特征圖的重要性,提升了網絡的表現能力[13]。該結構首先進行信息壓縮(squeeze)操作,通過全局平均池化(global average pooling,GAP)得到各通道特征圖的激活平均值,利用統計特性描述了通道的激活程度。然后,作者設計了激活(excitation)操作,以利用通道之間的依賴關系。具體實現時,重標定的網絡支路中,GAP 操作之后得到了 1 × 1 × C(其中,C 為通道數)大小的特征圖,先使用 1 × 1 conv 對特征圖進行降維操作,得到 1 × 1 × C/r(其中,C 為通道數,r 是降維因子)大小的全連接層(fully connected layer,FC);再使用限制線性單元(rectified linear units,ReLU)激活函數得到 1 × 1 × C/r 大小的特征圖;接著使用 1 × 1 conv 做升維操作,把降維的特征圖還原到 1 × 1 × C 大小;最后使用了 S 狀彎曲函數(sigmoid)作激活函數,實現了參數化的門限機制,得到與每個通道對應的 0~1 之間的激活值,該值重標定了特征圖的重要性[13]。激活值與對應通道相乘得到重新標定的縮放(scale)層。
1.2 稠密網絡
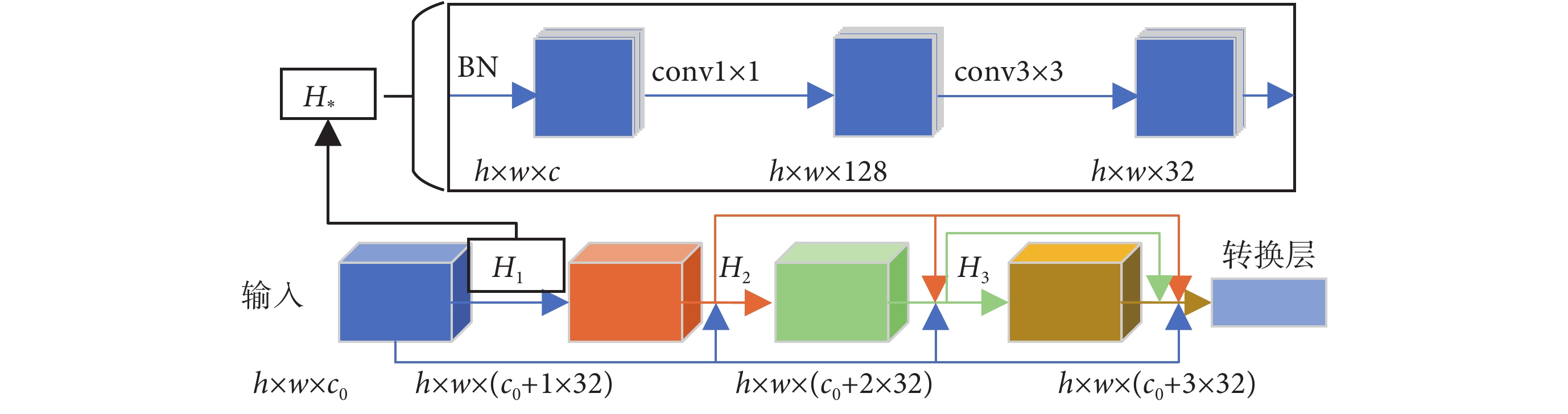
自 He 等[12]提出殘差網絡(residual network,ResNet)以來,神經網絡層與層之間的“捷徑”(shortcut)結構被廣泛使用。DenseNet 進一步發展了這種技術,設計了層與層兩兩之間通過捷徑相互連接的局部網絡結構,作者稱之為稠密塊(dense block)[14]。如圖 1 下部所示,稠密塊中每一層,都匯集其前面各層的特征圖作為輸入。這些特征圖先經批歸一化(batch normalization,BN)[22]操作之后,再通過卷積層調整其通道數。第 l 層特征圖與前面層的關系如式(1)所示[14]:
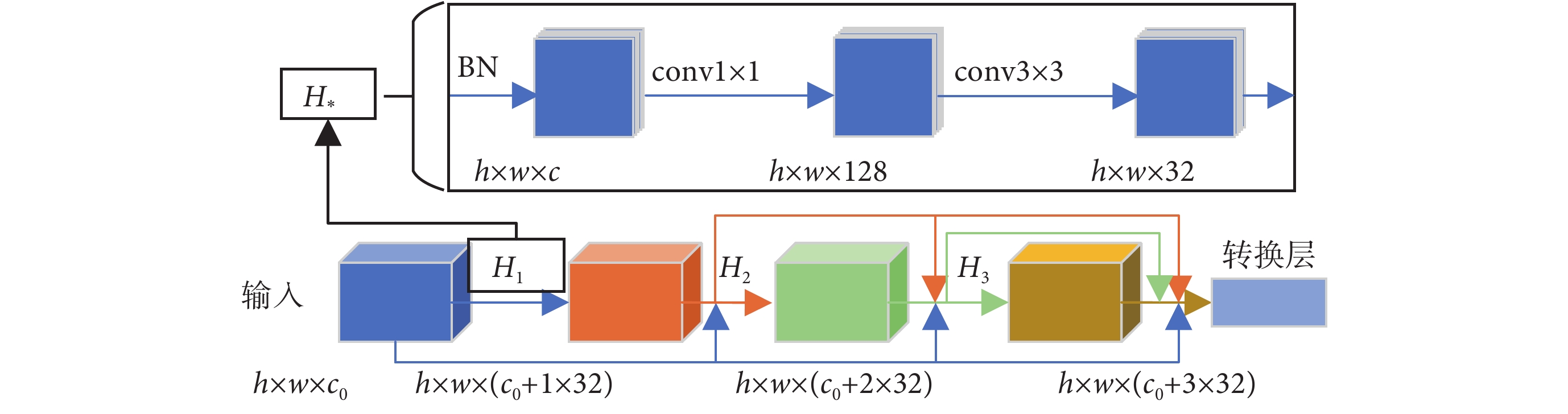 圖1
稠密連接模塊圖示
Figure1.
The schema of dense block module
圖1
稠密連接模塊圖示
Figure1.
The schema of dense block module
 |
其中, 代表第 l 層的特征圖,
代表第 l 層的特征圖, 表示對 0 到 l ? 1 層的特征進行平鋪連接(concatenate,concat),
表示對 0 到 l ? 1 層的特征進行平鋪連接(concatenate,concat), 代表非線性變換函數,其結構如圖 1 上部
代表非線性變換函數,其結構如圖 1 上部  所示。圖 1 中,h、w、c 分別特征圖的高、寬、通道數,c0 表示稠密塊第一層特征圖的通道數。
所示。圖 1 中,h、w、c 分別特征圖的高、寬、通道數,c0 表示稠密塊第一層特征圖的通道數。
這種方式的連接比簡單的通道相加保留了更多的信息,并促使網絡選擇更有表現力的通道,同時鼓勵網絡重利用前面層學到的特征,減輕了神經網絡中梯度消失的問題,使得網絡的訓練更加容易。
1.3 SR-DenseNet 網絡設計
參考醫師既結合不同層次特征又以帶型特征為主的分析方法,本文提出了 SR-DenseNet 模型,充分利用了稠密網絡的信息融合能力和 SE-Net 的通道重要性的重標定特點,提升了網絡的特征表現能力,有利于進一步提高網絡對染色體的識別能力。
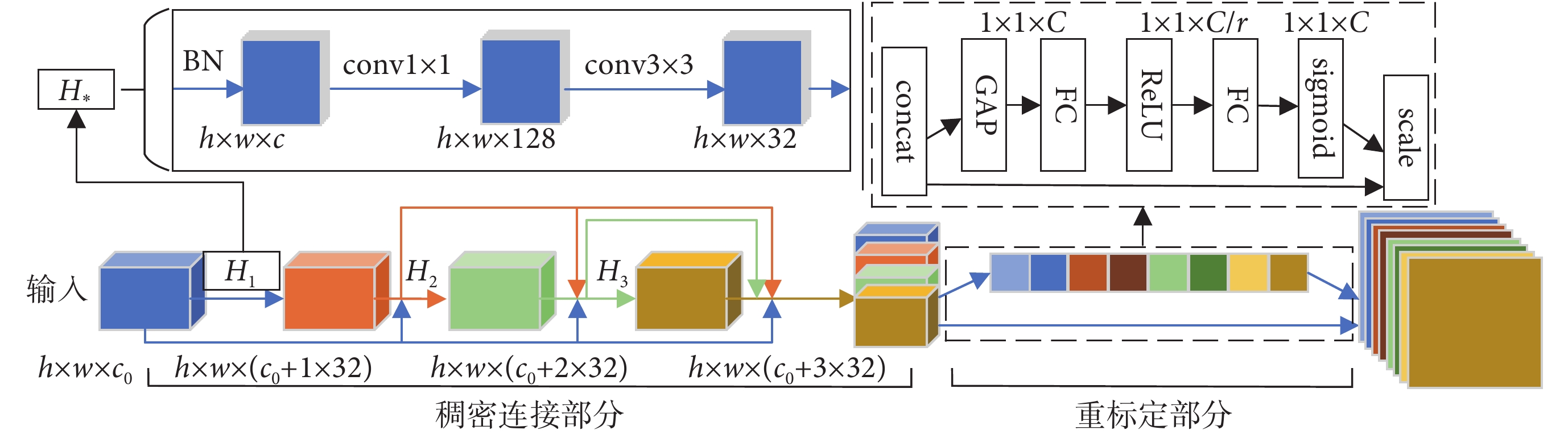
受到神經網絡模塊化思想的啟發[10, 12-14],本文設計了構建網絡的基本模塊,稱之為重標定的稠密模塊。模塊結構如圖 2 所示,前半部分包含稠密連接的卷積層,后半部分包含重標定部分。其中,稠密連接部分各字母含義與圖 1 相同,重標定部分 C 表示通道數,r 表示降維因子。稠密連接部分,前若干層特征圖輸入該結構后,先經 BN 操作,然后由 1 × 1 大小卷積核調整特征圖通道數,接著再進行 BN 操作,并由 3 × 3 大小卷積核提取特征并輸出 32 個特征圖,形成瓶頸結構;若干 BN-conv1 × 1-BN-conv3 × 3 瓶頸結構再以稠密連接的形式重復堆疊。重標定部分利用壓縮激活結構自適應地對各通道特征的重要性進行評價,充分利用對識別有利的特征。
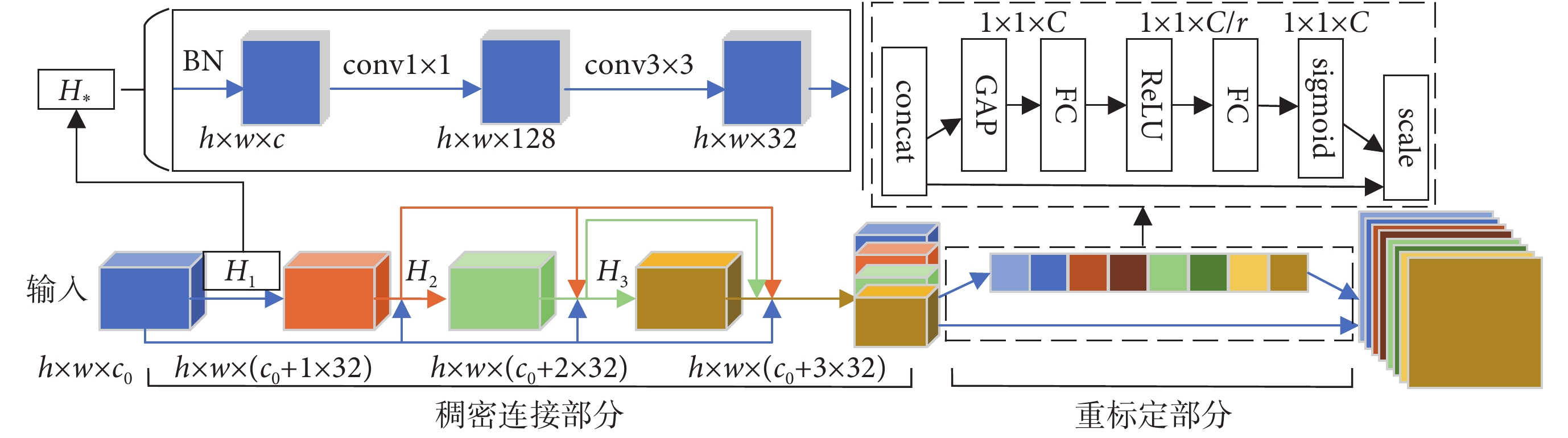 圖2
重標定的稠密模塊
Figure2.
The schema of recalibrated dense module
圖2
重標定的稠密模塊
Figure2.
The schema of recalibrated dense module
基于上述模塊,本文設計了 125 層的 SR-DenseNet 網絡模型。網絡第一層是包含 64 個 7 × 7 大小卷積核的卷積層。隨后是 4 個階段重標定的稠密模塊,各階段稠密模塊包含的瓶頸結構數量分別為 6、12、24、16。每個模塊的低層特征圖與高層特征圖在稠密部分的頂層平鋪連接后,再自適應地重標定。需要注意的是,與 SE-Net 不同,稠密部分頂層包含了該模塊不同層次的特征,本文網絡不對稠密部分中間層的特征圖實施重標定。最后的分類層包含 24 個神經元,對應染色體的 24 個類別。
1.4 多模型融合
本文希望得到一個模型,它對各類染色體的識別精度都達到最高。但在實驗過程中,本文發現不同的模型對不同類別染色體的識別錯誤率有差異。由于訓練過程的隨機性,重復實驗時這些相對差異不能穩定不變,為避免讀者誤解為某個模型能一直保持相同的識別偏好,后文以 A、B、C、D、E 指代本次實驗中的各模型,并對其進行分析。如表 1 所示,模型 A 對 0 號染色體識別較差,但對 1 號染色體較好;模型 D 對 0 號染色體和 1 號染色體識別錯誤率都達到最低,但模型 D 對 5 號染色體識別錯誤率卻很高。出現該問題的原因是由于在神經網絡訓練過程中的隨機性造成了不同模型學習到的特征偏好于部分類別。由以上可知,單一模型同時面對不同類別難識別樣本時,很難完全平衡。當醫師面臨難以識別的染色體圖像時,會由幾名醫師商議后共同決策。受此啟發,本文推測,在染色體圖像識別時,如果能綜合利用各模型各自的優勢,可以進一步降低模型的識別錯誤率。以表 1 所示,對 0 號染色體識別,模型 B、C、D 更有優勢。對新的樣本,如果識別結果為 0 號染色體,B、C、D 這三個模型對該類識別結果的置信度更高。模型融合時,以識別結果的置信度形成綜合意見,如果多數模型都識別正確(類似投票策略)或少數模型識別的置信度相對更高,都有可能糾正其中某個模型對個別染色體圖像的識別錯誤,從而可降低識別錯誤率。當然,也存在染色體圖像被所有模型都識別錯誤,此時模型融合就會失效。
由以上分析,本文提出了以識別置信度為基礎的多模型融合方法,形成染色體圖像識別模型專家組。組中各模型識別染色體圖像后,各自得到一個 24 維的識別置信度向量,該向量的 0~24 維分別描述了圖像歸屬 0~24 類染色體的置信度。各向量對應維度相加,得到模型融合的綜合置信度向量,如式(2)所示:
 |
其中,I 表示送入模型的單條染色體圖像, 是模型專家組中單個模型對圖像 I 的識別置信度向量
是模型專家組中單個模型對圖像 I 的識別置信度向量  (
( 表示圖像屬于第 i 類的置信度,
表示圖像屬于第 i 類的置信度, ),M 表示專家組中模型的數量,在本文中實驗中 M = 5。
),M 表示專家組中模型的數量,在本文中實驗中 M = 5。 體現了模型專家組對圖像 I 識別置信度的綜合意見。
體現了模型專家組對圖像 I 識別置信度的綜合意見。
得到綜合的識別置信度后,如式(3)所示,計算模型融合后的識別結果。
 |
其中, 是模型融合后圖像 I 被模型識別為類別 i 的置信度,type 是圖像 I 最終被識別的類別。
是模型融合后圖像 I 被模型識別為類別 i 的置信度,type 是圖像 I 最終被識別的類別。
2 實驗數據集和結果
2.1 數據集
本文選擇在國際上可公開使用的人類染色體識別數據集 CCD 和 PCDC 上進行對比實驗。實驗中采用的圖像數據均以編號命名,核型圖像中也不存在任何與送檢人員直接相關的信息,且數據僅供圖像識別研究使用,保證了樣本來源者的個人隱私。
2.1.1 CCD 染色體圖像數據集
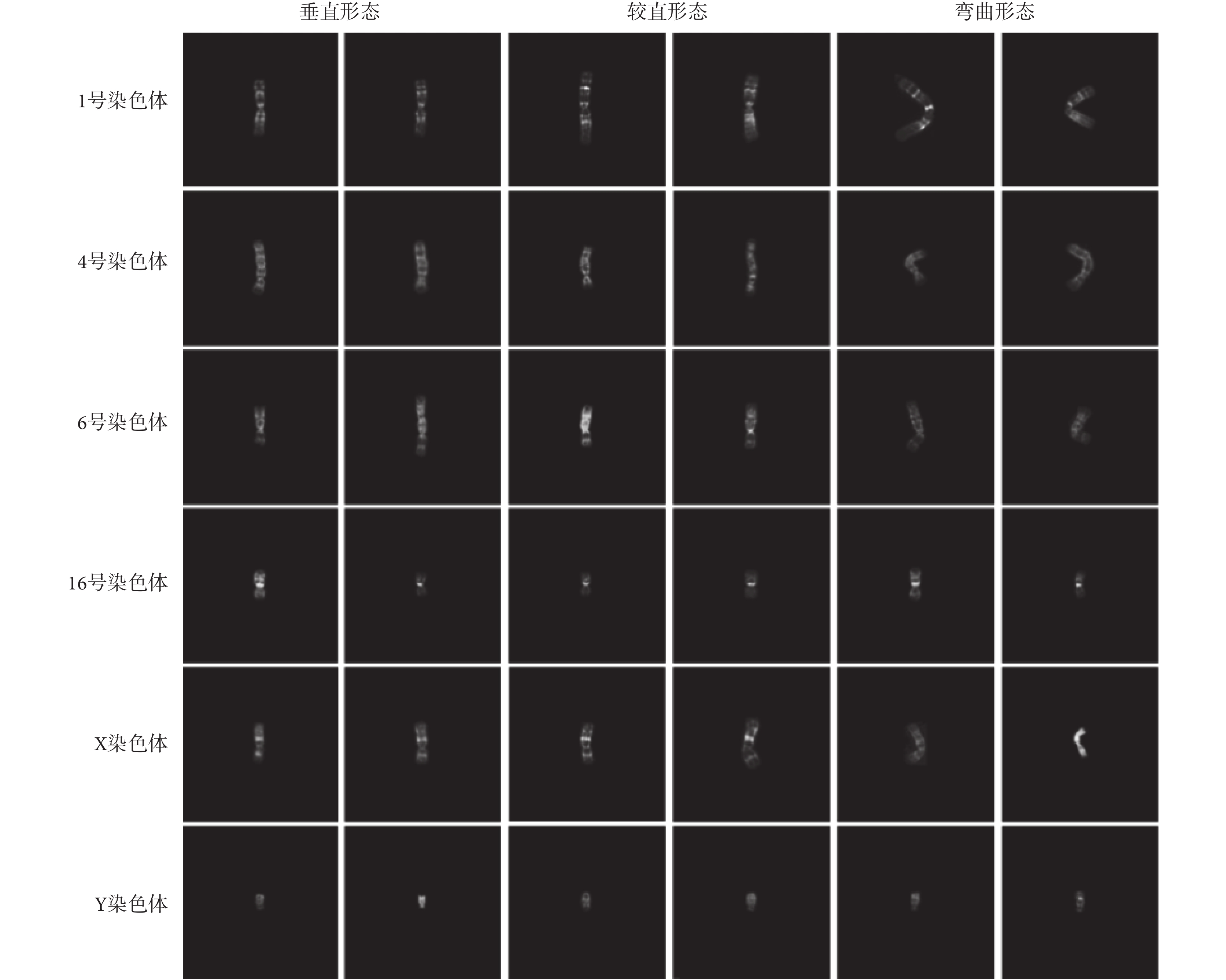
CCD 數據集,由 Lundsteen 等[23]在哥本哈根的瑞斯醫院(Rigshospitalet)收集樣本并標注類別[24]。該數據集包含 180 個細胞的染色體核型圖像,經分割得到 8 106 條染色體的圖像[25]。在本文的實驗中,將該數據集按 9∶1 的比例分為訓練集和驗證集兩部分,分別包含 7 295 張和 811 張已標注的染色體圖像樣本;由于樣本數量有限,驗證集同時也作為測試集。本文將原圖像樣本置于黑色背景中,統一制作成分辨率為 200 × 200 的圖像樣本,以滿足神經網絡需要固定大小輸入圖像的條件。部分調整后的數據集樣本如圖 3 所示。
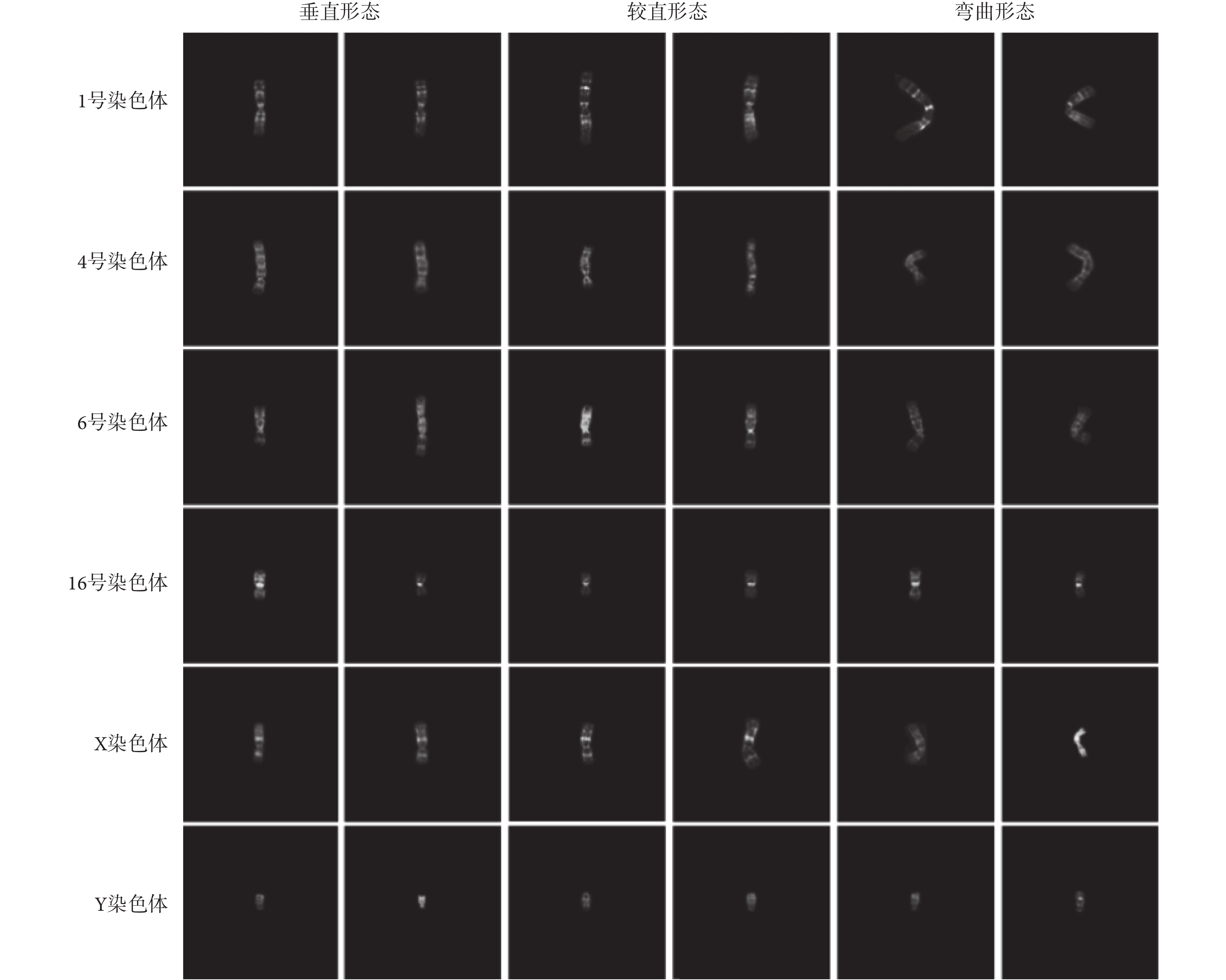 圖3
CCD 數據集部分樣本
Figure3.
Partial samples of CCD
圖3
CCD 數據集部分樣本
Figure3.
Partial samples of CCD
2.1.2 PCDC 染色體分類數據集
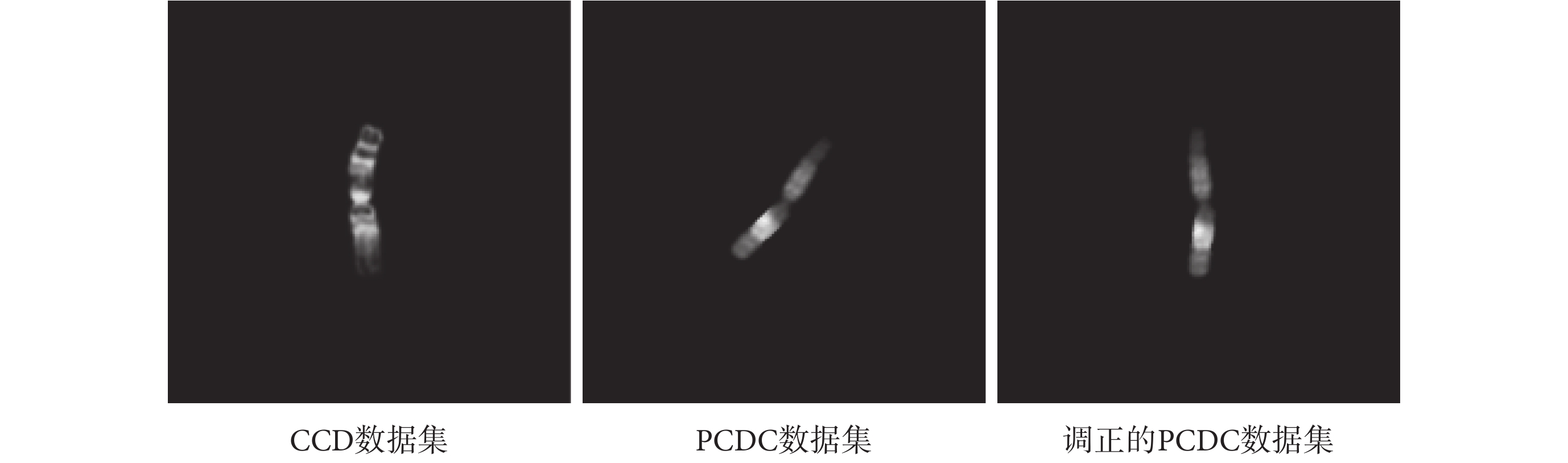
PCDC 染色體數據集的顯帶方式是 Q 顯帶[21]。如圖 4 所示,Q 顯帶的染色體條帶特征不如 G 顯帶的條帶特征明顯。該數據集來源于 119 張符合 ISCN 標準的染色體核型圖像,經分割標注,得到總計 5 474 張染色體圖像樣本。與 CCD 數據集類似,按 9∶1 比例將 PCDC 數據集分為訓練集和驗證集(測試集),分別包含 4 922 張和 552 張圖像樣本。本文按識別需要重新組織為 24 類,且對方向不正的樣本進行了調整。最終得到了類別組織、染色體方向、圖像大小與 CCD 保持一致的數據集。
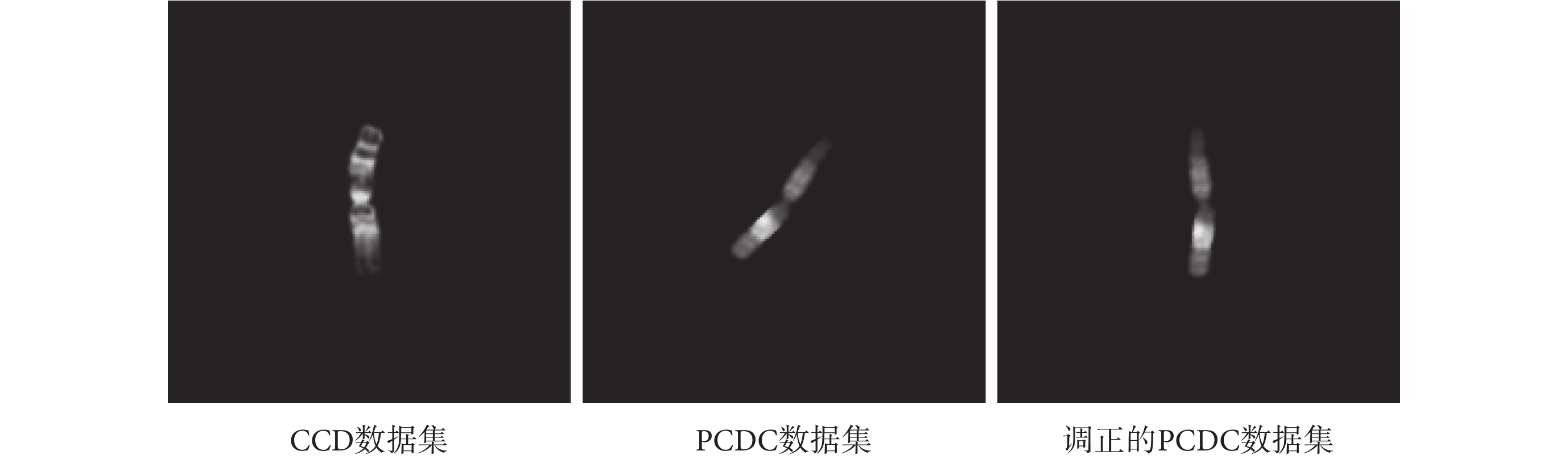 圖4
兩種數據集 1 號染色體樣本對比
Figure4.
The comparison of type 1 chromosome from two datasets
圖4
兩種數據集 1 號染色體樣本對比
Figure4.
The comparison of type 1 chromosome from two datasets
2.2 實驗結果對比與分析
2.2.1 評價指標
為了定量地評價各模型和模型融合方法,本文采用了在測試集上的總體識別錯誤率和各類別染色體識別錯誤率來評價模型的識別能力。模型總體識別錯誤率如式(4)所示:
 |
其中, 表示總體識別錯誤率,
表示總體識別錯誤率, 表示測試集第 i 類染色體的數量,
表示測試集第 i 類染色體的數量, 表示第 j 類染色體被錯分的數量,N 為染色體類別總數(N = 24,為常數)。該指標以數據集染色體的標注類別為準,按類別 j 比較識別結果與標注類別是否一致。若二者不同,
表示第 j 類染色體被錯分的數量,N 為染色體類別總數(N = 24,為常數)。該指標以數據集染色體的標注類別為準,按類別 j 比較識別結果與標注類別是否一致。若二者不同, 計數加一。然后,累加各類別錯分計數
計數加一。然后,累加各類別錯分計數  ,以累加值比測試集樣本總數得到總體識別錯誤率。
,以累加值比測試集樣本總數得到總體識別錯誤率。
模型各類識別錯誤率如式(5)所示:
 |
其中,type 表示染色體類別, 表示第 type 類的錯誤識別率,
表示第 type 類的錯誤識別率, 為第 type 類被模型錯分的樣本數量,
為第 type 類被模型錯分的樣本數量, 為第 type 類的樣本數量。該指標以數據集染色體的標注類別為準,按類別 type 比較識別結果與標注類別是否一致,若二者不同,
為第 type 類的樣本數量。該指標以數據集染色體的標注類別為準,按類別 type 比較識別結果與標注類別是否一致,若二者不同, 計數加一。然后,各類別分別以錯分樣本數比該類樣本數量得到 type 類的識別錯誤率。
計數加一。然后,各類別分別以錯分樣本數比該類樣本數量得到 type 類的識別錯誤率。
2.2.2 CCD 數據集識別效果對比
2012 年以來,圖像識別主要是基于深度神經網絡模型,期間在自然圖像識別數據集(ImageNet)上出現了大量優秀的模型,如亞歷克斯網絡模型(AlexNet)[9]、谷歌網絡模型(GoogLenet)[10]和視覺幾何組網絡模型(visual geometry group,VGG)[11]等,這些模型代表了通用分類模型的最高水平。Sharma 等[15]設計的模型(Sharma 模型)發表在計算機視覺與模式識別會議(conference on computer vision and pattern recognition,CVPR),該會議是計算機視覺頂級會議,代表了染色體識別的較高水平。Varifocal-Net 是一個二階段的模型,為了公平地比較,本文在單模型對比時僅采用 Varifocal-Net 中的 G-Net 部分進行比較。
模型訓練階段,本文分別按初始學習率為 0.1、0.01、0.001 和 0.000 1 從頭開始訓練,每 25 個周期學習率減小 10 倍,共訓練 100 個周期。訓練完成后,選擇各模型表現最好的結果記入表 2。各模型在測試集上的總體識別錯誤率如表 2 所示。
如表 2 所示,本文提出的模型在染色體識別數據集 CCD 上識別錯誤率僅為 1.60%,與 Sharma 模型相比,在其基礎上降低了 35.2%,略遜于 G-net;與 DenseNet 相比,總體識別錯誤率降低了 0.37%;與 SE-Net 相比,總體識別錯誤率降低了 0.74%。從實驗結果來看,隨著模型層數的加深,模型的識別錯誤率呈下降趨勢。這說明染色體圖像的形態變化十分復雜,需要較深的神經網絡模型來逐層完成從低級局部形態特征到高層類別語義特征的提取。與 DenseNet 相比,本文提出的 SR 稠密模塊具有更強的特征表現能力。該模塊既匯集了不同層次的局部特征,又自適應地標定了各通道特征的重要性,使網絡具備了專業遺傳學醫師的分析能力。在識別染色體圖像時,該模塊更多地關注最具辨識度的特征,并按權重同時兼顧其他特征。各個模型在 CCD 數據集上的單類別識別錯誤率比較結果如表 3 所示。
2.2.3 PCDC 數據集實驗結果
在 PCDC 數據集上,本文采用了與 CCD 數據集上相同的實驗方法。在該測試集上,各模型的總體識別錯誤率如表 4 所示。
如表 4 所示,本文提出的模型取得了最低的識別錯誤率,僅為 6.67%,比 DenseNet 提高了 0.21%;與 Sharma 模型相比,提升了 1.48%,與 G-Net 相比提升了 0.11%。實驗說明本文設計的模塊,既匯集了不同抽象層次特征又關注其中的重要特征,在面臨復雜的染色體樣本圖像時,模型特征表現能力更強,更有利于識別任務。與 CCD 數據集相比,各模型的表現都要差一點,這是因為 PCDC 數據集樣本圖像條帶特征不明顯所致,與前文對比兩數據集樣本特點的結論一致。
2.3 多模型融合方法的實驗結果
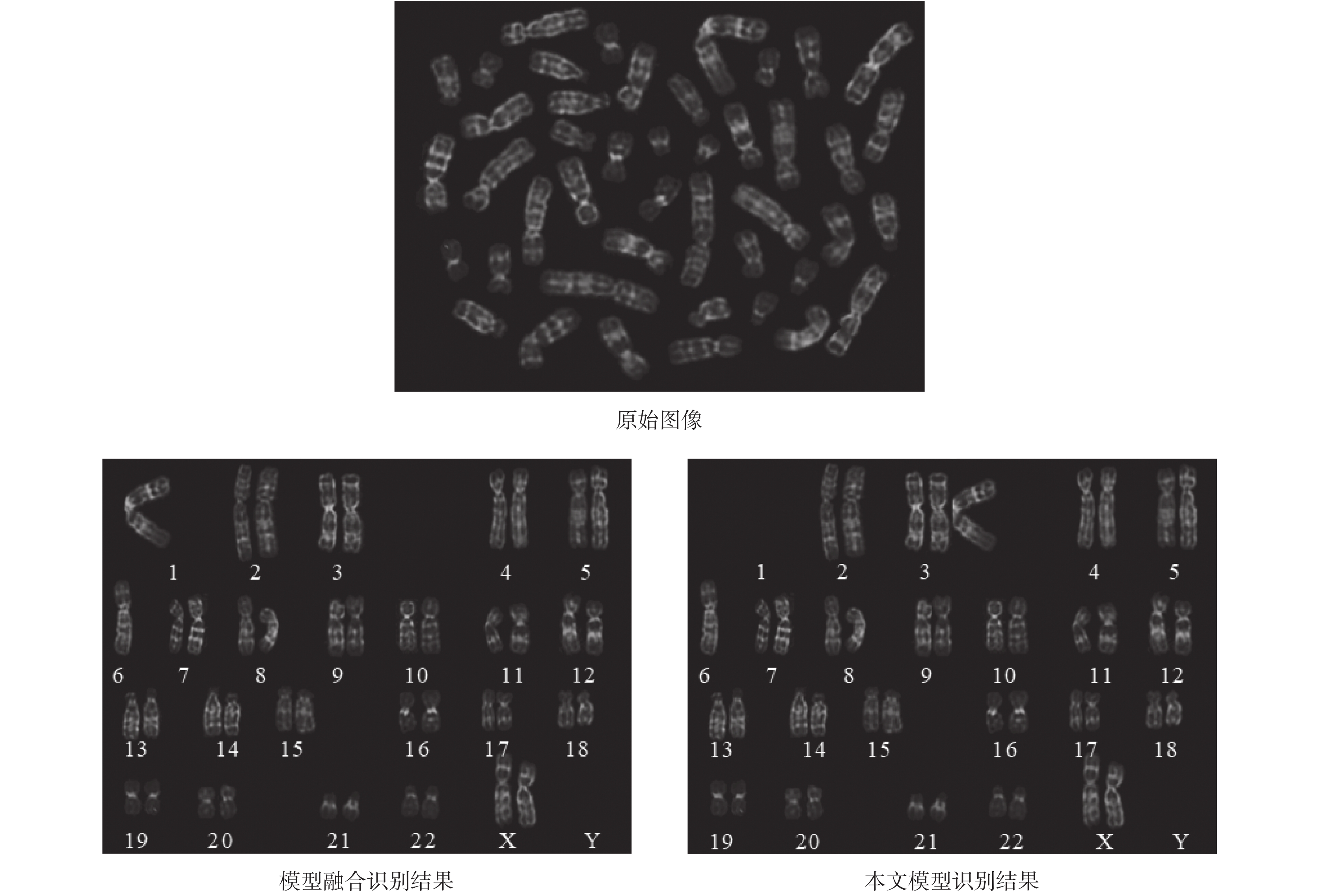
模型融合后,CCD 數據集上的整體識別錯誤率僅為 0.99%(如表 2 所示),在本文模型的總體識別錯誤率基礎上進一步降低了 38.12%。如表 3 所示,模型融合后,各類別的識別錯誤率均能達到最低。通過模型融合,PCDC 數據集上的整體識別錯誤率僅為 5.98%(如表 4 所示),在本文模型基礎上降低了 10.3%。以圖 5 為例,模型融合前,本文設計的模型把彎曲的 1 號染色體錯誤地識別為 3 號染色體;模型融合后,正確識別了彎曲的 1 號染色體,減小了錯誤率。從實驗結果看,本文提出的模型融合方法,充分挖掘了不同模型對識別任務有利的建模能力,構建了識別模型專家組,降低了總體識別錯誤率和單類別識別錯誤率。
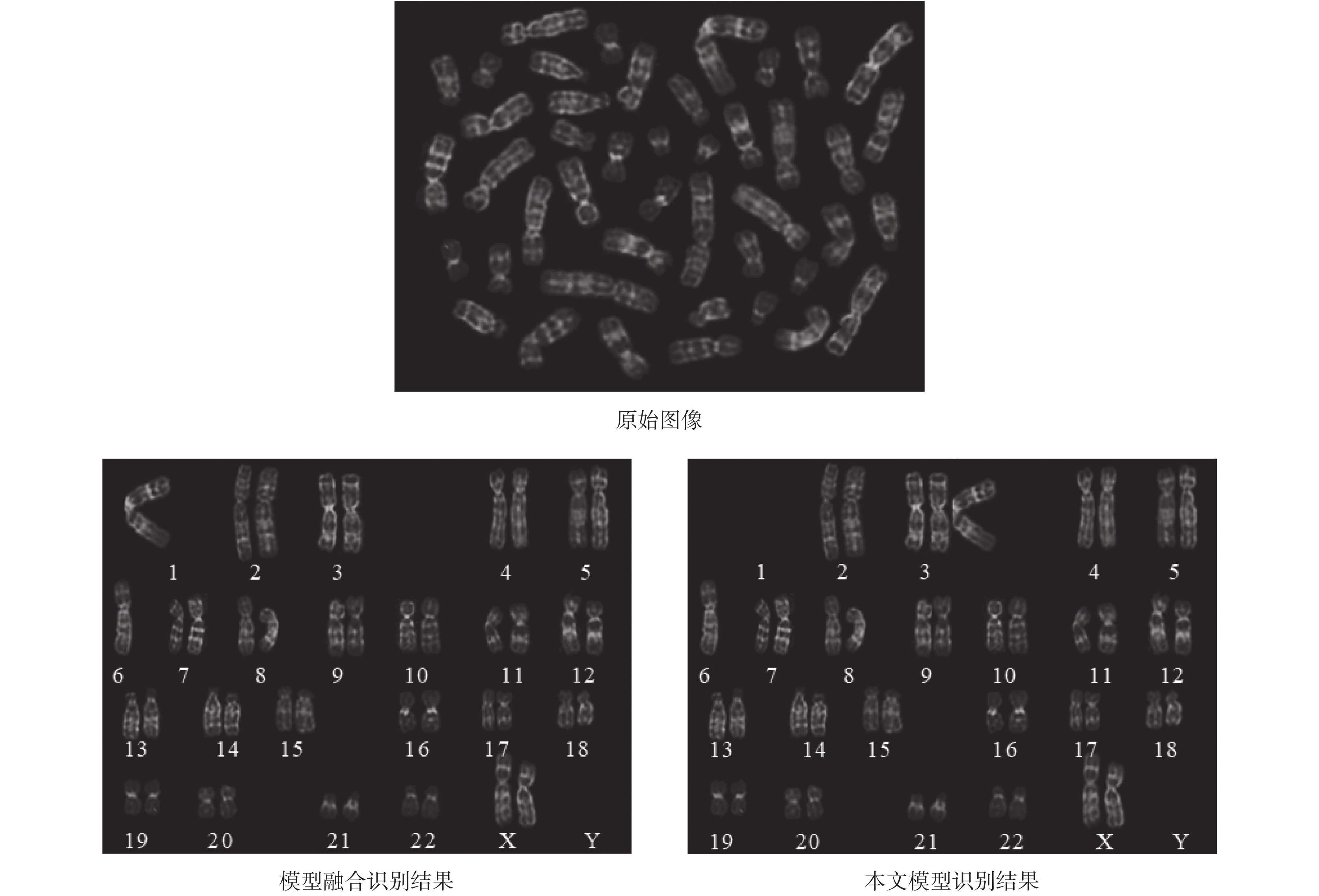 圖5
模型融合效果示例
Figure5.
Example of model fusion
圖5
模型融合效果示例
Figure5.
Example of model fusion
3 討論
從實驗結果來看,本文提出的方法在基線方法(DenseNet 和 SE-Net)上取得了一定的提升,但仍存在需繼續研究的問題。存在部分樣本,本文的方法重復實驗多次都不能正確識別(對比方法也存在相同的問題),如圖 6 所示。這些染色體樣本主要有兩個特點,分別是呈彎曲形態和成像質量欠佳。
 圖6
CCD 數據集中錯誤識別樣本
Figure6.
Misrecognition samples from CCD
圖6
CCD 數據集中錯誤識別樣本
Figure6.
Misrecognition samples from CCD
染色體是屬于非剛性物體,成像后的形態變化多樣(如圖 3 所示),其中尤其以彎曲形態最難識別。造成識別困難的本質原因有兩個:第一,數據集中彎曲的樣本的數量不足,造成卷積神經網絡不能充分學習這些彎曲樣本的特征;第二,卷積神經網絡的卷積核以滑窗形式在圖像中提取特征,且卷積核有內在的方向性,其從豎直形態染色體學習到的特征不能適用于彎曲形態染色體。如圖 6 所示,成像質量欠佳的染色體條帶特征難以辨認,網絡也難以識別。綜上,本文認為未來研究可從以下兩方面著手:① 從數據集的角度看,未來的研究應該構建規模更大的公開數據集,并盡可能地包含各種彎曲形態的染色體圖像樣本,以適應當前深度學習技術的數據需求并促進該領域的發展。② 從識別技術角度看,未來的研究或需要先識別出彎曲形態的染色體,轉換為豎直形態后再進行染色體識別;對成像質量欠佳的染色體識別,未來的研究可綜合核型圖中其他染色體的識別結果,推斷出可能的類別。
4 結論
本文提出了一種 SR-DenseNet 模型,該網絡包含 4 個階段重標定的稠密模塊。每個模塊前面部分包含若干稠密連接的卷積層,以提取不同層次的特征,后面部分以壓縮激活結構重標定提取到特征。該模塊既能充分提取不同層次的特征,又重點利用了更具表現力的特征。實驗結果表明,該網絡結構有更好的特征提取能力,識別錯誤率更低。以本文設計的 SR-DenseNet 為主,構建了識別模型專家組,進一步降低了識別錯誤率。實驗發現,識別錯誤的樣本大多為形態各異的彎曲樣本,未來的工作需要更加關注這些樣本。
利益沖突聲明:本文全體作者均聲明不存在利益沖突。
致謝:誠摯地感謝愛丁堡西部綜合醫院(Western General Hospital,Edinburgh)的 Jim Piper 和奧爾堡大學(Aalborg University)的 Erik Granum 名譽教授,兩位學者在本文獲取 CCD 數據集時提供了熱情幫助和詳細的解答。
引言
染色體是人類遺傳物質的重要載體。正常情況下,體細胞包含 23 對(46 條)無結構改變的染色體。染色體在細胞分裂過程中出現染色體結構或數目異常的疾病,稱為染色體病。如果這種情況出現在胚胎發育階段,嚴重者會停止發育并流產,少數存活者可能出現機體畸變、智力低下、發育遲緩等情況[1-2]。目前這種疾病無有效的治療方法,因此對染色體病的遺傳分析和產前診斷是重要的預防手段。
染色體核型分析,是臨床上診斷染色體是否存在異常的常用方法。該技術以細胞分裂中期的染色體為研究對象,并借助顯帶技術對染色體進行數字成像。成像后由專業的遺傳學醫師,人工從圖像中分割染色體與其他雜質。再根據圖像中各染色體如長短、染色體帶型、著絲粒位置、有無隨體、長短臂比例特點對染色體半自動分類配對。其中,把分割得到的單條染色體的圖像分為 24 類(0~22 號常染色體和 X/Y 性染色體)之一的工作即為染色體圖像類型識別(以下簡稱染色體識別)。得到配對的染色體圖像后,按國際人類細胞遺傳命名系統(international system for human cytogenetic nomenclature,ISCN)組織排列,再計數、分析以確定是否存在數目和結構異常。
傳統的染色體核型分析主要依靠專業醫師對采集到的染色體圖像進行預處理、識別配對和異常分析[3]。在分割、識別、計數任務中,以識別配對在臨床操作中最為耗時;且受染色體圖像質量、醫師技術經驗和操作時精力集中程度等一系列因素的影響,識別任務是染色體核型分析效率較低、容易出錯的環節。因而準確高效的染色體識別對自動核型分析具有重大意義。
隨著計算機圖像識別技術的發展,染色體核型自動分析技術受到了國內外眾多研究者的關注[4-8]。2012 年以前的技術大都采用人工設計的特征或淺層人工神經網絡提取特征再進行分類識別的方式。郭宏宇等[4]將模糊理論運用到染色體自動識別系統,并與神經網絡相結合,提出了一種模糊神經網絡模型。蔣欣[5]基于中點法提取染色體中軸,基于幾何特征和灰度分布定位著絲粒,采用了平均灰度投影曲線、灰度梯度投影曲線和形狀投影曲線提取帶紋,再用加權的密度分布(weighted density distribution,WDD)[5]計算和表示帶紋特征;然后采用兩層分類器進行染色體識別。
深度學習技術的出現,促進了計算機圖像研究領域長足的進步,并在大規模自然圖像識別競賽中取得了優異的成績[9-14]。很多學者把相關技術應用在染色體識別任務中,自動提取圖像特征并進行識別,取得了較好的結果[15-20]。Sharma 等[15]提出了結合眾包、預處理和深度學習技術的方法,分割并識別染色體。該研究使用眾包的方法分割出單條染色體后,對彎曲的染色體進行了預處理,但其預處理方法為填充的部分引入了非真實的像素。Qin 等[16]提出了變焦網絡(Varifocal-Net)對染色體進行識別。該網絡是一個二階段的網絡結構,包含全局尺度的網絡(global-scale network,G-Net)和局部尺度的網絡(local-scale network,L-Net)。G-Net 提取全局特征并檢測可用于提取精細特征的區域,L-Net 變焦到 G-Net 定位的區域并進一步提取精細的局部特征。譚凱[19]提出了單條染色體圖像的伸直處理算法,對彎曲染色體圖像進行預處理,以提升后續網絡對染色體的識別效果。Lin 等[20]為染色體識別任務設計了改進的開端(Inception)網絡,結合提出的數據增強方法,取得了較好的識別效果。
本文充分結合壓縮激活網絡(squeeze-and-excitation networks,SE-Net)[13]和稠密卷積網絡(dense convolutional network,DenseNet)[14]的優點,設計了分段重標定(segmental recalibration,SR)的 DenseNet(SR-DenseNet)模型。該網絡利用單條染色體圖像和對應的類別標注信息,自動提取染色體特征,并完成染色體圖像識別任務。然后,本文選擇國際上可公開使用的兩個數據集進行實驗,分別是:哥本哈根染色體數據集(Copenhagen chromosome dataset,CCD)(網址:ftp://ftp.igmm.ed.ac.uk/pub/CromData/gbands/CPR.data/)和意大利帕多瓦大學染色體分類數據集(Padova chromosome dataset for classification,PCDC)(網址:http://bioimlab.dei.unipd.it/Chromosome%20Data%20Set%204Class.htm)[21]。在這兩個數據集上,本文與先進的通用卷積神經網絡[9-14]以及 Sharma 等[15]、Qin 等[16]設計的染色體識別網絡進行了對比實驗。最后,提出了一種模型融合算法,在兩個數據集上均進行了實驗,進一步降低了染色體識別任務的錯誤率。實驗結果表明,本文所提方法是有效的,具備實現染色體識別自動化的潛力,為進一步實現染色體核型分析的自動化打下基礎。
1 SR-DenseNet 網絡模型
1.1 SE-Net 的壓縮激活結構
傳統的卷積神經網絡,在局部感受野上同時對不同通道進行卷積運算(convolution,conv),以提取空間和通道之間的信息。SE-Net 的壓縮激活結構顯式地對通道之間的相關性進行建模,重新標定了各通道之間特征圖的重要性,提升了網絡的表現能力[13]。該結構首先進行信息壓縮(squeeze)操作,通過全局平均池化(global average pooling,GAP)得到各通道特征圖的激活平均值,利用統計特性描述了通道的激活程度。然后,作者設計了激活(excitation)操作,以利用通道之間的依賴關系。具體實現時,重標定的網絡支路中,GAP 操作之后得到了 1 × 1 × C(其中,C 為通道數)大小的特征圖,先使用 1 × 1 conv 對特征圖進行降維操作,得到 1 × 1 × C/r(其中,C 為通道數,r 是降維因子)大小的全連接層(fully connected layer,FC);再使用限制線性單元(rectified linear units,ReLU)激活函數得到 1 × 1 × C/r 大小的特征圖;接著使用 1 × 1 conv 做升維操作,把降維的特征圖還原到 1 × 1 × C 大小;最后使用了 S 狀彎曲函數(sigmoid)作激活函數,實現了參數化的門限機制,得到與每個通道對應的 0~1 之間的激活值,該值重標定了特征圖的重要性[13]。激活值與對應通道相乘得到重新標定的縮放(scale)層。
1.2 稠密網絡
自 He 等[12]提出殘差網絡(residual network,ResNet)以來,神經網絡層與層之間的“捷徑”(shortcut)結構被廣泛使用。DenseNet 進一步發展了這種技術,設計了層與層兩兩之間通過捷徑相互連接的局部網絡結構,作者稱之為稠密塊(dense block)[14]。如圖 1 下部所示,稠密塊中每一層,都匯集其前面各層的特征圖作為輸入。這些特征圖先經批歸一化(batch normalization,BN)[22]操作之后,再通過卷積層調整其通道數。第 l 層特征圖與前面層的關系如式(1)所示[14]:
圖1
稠密連接模塊圖示
Figure1.
The schema of dense block module
|
其中, 代表第 l 層的特征圖, 表示對 0 到 l ? 1 層的特征進行平鋪連接(concatenate,concat), 代表非線性變換函數,其結構如圖 1 上部 所示。圖 1 中,h、w、c 分別特征圖的高、寬、通道數,c0 表示稠密塊第一層特征圖的通道數。
這種方式的連接比簡單的通道相加保留了更多的信息,并促使網絡選擇更有表現力的通道,同時鼓勵網絡重利用前面層學到的特征,減輕了神經網絡中梯度消失的問題,使得網絡的訓練更加容易。
1.3 SR-DenseNet 網絡設計
參考醫師既結合不同層次特征又以帶型特征為主的分析方法,本文提出了 SR-DenseNet 模型,充分利用了稠密網絡的信息融合能力和 SE-Net 的通道重要性的重標定特點,提升了網絡的特征表現能力,有利于進一步提高網絡對染色體的識別能力。
受到神經網絡模塊化思想的啟發[10, 12-14],本文設計了構建網絡的基本模塊,稱之為重標定的稠密模塊。模塊結構如圖 2 所示,前半部分包含稠密連接的卷積層,后半部分包含重標定部分。其中,稠密連接部分各字母含義與圖 1 相同,重標定部分 C 表示通道數,r 表示降維因子。稠密連接部分,前若干層特征圖輸入該結構后,先經 BN 操作,然后由 1 × 1 大小卷積核調整特征圖通道數,接著再進行 BN 操作,并由 3 × 3 大小卷積核提取特征并輸出 32 個特征圖,形成瓶頸結構;若干 BN-conv1 × 1-BN-conv3 × 3 瓶頸結構再以稠密連接的形式重復堆疊。重標定部分利用壓縮激活結構自適應地對各通道特征的重要性進行評價,充分利用對識別有利的特征。
圖2
重標定的稠密模塊
Figure2.
The schema of recalibrated dense module
基于上述模塊,本文設計了 125 層的 SR-DenseNet 網絡模型。網絡第一層是包含 64 個 7 × 7 大小卷積核的卷積層。隨后是 4 個階段重標定的稠密模塊,各階段稠密模塊包含的瓶頸結構數量分別為 6、12、24、16。每個模塊的低層特征圖與高層特征圖在稠密部分的頂層平鋪連接后,再自適應地重標定。需要注意的是,與 SE-Net 不同,稠密部分頂層包含了該模塊不同層次的特征,本文網絡不對稠密部分中間層的特征圖實施重標定。最后的分類層包含 24 個神經元,對應染色體的 24 個類別。
1.4 多模型融合
本文希望得到一個模型,它對各類染色體的識別精度都達到最高。但在實驗過程中,本文發現不同的模型對不同類別染色體的識別錯誤率有差異。由于訓練過程的隨機性,重復實驗時這些相對差異不能穩定不變,為避免讀者誤解為某個模型能一直保持相同的識別偏好,后文以 A、B、C、D、E 指代本次實驗中的各模型,并對其進行分析。如表 1 所示,模型 A 對 0 號染色體識別較差,但對 1 號染色體較好;模型 D 對 0 號染色體和 1 號染色體識別錯誤率都達到最低,但模型 D 對 5 號染色體識別錯誤率卻很高。出現該問題的原因是由于在神經網絡訓練過程中的隨機性造成了不同模型學習到的特征偏好于部分類別。由以上可知,單一模型同時面對不同類別難識別樣本時,很難完全平衡。當醫師面臨難以識別的染色體圖像時,會由幾名醫師商議后共同決策。受此啟發,本文推測,在染色體圖像識別時,如果能綜合利用各模型各自的優勢,可以進一步降低模型的識別錯誤率。以表 1 所示,對 0 號染色體識別,模型 B、C、D 更有優勢。對新的樣本,如果識別結果為 0 號染色體,B、C、D 這三個模型對該類識別結果的置信度更高。模型融合時,以識別結果的置信度形成綜合意見,如果多數模型都識別正確(類似投票策略)或少數模型識別的置信度相對更高,都有可能糾正其中某個模型對個別染色體圖像的識別錯誤,從而可降低識別錯誤率。當然,也存在染色體圖像被所有模型都識別錯誤,此時模型融合就會失效。
由以上分析,本文提出了以識別置信度為基礎的多模型融合方法,形成染色體圖像識別模型專家組。組中各模型識別染色體圖像后,各自得到一個 24 維的識別置信度向量,該向量的 0~24 維分別描述了圖像歸屬 0~24 類染色體的置信度。各向量對應維度相加,得到模型融合的綜合置信度向量,如式(2)所示:
|
其中,I 表示送入模型的單條染色體圖像, 是模型專家組中單個模型對圖像 I 的識別置信度向量 ( 表示圖像屬于第 i 類的置信度,),M 表示專家組中模型的數量,在本文中實驗中 M = 5。 體現了模型專家組對圖像 I 識別置信度的綜合意見。
得到綜合的識別置信度后,如式(3)所示,計算模型融合后的識別結果。
|
其中, 是模型融合后圖像 I 被模型識別為類別 i 的置信度,type 是圖像 I 最終被識別的類別。
2 實驗數據集和結果
2.1 數據集
本文選擇在國際上可公開使用的人類染色體識別數據集 CCD 和 PCDC 上進行對比實驗。實驗中采用的圖像數據均以編號命名,核型圖像中也不存在任何與送檢人員直接相關的信息,且數據僅供圖像識別研究使用,保證了樣本來源者的個人隱私。
2.1.1 CCD 染色體圖像數據集
CCD 數據集,由 Lundsteen 等[23]在哥本哈根的瑞斯醫院(Rigshospitalet)收集樣本并標注類別[24]。該數據集包含 180 個細胞的染色體核型圖像,經分割得到 8 106 條染色體的圖像[25]。在本文的實驗中,將該數據集按 9∶1 的比例分為訓練集和驗證集兩部分,分別包含 7 295 張和 811 張已標注的染色體圖像樣本;由于樣本數量有限,驗證集同時也作為測試集。本文將原圖像樣本置于黑色背景中,統一制作成分辨率為 200 × 200 的圖像樣本,以滿足神經網絡需要固定大小輸入圖像的條件。部分調整后的數據集樣本如圖 3 所示。
圖3
CCD 數據集部分樣本
Figure3.
Partial samples of CCD
2.1.2 PCDC 染色體分類數據集
PCDC 染色體數據集的顯帶方式是 Q 顯帶[21]。如圖 4 所示,Q 顯帶的染色體條帶特征不如 G 顯帶的條帶特征明顯。該數據集來源于 119 張符合 ISCN 標準的染色體核型圖像,經分割標注,得到總計 5 474 張染色體圖像樣本。與 CCD 數據集類似,按 9∶1 比例將 PCDC 數據集分為訓練集和驗證集(測試集),分別包含 4 922 張和 552 張圖像樣本。本文按識別需要重新組織為 24 類,且對方向不正的樣本進行了調整。最終得到了類別組織、染色體方向、圖像大小與 CCD 保持一致的數據集。
圖4
兩種數據集 1 號染色體樣本對比
Figure4.
The comparison of type 1 chromosome from two datasets
2.2 實驗結果對比與分析
2.2.1 評價指標
為了定量地評價各模型和模型融合方法,本文采用了在測試集上的總體識別錯誤率和各類別染色體識別錯誤率來評價模型的識別能力。模型總體識別錯誤率如式(4)所示:
|
其中, 表示總體識別錯誤率, 表示測試集第 i 類染色體的數量, 表示第 j 類染色體被錯分的數量,N 為染色體類別總數(N = 24,為常數)。該指標以數據集染色體的標注類別為準,按類別 j 比較識別結果與標注類別是否一致。若二者不同, 計數加一。然后,累加各類別錯分計數 ,以累加值比測試集樣本總數得到總體識別錯誤率。
模型各類識別錯誤率如式(5)所示:
|
其中,type 表示染色體類別, 表示第 type 類的錯誤識別率, 為第 type 類被模型錯分的樣本數量, 為第 type 類的樣本數量。該指標以數據集染色體的標注類別為準,按類別 type 比較識別結果與標注類別是否一致,若二者不同, 計數加一。然后,各類別分別以錯分樣本數比該類樣本數量得到 type 類的識別錯誤率。
2.2.2 CCD 數據集識別效果對比
2012 年以來,圖像識別主要是基于深度神經網絡模型,期間在自然圖像識別數據集(ImageNet)上出現了大量優秀的模型,如亞歷克斯網絡模型(AlexNet)[9]、谷歌網絡模型(GoogLenet)[10]和視覺幾何組網絡模型(visual geometry group,VGG)[11]等,這些模型代表了通用分類模型的最高水平。Sharma 等[15]設計的模型(Sharma 模型)發表在計算機視覺與模式識別會議(conference on computer vision and pattern recognition,CVPR),該會議是計算機視覺頂級會議,代表了染色體識別的較高水平。Varifocal-Net 是一個二階段的模型,為了公平地比較,本文在單模型對比時僅采用 Varifocal-Net 中的 G-Net 部分進行比較。
模型訓練階段,本文分別按初始學習率為 0.1、0.01、0.001 和 0.000 1 從頭開始訓練,每 25 個周期學習率減小 10 倍,共訓練 100 個周期。訓練完成后,選擇各模型表現最好的結果記入表 2。各模型在測試集上的總體識別錯誤率如表 2 所示。
如表 2 所示,本文提出的模型在染色體識別數據集 CCD 上識別錯誤率僅為 1.60%,與 Sharma 模型相比,在其基礎上降低了 35.2%,略遜于 G-net;與 DenseNet 相比,總體識別錯誤率降低了 0.37%;與 SE-Net 相比,總體識別錯誤率降低了 0.74%。從實驗結果來看,隨著模型層數的加深,模型的識別錯誤率呈下降趨勢。這說明染色體圖像的形態變化十分復雜,需要較深的神經網絡模型來逐層完成從低級局部形態特征到高層類別語義特征的提取。與 DenseNet 相比,本文提出的 SR 稠密模塊具有更強的特征表現能力。該模塊既匯集了不同層次的局部特征,又自適應地標定了各通道特征的重要性,使網絡具備了專業遺傳學醫師的分析能力。在識別染色體圖像時,該模塊更多地關注最具辨識度的特征,并按權重同時兼顧其他特征。各個模型在 CCD 數據集上的單類別識別錯誤率比較結果如表 3 所示。
2.2.3 PCDC 數據集實驗結果
在 PCDC 數據集上,本文采用了與 CCD 數據集上相同的實驗方法。在該測試集上,各模型的總體識別錯誤率如表 4 所示。
如表 4 所示,本文提出的模型取得了最低的識別錯誤率,僅為 6.67%,比 DenseNet 提高了 0.21%;與 Sharma 模型相比,提升了 1.48%,與 G-Net 相比提升了 0.11%。實驗說明本文設計的模塊,既匯集了不同抽象層次特征又關注其中的重要特征,在面臨復雜的染色體樣本圖像時,模型特征表現能力更強,更有利于識別任務。與 CCD 數據集相比,各模型的表現都要差一點,這是因為 PCDC 數據集樣本圖像條帶特征不明顯所致,與前文對比兩數據集樣本特點的結論一致。
2.3 多模型融合方法的實驗結果
模型融合后,CCD 數據集上的整體識別錯誤率僅為 0.99%(如表 2 所示),在本文模型的總體識別錯誤率基礎上進一步降低了 38.12%。如表 3 所示,模型融合后,各類別的識別錯誤率均能達到最低。通過模型融合,PCDC 數據集上的整體識別錯誤率僅為 5.98%(如表 4 所示),在本文模型基礎上降低了 10.3%。以圖 5 為例,模型融合前,本文設計的模型把彎曲的 1 號染色體錯誤地識別為 3 號染色體;模型融合后,正確識別了彎曲的 1 號染色體,減小了錯誤率。從實驗結果看,本文提出的模型融合方法,充分挖掘了不同模型對識別任務有利的建模能力,構建了識別模型專家組,降低了總體識別錯誤率和單類別識別錯誤率。
圖5
模型融合效果示例
Figure5.
Example of model fusion
3 討論
從實驗結果來看,本文提出的方法在基線方法(DenseNet 和 SE-Net)上取得了一定的提升,但仍存在需繼續研究的問題。存在部分樣本,本文的方法重復實驗多次都不能正確識別(對比方法也存在相同的問題),如圖 6 所示。這些染色體樣本主要有兩個特點,分別是呈彎曲形態和成像質量欠佳。
圖6
CCD 數據集中錯誤識別樣本
Figure6.
Misrecognition samples from CCD
染色體是屬于非剛性物體,成像后的形態變化多樣(如圖 3 所示),其中尤其以彎曲形態最難識別。造成識別困難的本質原因有兩個:第一,數據集中彎曲的樣本的數量不足,造成卷積神經網絡不能充分學習這些彎曲樣本的特征;第二,卷積神經網絡的卷積核以滑窗形式在圖像中提取特征,且卷積核有內在的方向性,其從豎直形態染色體學習到的特征不能適用于彎曲形態染色體。如圖 6 所示,成像質量欠佳的染色體條帶特征難以辨認,網絡也難以識別。綜上,本文認為未來研究可從以下兩方面著手:① 從數據集的角度看,未來的研究應該構建規模更大的公開數據集,并盡可能地包含各種彎曲形態的染色體圖像樣本,以適應當前深度學習技術的數據需求并促進該領域的發展。② 從識別技術角度看,未來的研究或需要先識別出彎曲形態的染色體,轉換為豎直形態后再進行染色體識別;對成像質量欠佳的染色體識別,未來的研究可綜合核型圖中其他染色體的識別結果,推斷出可能的類別。
4 結論
本文提出了一種 SR-DenseNet 模型,該網絡包含 4 個階段重標定的稠密模塊。每個模塊前面部分包含若干稠密連接的卷積層,以提取不同層次的特征,后面部分以壓縮激活結構重標定提取到特征。該模塊既能充分提取不同層次的特征,又重點利用了更具表現力的特征。實驗結果表明,該網絡結構有更好的特征提取能力,識別錯誤率更低。以本文設計的 SR-DenseNet 為主,構建了識別模型專家組,進一步降低了識別錯誤率。實驗發現,識別錯誤的樣本大多為形態各異的彎曲樣本,未來的工作需要更加關注這些樣本。
利益沖突聲明:本文全體作者均聲明不存在利益沖突。
致謝:誠摯地感謝愛丁堡西部綜合醫院(Western General Hospital,Edinburgh)的 Jim Piper 和奧爾堡大學(Aalborg University)的 Erik Granum 名譽教授,兩位學者在本文獲取 CCD 數據集時提供了熱情幫助和詳細的解答。