由于數據的高維和復雜性,空間轉錄組數據的分析一直是一個具有挑戰性的問題,而聚類分析則是空間轉錄組數據分析的核心問題。本文提出了一種基于圖注意力網絡的深度學習方法,用于空間轉錄組數據的聚類分析。首先,對空間轉錄組數據進行增強,然后使用圖注意力網絡對節點進行特征提取,最后使用萊頓(Leiden)算法進行聚類分析。通過聚類的評價指標發現,與傳統的非空間及空間聚類方法相比,本文提出的方法具有更好的數據分析性能。實驗結果表明,本文所提方法可以有效地聚類空間轉錄組數據,從而能夠識別不同的空間區域,為研究空間轉錄組數據提供了新的工具。
引用本文: 吳瀚文, 高潔. 通過圖注意力網絡識別空間轉錄組中的空間域. 生物醫學工程學雜志, 2024, 41(2): 246-252. doi: 10.7507/1001-5515.202304030 復制
版權信息: ?四川大學華西醫院華西期刊社《生物醫學工程學雜志》版權所有,未經授權不得轉載、改編
0 引言
空間轉錄組學是一種結合了組織學和轉錄組學的新興技術,它可以研究細胞在組織中的分布和表達方式。空間轉錄組(spatial transcriptome,ST)數據通常是由高通量成像技術生成,如空間轉錄組學和空間RNA測序(spatial RNA sequencing,sRNA-seq)。這些技術可以在組織中捕獲數百萬個單細胞的轉錄組數據,并提供高分辨率的空間信息。然而,由于數據的高維和復雜性,空間轉錄組數據的分析一直是一個具有挑戰性的問題。識別空間域(即,在基因表達和組織學上都具有空間一致性的區域)是空間轉錄組學中最重要的課題之一。目前,空間域識別方法主要分為非空間聚類和空間聚類兩類。傳統的非空間聚類算法,如K均值聚類算法(k-means clustering algorithm)、魯汶算法(Louvain method),以基因表達數據作為輸入,導致聚類難以符合組織切片[1-2]。而空間聚類方法結合了基因表達、空間位置和形態學,可以解釋基因表達的空間依賴性,以更好地匹配空間位置。例如,Zhao等[3]提出的空間貝葉斯方法(BayesSpace)即是基于貝葉斯統計方法,通過引入空間鄰居結構來鼓勵相鄰點屬于同一聚類,然后將捕獲區域劃分為更小的亞區域,實現了聚類精度的提高。Xu等[4]提出的用于空間轉錄組分析的卷積神經網絡(convolutional neural networks,CNN)學習策略(CNN learning strategy for spatial transcriptomics analysis,CosTA)使用CNN聚類結構,通過CNN生成特征,再通過高斯混合模型聚類生成軟分配[5],并使用軟分配來更新CNN。Yuan等[6]提出的用于基因的圖CNN(graph CNN for genes,GCNG)方法將空間信息編碼為圖形,使用監督訓練將其與表達數據組合,并用圖卷積網絡對圖數據進行特征嵌入,從而實現聚類。Hu等[7]提出的整合基因表達、空間位置和組織學,通過圖卷積網絡識別空間域和空間可變基因(integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network,SpaGCN)方法應用圖卷積網絡來整合基因表達和空間位置,并進一步與自監督模塊相結合來識別空間域。Pham等[8]提出的空間轉錄組學數據的下游分析工具包(stLearn)提供了一種組織內歸一化技術,該技術基于從形態學圖像中收集到的特征和空間位置,通過形態學距離對基因表達進行歸一化。Xu等[9]提出的無監督空間嵌入深度表示方法(unsupervised spatially embedded deep representation of spatial transcriptomics,SEDR)采用深度自動編碼器網絡和圖形自動編碼器來嵌入空間信息。Teng等[10]提出可用于熒光原位雜交技術的迭代細胞分型(fluorescence in situ hybridization iterative cell typing,FICT)方法,能同時利用空間轉錄組數據中的表達信息和空間信息來識別細胞類型和亞型。盡管這些空間聚類方法可以在一定程度上識別空間域,但往往是通過節點的空間位置計算節點之間的距離,并由此預先定義節點之間的相似性,而這些預先定義的方法沒能利用深度學習的靈活性,故存在一定局限。
為了解決這個問題,本文提出了一種基于圖注意力網絡(graph attention networks,GAT)的深度學習方法,并將其命名為用圖注意力自動編碼器進行空間轉錄組聚類分析(spatial transcriptomics clustering analysis with graph attention auto-encoders,STCAGAE),用于空間轉錄組數據的聚類分析[11]。本文使用的圖注意力網絡可以自適應地學習節點之間的相似性,從而更好地聚類空間域。本文利用預先訓練好的深度神經網絡模型從形態圖像塊中提取特征向量,然后將提取的特征與基因表達和空間位置數據相結合,以表征空間相鄰點的相關性,并創建空間增強基因表達矩陣。同時使用圖注意力自動編碼器和去噪自動編碼器聯合生成增強空間轉錄組數據的潛在表示,最后使用萊頓(Leiden)算法最終的嵌入進行聚類[12]。本文旨在識別不同的空間域,并為研究空間轉錄組數據提供新的方法。
1 方法
1.1 數據來源及預處理
本文研究所用數據來自十倍基因組學(10 × Genomics)生物技術公司利用維希姆(Visium)技術發布的高通量空間轉錄組測序平臺(10 × Genomics Visium)。該平臺可以利用Visium技術,從整個組織切片和各種各樣的樣本類型中獲得整個轉錄組高通量基因表達分析結果。本文選取了該平臺的一組可公開下載的數據集,即人類背外側前額葉皮層(dorsolateral prefrontal cortex dataset,DLPFC)數據集。DLPFC數據集包含了12個不同的切片數據,其中每個切片都包含6個皮質層和1個白質層。首先,從DLPFC數據集中移除主要組織區域之外的區域。然后使用計算機編程語言Python 3.8(Guido van Rossum,荷蘭)中分析單細胞數據的軟件包Scanpy 1.9.1(Theis Lab,德國),根據文庫大小對原始基因表達數據進行過濾、對數轉換和標準化[13]。最后,使用主成分分析(principal components analysis,PCA)對增廣基因表達數據進行降維,并將降維數據作為下一個模型訓練的輸入。
1.2 STCAGAE算法
1.2.1 增強空間數據
空間基因表達技術提供了具有額外空間位置信息和組織形態的轉錄組范圍的基因表達譜。本文使用這兩個額外的空間數據實現了節點的基因表達數據增強,得到了對節點更真實的表示,從而提高了STCAGAE的性能。計算點i和點j之間的空間基因表達權重GCij,如式(1)所示:
 |
其中,Si代表節點i的基因表達向量, 代表節點i的基因表達向量的均值,Sj代表節點j的基因表達向量,
代表節點i的基因表達向量的均值,Sj代表節點j的基因表達向量, 代表節點j的基因表達向量的均值。對于具有形態學信息的空間轉錄組數據,本文首先根據每個點的坐標對一個圖像進行分割,得到其部分圖像。然后,使用深度學習框架Pytorch 1.8.0(Facebook Inc,美國)中的圖像預處理模塊轉換器(transform)對圖像進行變換和增強,包括歸一化、旋轉、調整銳度等[14]。每個點的特征從預先訓練的CNN模型得出,該模型可以將每個節點的圖像轉換為2 048維的潛在變量。為了更好地表示節點的形態,本文進行了PCA,提取了前50個主成分作為潛在特征。最后,利用余弦距離計算點i與相鄰點j之間的形態相似性權重MSij,如式(2)所示:
代表節點j的基因表達向量的均值。對于具有形態學信息的空間轉錄組數據,本文首先根據每個點的坐標對一個圖像進行分割,得到其部分圖像。然后,使用深度學習框架Pytorch 1.8.0(Facebook Inc,美國)中的圖像預處理模塊轉換器(transform)對圖像進行變換和增強,包括歸一化、旋轉、調整銳度等[14]。每個點的特征從預先訓練的CNN模型得出,該模型可以將每個節點的圖像轉換為2 048維的潛在變量。為了更好地表示節點的形態,本文進行了PCA,提取了前50個主成分作為潛在特征。最后,利用余弦距離計算點i與相鄰點j之間的形態相似性權重MSij,如式(2)所示:
 |
本文使用空間坐標來確定每個點和所有其他點之間的距離,然后對前4個相鄰點之間的距離進行排序,以計算半徑r。對于一個給定的點i,當且僅當兩個點之間的距離小于r時,認為點j是點i的相鄰點,則設表示點i與點j位置關系的權重參數SWij = 1,否則SWij = 0。然后,增強每個點i的基因表達,如果是10 × Genomics Visium平臺的數據,將數據增強為如式(3)所示;否則,將數據增強為如式(4)所示:
 |
 |
其中,GEi和GEj代表點i和點i的相鄰點j的原始基因表達。
1.2.2 構造圖
為了結合給定點的相鄰點的相似性,本文使用空間坐標來計算點之間的距離,并使用前6個最近鄰構建一個無向連接圖。對于無向連接圖,在每個點上都添加了自循環。A是鄰接矩陣,那么如果i和j是相鄰點,則在點i和點j處的值Aij = Aji = 1,否則Aij = 0。
1.2.3 去噪自動編碼器
使用Pytorch 1.8.0(Facebook Inc,美國)的線性層實現了一個去噪自動編碼器,用于基因表達的潛在表示[15]。編碼器E由用戶設置的多個完全連接的堆疊線性層組成,將集成的基因表達式X轉換為低維表示,如式(5)所示:
 |
其中,Zg代表經過編碼器得到的低維表示,R代表實數集,N代表節點數量,M代表輸入基因數量,d代表最后一個編碼器層的維數。然后,向Zg加入噪聲得到重構的低維表示,如式(6)所示。接下來,解碼器D將重構后的低維表示轉換為重構的基因表達矩陣,如式(7)所示:
 '/> '/> |
 '/> '/> |
其中, 代表重構的低維表示,Z代表噪聲向量,D代表解碼器,X'代表重構的基因表達矩陣,d''代表最終層的維數,N、M和d與上述相同。輸入基因和重建表達式的類似性用均方誤差來計算,如式(8)所示:
代表重構的低維表示,Z代表噪聲向量,D代表解碼器,X'代表重構的基因表達矩陣,d''代表最終層的維數,N、M和d與上述相同。輸入基因和重建表達式的類似性用均方誤差來計算,如式(8)所示:
 |
其中,Ll代表去噪自編碼器的損失函數,Xi代表節點i的基因表達向量,E(Xi)代表節點i的基因表達向量經過編碼器層得到的低維表示,D(?)代表經過解碼器層重構后的表示。
1.2.4 變分圖自動編碼器
變分圖自動編碼器使用了基于Pytorch 1.8.0(Facebook Inc,美國)構建的用于編寫和訓練圖神經網絡的庫Pytorch Geometric 2.2.0(Technische University Dortmund,德國) [16],其中編碼器由圖注意力網絡組成。圖注意力網絡將特征矩陣X作為輸入,首先通過一個線性變換W做維度轉換,如式(9)所示:
 |
其中, 代表特征矩陣經過線性變換之后的列數。接著使用自注意力(self-attention)為每個節點分配注意力,對于點i和點j,它們的初始特征為hi和hj,于是它們經過線性變換W后變為Whi和Whj,為它們分配注意力,如式(10)所示:
代表特征矩陣經過線性變換之后的列數。接著使用自注意力(self-attention)為每個節點分配注意力,對于點i和點j,它們的初始特征為hi和hj,于是它們經過線性變換W后變為Whi和Whj,為它們分配注意力,如式(10)所示:
 |
其中 ,eij 代表節點 i 對節點 j 的影響力系數,a (·)代表計算節點相關度的函數,如式(11)所示:
 '/> '/> |
在本文中,對于節點i,只計算其鄰域內節點j對節點i的影響力系數。為了更好地在不同節點之間分配權重,本文將目標節點與所有相鄰點計算出來的相關度進行統一的歸一化處理,使用線性整流函數(rectified linear unit,ReLU),如式(12)所示:
 |
其中, 代表歸一化后的相關度權重系數,Ni代表節點i的鄰域,eik代表節點 i 對節點 i鄰域內的節點 k 的影響力系數。每個節點的最終輸出特征向量如式(13)所示:
代表歸一化后的相關度權重系數,Ni代表節點i的鄰域,eik代表節點 i 對節點 i鄰域內的節點 k 的影響力系數。每個節點的最終輸出特征向量如式(13)所示:
 '/> '/> |
其中,hi'代表節點的最終輸出特征,σ代表激活函數。損失函數包括生成圖與原始圖之間的重構損失,節點表示的向量分布和正態分布的相對熵(relative entropy),如式(14)所示:
 |
其中,Lg代表變分圖自動編碼器的損失函數, 代表二進制交叉熵函數,
代表二進制交叉熵函數, 代表相對熵,
代表相對熵, ,代表正態分布。
,代表正態分布。
同時,本文利用多頭注意力以穩定學習過程和增加模型容量。多頭注意力就是將同一個輸入分別送入不同的注意力層,分別計算隱藏狀態,然后將不同的特征表示連接起來(或相加)。經過不斷調整參數并實驗,本文發現模型中的參數取8個注意力頭和特征相加時,DLPFC數據集的不同切片聚類得到的調整蘭德系數(adjusted Rand index,ARI)值都最大,因此模型達到最佳效果。
1.2.5 聚類與可視化
在得到嵌入之后,使用Leiden算法來識別空間域,以識別相似的細胞和模式。為了獲得最佳分辨率,在0.1~2.5之間進行網格搜索,步長為0.01,直到達到必要的簇數。為了可視化,使用了統一流形逼近和投影[17]。
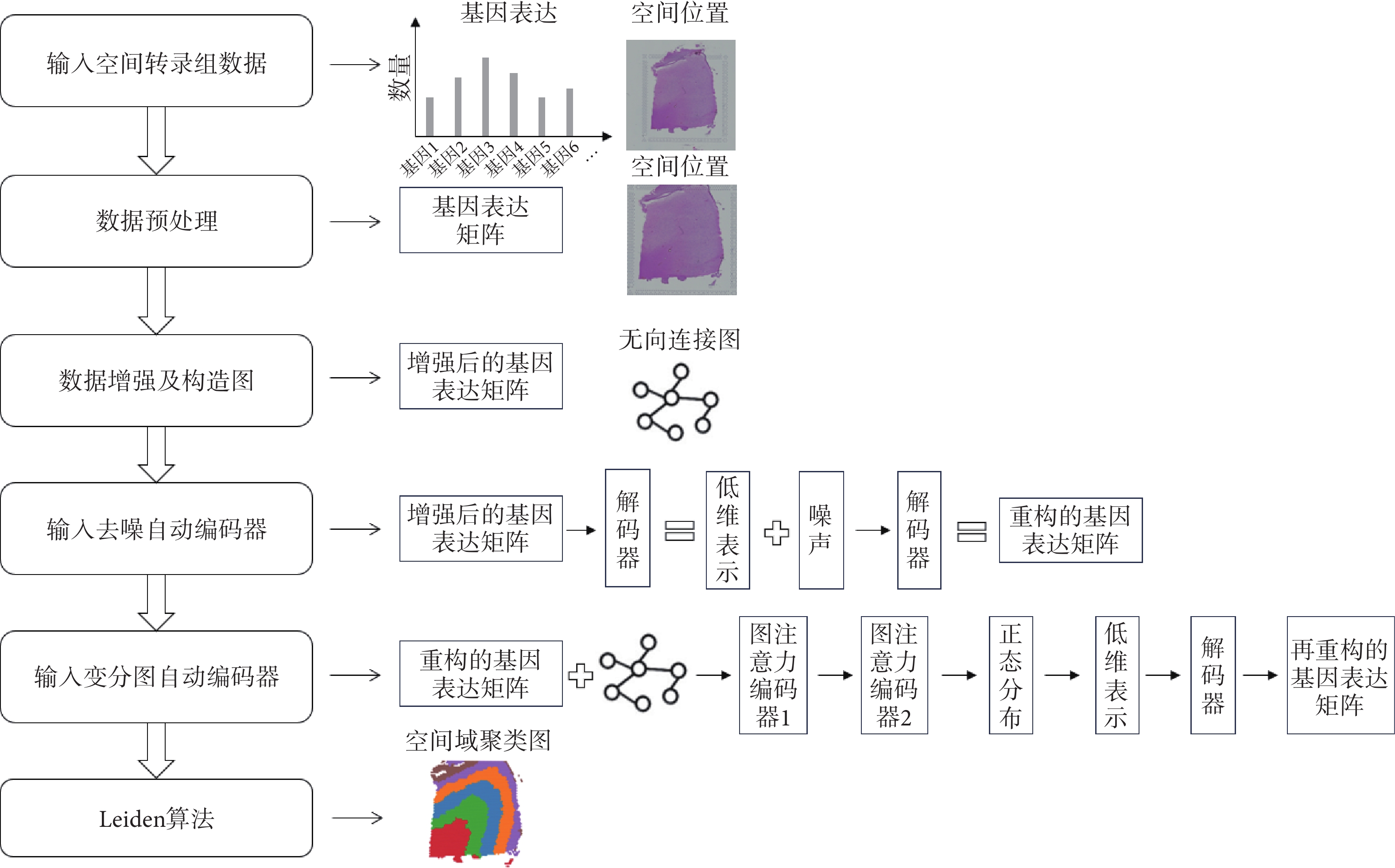
綜上,本文提出STCAGAE算法,如圖1所示。通過對整合基因表達、空間位置和組織形態信息的低維表示進行建模來表征空間域。為了建立形態學特征矩陣,首先使用預先訓練好的深度學習網絡對蘇木精-伊紅(hematoxylin-eosin,H&E)染色的組織地形圖數據進行處理。結合形態特征和空間相鄰點信息,每個點的基因表達均得到增強。然后,利用去噪自編碼器學習從集成特征空間到低維表示空間的非線性映射,以減少模型的過擬合。同時,基于節點之間的歐氏距離計算圖鄰接矩陣。將增強后的基因表達矩陣和圖鄰接矩陣輸入到一個變分圖自動編碼器中,從而通過與相應的空間相鄰點的集成表示來生成空間嵌入。最終的潛在嵌入是通過連接集成表示和空間嵌入來形成的。最后使用Leiden算法進行空間域聚類,識別出從右上到左下依次是皮質層1~6、白質層的不同空間域。
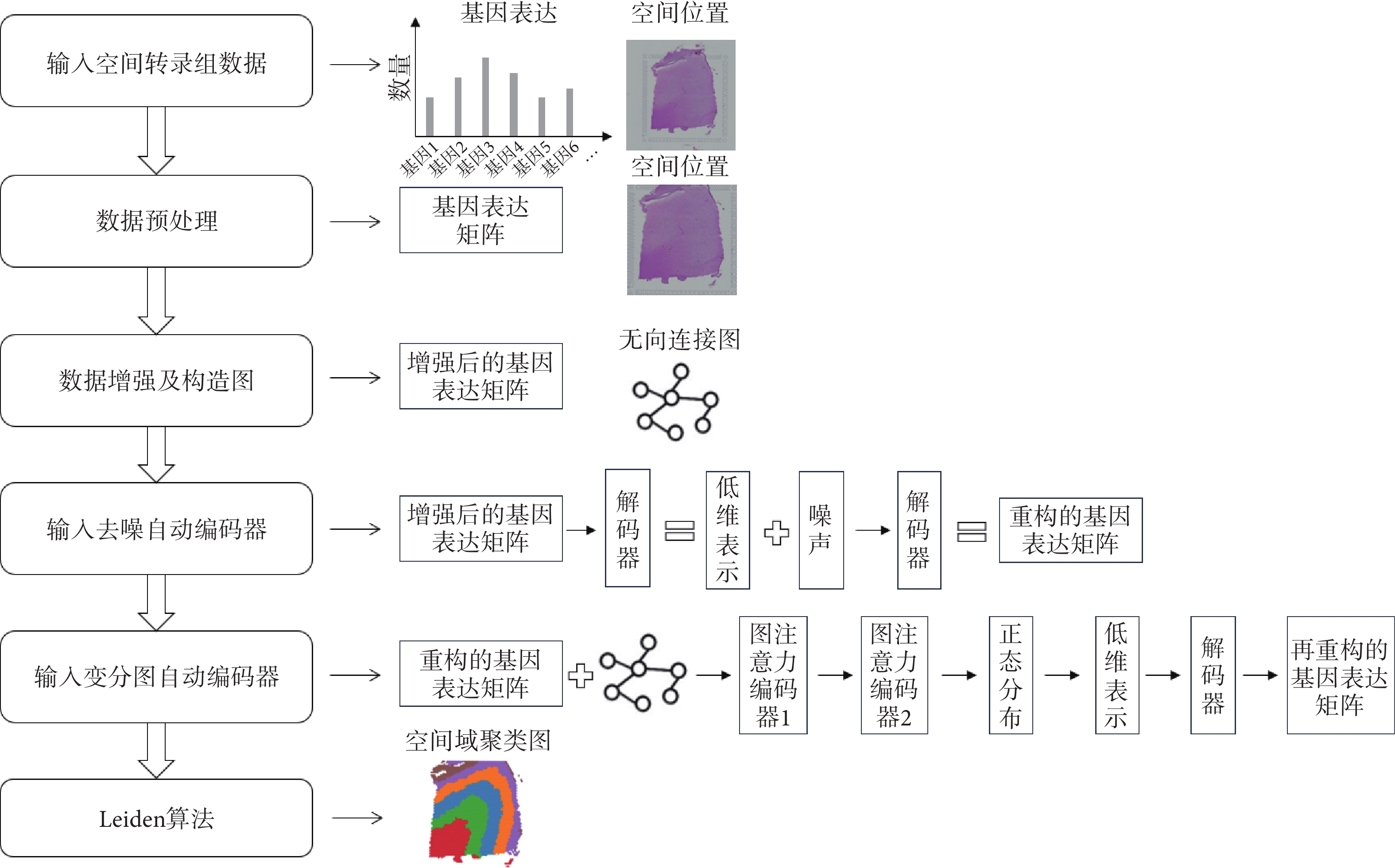 圖1
STCAGAE算法的流程示意圖
Figure1.
The flow chart of STCAGAE algorithm
圖1
STCAGAE算法的流程示意圖
Figure1.
The flow chart of STCAGAE algorithm
2 結果
本文在一個經典空間轉錄組數據集——DLPFC數據集上進行了實驗,驗證了STCAGAE算法的有效性。STCAGAE算法可以將細胞聚類成多個簇,并且簇之間具有明顯的空間分布和表達差異。與傳統的聚類方法相比,STCAGAE算法在聚類性能上更有優勢。
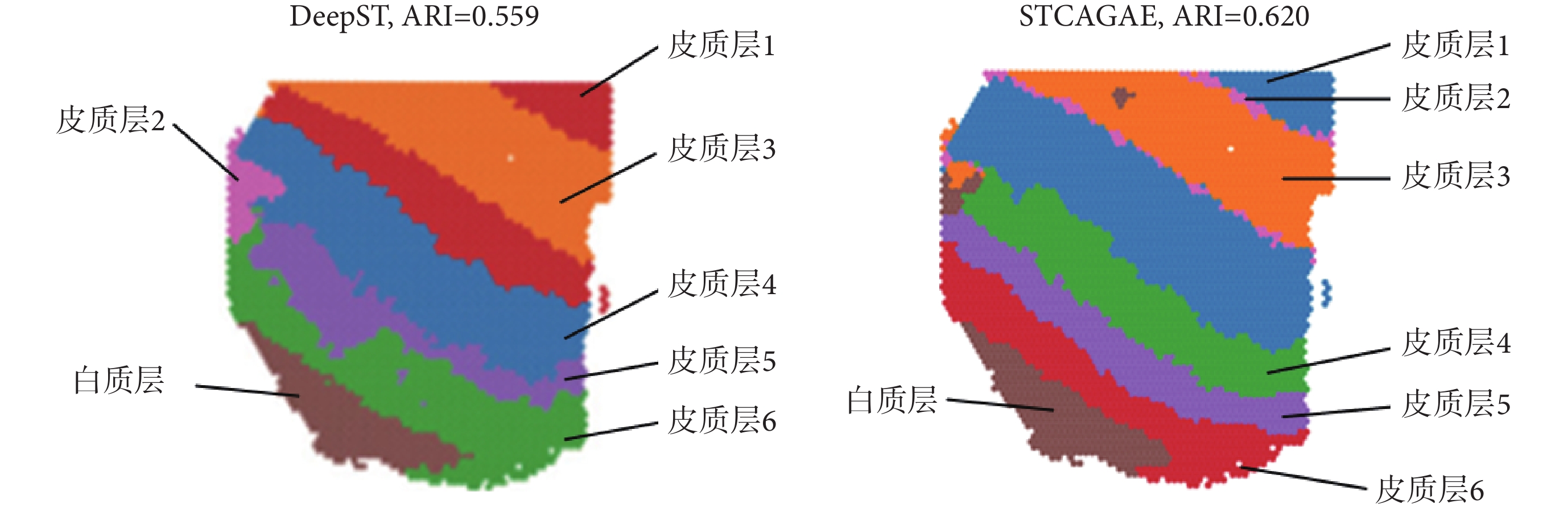
本文在DLPFC數據集的編號為151507的切片上通過STCAGAE算法對空間域進行聚類。同時,比較了其他空間算法在該數據集上的聚類性能。通過ARI值計算識別空間域的精度,如圖2所示,STCAGAE算法的ARI值達到了0.620,優于通過深度學習識別空間轉錄組學中的空間域(identifying spatial domains in spatial transcriptomics by deep learning,DeepST)方法的ARI值0.559[18-19]。
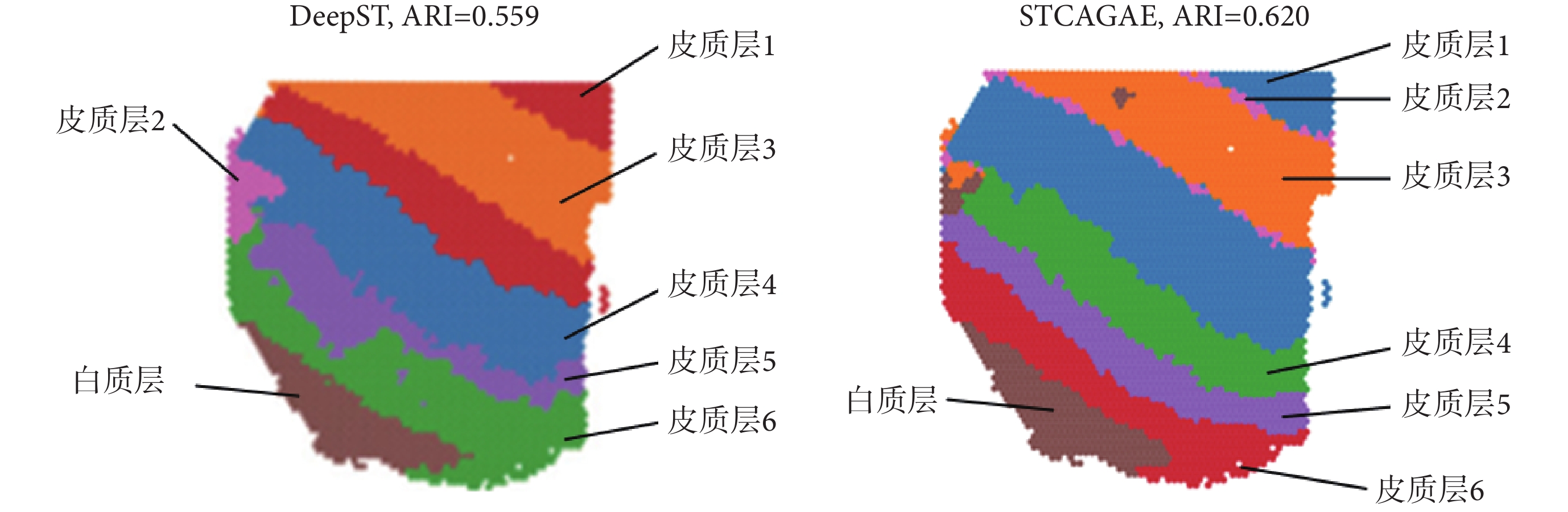 圖2
DLPFC數據集151507切片上的ARI值對比
Figure2.
The comparison of ARI on section 151507 in the DLPFC dataset
圖2
DLPFC數據集151507切片上的ARI值對比
Figure2.
The comparison of ARI on section 151507 in the DLPFC dataset
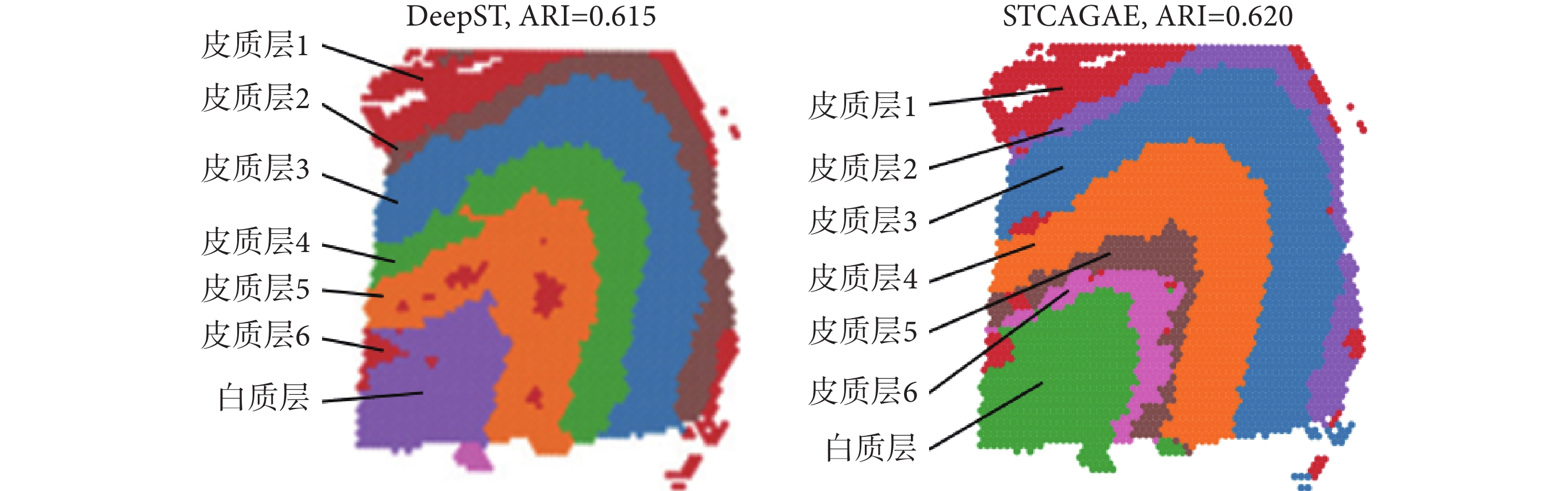
通過STCAGAE算法,又在DLPFC數據集的編號為151673的切片上進行了空間域的聚類。同時也比較了其他空間算法在該數據集上的聚類性能。通過ARI值計算識別空間域的精度,如圖3所示ARI值達到了0.620,優于DeepST算法的ARI值0.615。
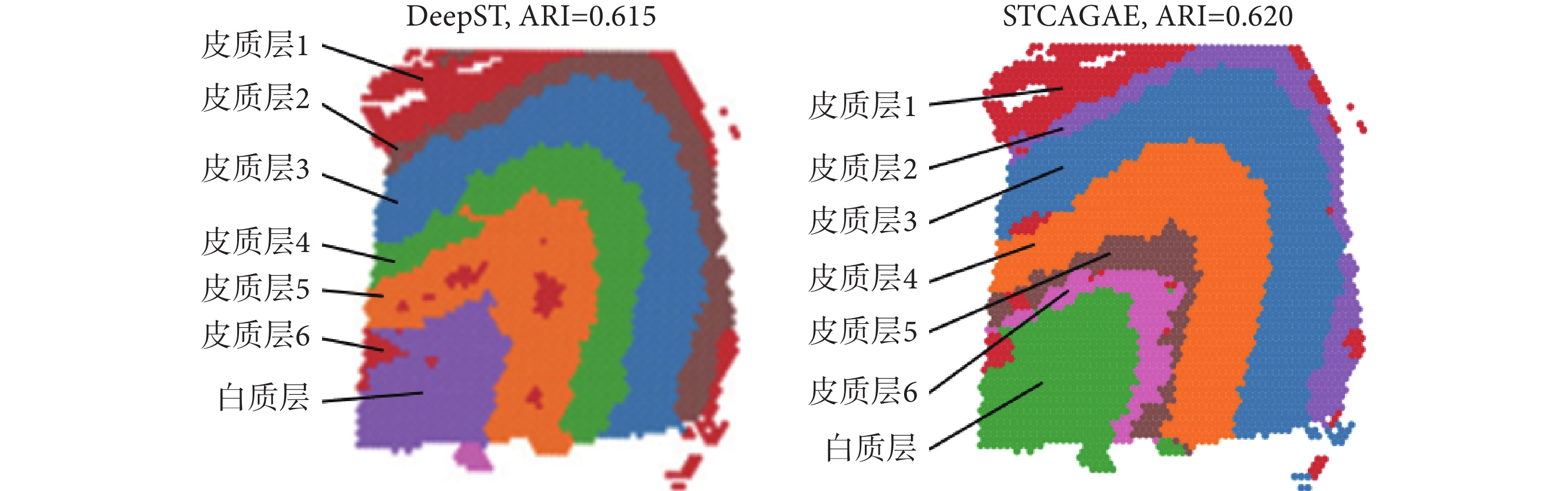 圖3
DLPFC數據集151673切片上ARI值的對比
Figure3.
The comparison of ARI on section 151507 in the DLPFC dataset
圖3
DLPFC數據集151673切片上ARI值的對比
Figure3.
The comparison of ARI on section 151507 in the DLPFC dataset
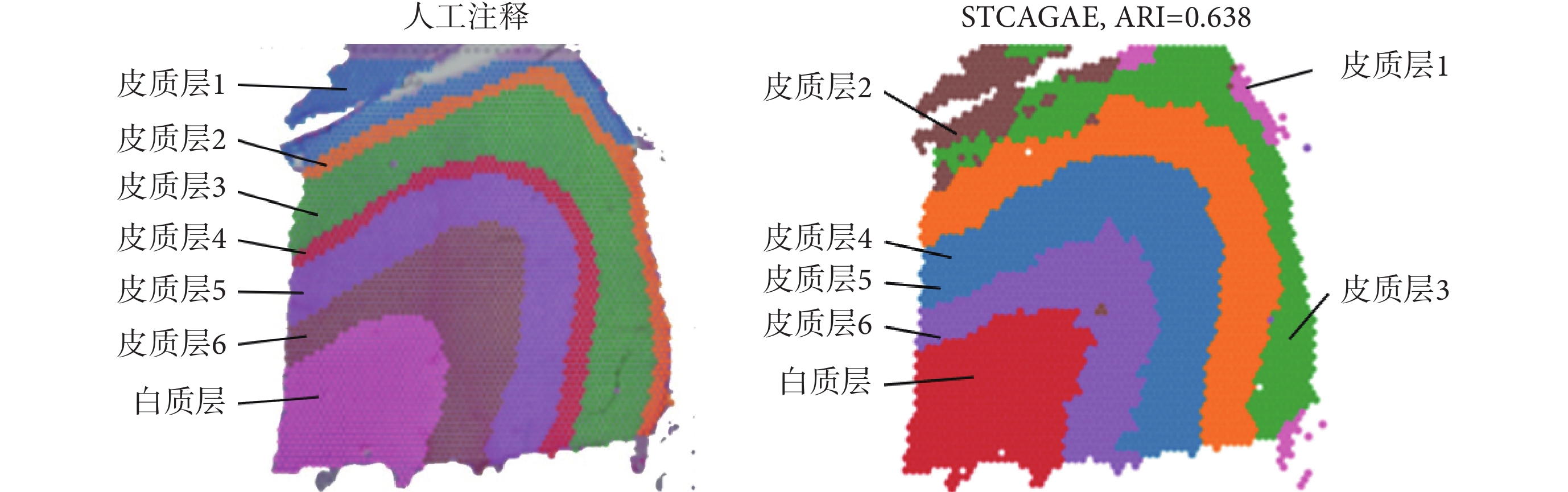
對于DLPFC數據集的151676切片,本文通過STCAGAE算法進行了空間域的聚類。通過ARI值計算識別空間域的精度,如圖4所示ARI值達到了0.638。然后,本文在該數據集上通過其他空間算法進行實驗,并比較各種聚類算法的性能,發現STCAGAE效果優于軟件包Scanpy 1.9.1(Theis Lab實驗室,德國)(ARI = 0.29)、SEDR(ARI = 0.42)、SpaGCN(ARI = 0.44)、BayesSpace(ARI = 0.44)和用于空間解析轉錄組學的自適應圖注意力自動編碼器(deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder,STAGATE)方法[20](ARI = 0.60)。
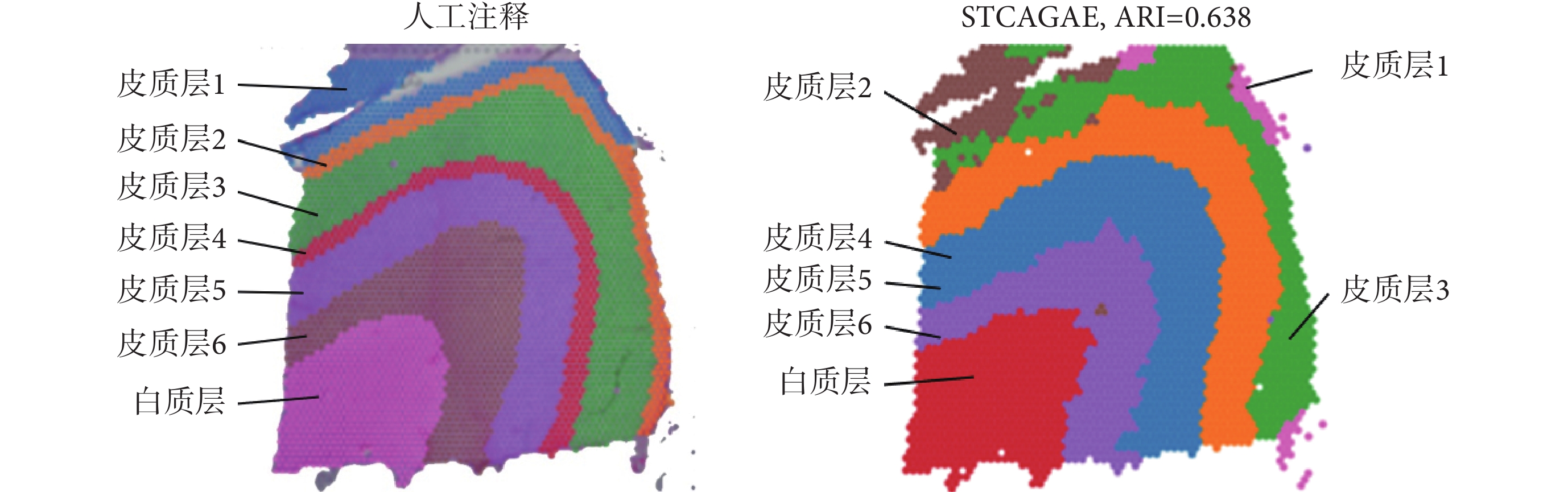 圖4
DLPFC數據集上151676切片的人工注釋以及STCAGAE聚類結果
Figure4.
Human annotation and clustering result of section 151676 in the DLPFC dataset
圖4
DLPFC數據集上151676切片的人工注釋以及STCAGAE聚類結果
Figure4.
Human annotation and clustering result of section 151676 in the DLPFC dataset
本文在DLPFC數據集上測試STCAGAE的性能,發現STCAGAE可以有效地將數據劃分為6個皮質層和1個白質層[21-22]。其中,皮質層1主要由神經膠質細胞、軸突、樹突和少量神經元組成,對神經元活動的調節非常敏感,功能涉及到信息處理、信號傳遞和空間感知等方面;皮質層2是DLPFC的主要輸入層,具有調節和控制信息傳遞的功能,在決策制定、工作記憶和靈活的認知控制等高級認知過程中扮演著重要的角色;皮質層3主要包含錐形神經元,在視覺加工和感知處理中扮演重要的角色;皮質層4主要包含大錐形神經元,參與計劃、決策和行動控制等許多高級認知過程;皮質層5主要由大錐形神經元和星形神經元等神經元組成,對調節神經元活動起到重要的作用,同時也與情緒調節、行動控制和認知過程等方面有關;皮質層6主要由星形神經元和橫向神經元等組成,在感知處理、計劃和動作控制等方面具有重要的作用;白質層是DLPFC的最內層,主要由神經元軸突組成。白質對于DLPFC與其他腦區的信息交流具有重要作用,同時也與情緒調節、決策制定和計劃等各種高級認知過程密切相關。與其他聚類方法相比,STCAGAE實現了顯著的改善。STCAGAE清晰地勾畫出了層的邊界,并獲得了最好的聚類精度。而通過聚類得到的空間域中的各種細胞類型的有機結合與相互作用,能夠行使特定的生物學功能。
3 結論
本文通過整合空間位置、組織學和基因表達來建模空間嵌入表示,提出了一種基于圖注意力網絡的深度學習方法,用于空間轉錄組數據的聚類分析。實驗結果表明,本文提出的方法可以有效地聚類空間轉錄組數據,并且與其他的聚類方法相比具有更好的性能。總之,本文所提方法可以幫助研究人員更好地理解細胞在組織中的分布和表達方式,并為相關領域的研究提供新的工具和思路。
本文通過圖注意力網絡對空間轉錄組數據進行了特征提取,并通過Leiden算法進行聚類。值得注意的是,圖注意力網絡中的注意力是靜態的,也就是說對于某個節點,它的相鄰點的注意力系數是固定不變的,這一點后續會加以改進。另外,進行特征提取之后,聚類分析的方法有很多種,包括基于傳統聚類算法的方法、基于自監督學習的方法、基于端對端訓練的方法等。不同聚類方法的效果不同,什么樣的方法能在不同的空間轉錄組數據上得到最好的效果,這一點還有待進一步探索。
重要聲明
利益沖突聲明:本文全體作者均聲明不存在利益沖突。
作者貢獻聲明:吳瀚文主要負責算法程序設計、數據記錄與分析、論文編寫;高潔主要負責項目主持、數據分析指導、論文審閱修訂。
0 引言
空間轉錄組學是一種結合了組織學和轉錄組學的新興技術,它可以研究細胞在組織中的分布和表達方式。空間轉錄組(spatial transcriptome,ST)數據通常是由高通量成像技術生成,如空間轉錄組學和空間RNA測序(spatial RNA sequencing,sRNA-seq)。這些技術可以在組織中捕獲數百萬個單細胞的轉錄組數據,并提供高分辨率的空間信息。然而,由于數據的高維和復雜性,空間轉錄組數據的分析一直是一個具有挑戰性的問題。識別空間域(即,在基因表達和組織學上都具有空間一致性的區域)是空間轉錄組學中最重要的課題之一。目前,空間域識別方法主要分為非空間聚類和空間聚類兩類。傳統的非空間聚類算法,如K均值聚類算法(k-means clustering algorithm)、魯汶算法(Louvain method),以基因表達數據作為輸入,導致聚類難以符合組織切片[1-2]。而空間聚類方法結合了基因表達、空間位置和形態學,可以解釋基因表達的空間依賴性,以更好地匹配空間位置。例如,Zhao等[3]提出的空間貝葉斯方法(BayesSpace)即是基于貝葉斯統計方法,通過引入空間鄰居結構來鼓勵相鄰點屬于同一聚類,然后將捕獲區域劃分為更小的亞區域,實現了聚類精度的提高。Xu等[4]提出的用于空間轉錄組分析的卷積神經網絡(convolutional neural networks,CNN)學習策略(CNN learning strategy for spatial transcriptomics analysis,CosTA)使用CNN聚類結構,通過CNN生成特征,再通過高斯混合模型聚類生成軟分配[5],并使用軟分配來更新CNN。Yuan等[6]提出的用于基因的圖CNN(graph CNN for genes,GCNG)方法將空間信息編碼為圖形,使用監督訓練將其與表達數據組合,并用圖卷積網絡對圖數據進行特征嵌入,從而實現聚類。Hu等[7]提出的整合基因表達、空間位置和組織學,通過圖卷積網絡識別空間域和空間可變基因(integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network,SpaGCN)方法應用圖卷積網絡來整合基因表達和空間位置,并進一步與自監督模塊相結合來識別空間域。Pham等[8]提出的空間轉錄組學數據的下游分析工具包(stLearn)提供了一種組織內歸一化技術,該技術基于從形態學圖像中收集到的特征和空間位置,通過形態學距離對基因表達進行歸一化。Xu等[9]提出的無監督空間嵌入深度表示方法(unsupervised spatially embedded deep representation of spatial transcriptomics,SEDR)采用深度自動編碼器網絡和圖形自動編碼器來嵌入空間信息。Teng等[10]提出可用于熒光原位雜交技術的迭代細胞分型(fluorescence in situ hybridization iterative cell typing,FICT)方法,能同時利用空間轉錄組數據中的表達信息和空間信息來識別細胞類型和亞型。盡管這些空間聚類方法可以在一定程度上識別空間域,但往往是通過節點的空間位置計算節點之間的距離,并由此預先定義節點之間的相似性,而這些預先定義的方法沒能利用深度學習的靈活性,故存在一定局限。
為了解決這個問題,本文提出了一種基于圖注意力網絡(graph attention networks,GAT)的深度學習方法,并將其命名為用圖注意力自動編碼器進行空間轉錄組聚類分析(spatial transcriptomics clustering analysis with graph attention auto-encoders,STCAGAE),用于空間轉錄組數據的聚類分析[11]。本文使用的圖注意力網絡可以自適應地學習節點之間的相似性,從而更好地聚類空間域。本文利用預先訓練好的深度神經網絡模型從形態圖像塊中提取特征向量,然后將提取的特征與基因表達和空間位置數據相結合,以表征空間相鄰點的相關性,并創建空間增強基因表達矩陣。同時使用圖注意力自動編碼器和去噪自動編碼器聯合生成增強空間轉錄組數據的潛在表示,最后使用萊頓(Leiden)算法最終的嵌入進行聚類[12]。本文旨在識別不同的空間域,并為研究空間轉錄組數據提供新的方法。
1 方法
1.1 數據來源及預處理
本文研究所用數據來自十倍基因組學(10 × Genomics)生物技術公司利用維希姆(Visium)技術發布的高通量空間轉錄組測序平臺(10 × Genomics Visium)。該平臺可以利用Visium技術,從整個組織切片和各種各樣的樣本類型中獲得整個轉錄組高通量基因表達分析結果。本文選取了該平臺的一組可公開下載的數據集,即人類背外側前額葉皮層(dorsolateral prefrontal cortex dataset,DLPFC)數據集。DLPFC數據集包含了12個不同的切片數據,其中每個切片都包含6個皮質層和1個白質層。首先,從DLPFC數據集中移除主要組織區域之外的區域。然后使用計算機編程語言Python 3.8(Guido van Rossum,荷蘭)中分析單細胞數據的軟件包Scanpy 1.9.1(Theis Lab,德國),根據文庫大小對原始基因表達數據進行過濾、對數轉換和標準化[13]。最后,使用主成分分析(principal components analysis,PCA)對增廣基因表達數據進行降維,并將降維數據作為下一個模型訓練的輸入。
1.2 STCAGAE算法
1.2.1 增強空間數據
空間基因表達技術提供了具有額外空間位置信息和組織形態的轉錄組范圍的基因表達譜。本文使用這兩個額外的空間數據實現了節點的基因表達數據增強,得到了對節點更真實的表示,從而提高了STCAGAE的性能。計算點i和點j之間的空間基因表達權重GCij,如式(1)所示:
|
其中,Si代表節點i的基因表達向量, 代表節點i的基因表達向量的均值,Sj代表節點j的基因表達向量, 代表節點j的基因表達向量的均值。對于具有形態學信息的空間轉錄組數據,本文首先根據每個點的坐標對一個圖像進行分割,得到其部分圖像。然后,使用深度學習框架Pytorch 1.8.0(Facebook Inc,美國)中的圖像預處理模塊轉換器(transform)對圖像進行變換和增強,包括歸一化、旋轉、調整銳度等[14]。每個點的特征從預先訓練的CNN模型得出,該模型可以將每個節點的圖像轉換為2 048維的潛在變量。為了更好地表示節點的形態,本文進行了PCA,提取了前50個主成分作為潛在特征。最后,利用余弦距離計算點i與相鄰點j之間的形態相似性權重MSij,如式(2)所示:
|
本文使用空間坐標來確定每個點和所有其他點之間的距離,然后對前4個相鄰點之間的距離進行排序,以計算半徑r。對于一個給定的點i,當且僅當兩個點之間的距離小于r時,認為點j是點i的相鄰點,則設表示點i與點j位置關系的權重參數SWij = 1,否則SWij = 0。然后,增強每個點i的基因表達,如果是10 × Genomics Visium平臺的數據,將數據增強為如式(3)所示;否則,將數據增強為如式(4)所示:
|
|
其中,GEi和GEj代表點i和點i的相鄰點j的原始基因表達。
1.2.2 構造圖
為了結合給定點的相鄰點的相似性,本文使用空間坐標來計算點之間的距離,并使用前6個最近鄰構建一個無向連接圖。對于無向連接圖,在每個點上都添加了自循環。A是鄰接矩陣,那么如果i和j是相鄰點,則在點i和點j處的值Aij = Aji = 1,否則Aij = 0。
1.2.3 去噪自動編碼器
使用Pytorch 1.8.0(Facebook Inc,美國)的線性層實現了一個去噪自動編碼器,用于基因表達的潛在表示[15]。編碼器E由用戶設置的多個完全連接的堆疊線性層組成,將集成的基因表達式X轉換為低維表示,如式(5)所示:
|
其中,Zg代表經過編碼器得到的低維表示,R代表實數集,N代表節點數量,M代表輸入基因數量,d代表最后一個編碼器層的維數。然后,向Zg加入噪聲得到重構的低維表示,如式(6)所示。接下來,解碼器D將重構后的低維表示轉換為重構的基因表達矩陣,如式(7)所示:
| '/> |
| '/> |
其中,代表重構的低維表示,Z代表噪聲向量,D代表解碼器,X'代表重構的基因表達矩陣,d''代表最終層的維數,N、M和d與上述相同。輸入基因和重建表達式的類似性用均方誤差來計算,如式(8)所示:
|
其中,Ll代表去噪自編碼器的損失函數,Xi代表節點i的基因表達向量,E(Xi)代表節點i的基因表達向量經過編碼器層得到的低維表示,D(?)代表經過解碼器層重構后的表示。
1.2.4 變分圖自動編碼器
變分圖自動編碼器使用了基于Pytorch 1.8.0(Facebook Inc,美國)構建的用于編寫和訓練圖神經網絡的庫Pytorch Geometric 2.2.0(Technische University Dortmund,德國) [16],其中編碼器由圖注意力網絡組成。圖注意力網絡將特征矩陣X作為輸入,首先通過一個線性變換W做維度轉換,如式(9)所示:
|
其中,代表特征矩陣經過線性變換之后的列數。接著使用自注意力(self-attention)為每個節點分配注意力,對于點i和點j,它們的初始特征為hi和hj,于是它們經過線性變換W后變為Whi和Whj,為它們分配注意力,如式(10)所示:
|
其中 ,eij 代表節點 i 對節點 j 的影響力系數,a (·)代表計算節點相關度的函數,如式(11)所示:
| '/> |
在本文中,對于節點i,只計算其鄰域內節點j對節點i的影響力系數。為了更好地在不同節點之間分配權重,本文將目標節點與所有相鄰點計算出來的相關度進行統一的歸一化處理,使用線性整流函數(rectified linear unit,ReLU),如式(12)所示:
|
其中,代表歸一化后的相關度權重系數,Ni代表節點i的鄰域,eik代表節點 i 對節點 i鄰域內的節點 k 的影響力系數。每個節點的最終輸出特征向量如式(13)所示:
| '/> |
其中,hi'代表節點的最終輸出特征,σ代表激活函數。損失函數包括生成圖與原始圖之間的重構損失,節點表示的向量分布和正態分布的相對熵(relative entropy),如式(14)所示:
|
其中,Lg代表變分圖自動編碼器的損失函數,代表二進制交叉熵函數,代表相對熵,,代表正態分布。
同時,本文利用多頭注意力以穩定學習過程和增加模型容量。多頭注意力就是將同一個輸入分別送入不同的注意力層,分別計算隱藏狀態,然后將不同的特征表示連接起來(或相加)。經過不斷調整參數并實驗,本文發現模型中的參數取8個注意力頭和特征相加時,DLPFC數據集的不同切片聚類得到的調整蘭德系數(adjusted Rand index,ARI)值都最大,因此模型達到最佳效果。
1.2.5 聚類與可視化
在得到嵌入之后,使用Leiden算法來識別空間域,以識別相似的細胞和模式。為了獲得最佳分辨率,在0.1~2.5之間進行網格搜索,步長為0.01,直到達到必要的簇數。為了可視化,使用了統一流形逼近和投影[17]。
綜上,本文提出STCAGAE算法,如圖1所示。通過對整合基因表達、空間位置和組織形態信息的低維表示進行建模來表征空間域。為了建立形態學特征矩陣,首先使用預先訓練好的深度學習網絡對蘇木精-伊紅(hematoxylin-eosin,H&E)染色的組織地形圖數據進行處理。結合形態特征和空間相鄰點信息,每個點的基因表達均得到增強。然后,利用去噪自編碼器學習從集成特征空間到低維表示空間的非線性映射,以減少模型的過擬合。同時,基于節點之間的歐氏距離計算圖鄰接矩陣。將增強后的基因表達矩陣和圖鄰接矩陣輸入到一個變分圖自動編碼器中,從而通過與相應的空間相鄰點的集成表示來生成空間嵌入。最終的潛在嵌入是通過連接集成表示和空間嵌入來形成的。最后使用Leiden算法進行空間域聚類,識別出從右上到左下依次是皮質層1~6、白質層的不同空間域。
圖1
STCAGAE算法的流程示意圖
Figure1.
The flow chart of STCAGAE algorithm
2 結果
本文在一個經典空間轉錄組數據集——DLPFC數據集上進行了實驗,驗證了STCAGAE算法的有效性。STCAGAE算法可以將細胞聚類成多個簇,并且簇之間具有明顯的空間分布和表達差異。與傳統的聚類方法相比,STCAGAE算法在聚類性能上更有優勢。
本文在DLPFC數據集的編號為151507的切片上通過STCAGAE算法對空間域進行聚類。同時,比較了其他空間算法在該數據集上的聚類性能。通過ARI值計算識別空間域的精度,如圖2所示,STCAGAE算法的ARI值達到了0.620,優于通過深度學習識別空間轉錄組學中的空間域(identifying spatial domains in spatial transcriptomics by deep learning,DeepST)方法的ARI值0.559[18-19]。
圖2
DLPFC數據集151507切片上的ARI值對比
Figure2.
The comparison of ARI on section 151507 in the DLPFC dataset
通過STCAGAE算法,又在DLPFC數據集的編號為151673的切片上進行了空間域的聚類。同時也比較了其他空間算法在該數據集上的聚類性能。通過ARI值計算識別空間域的精度,如圖3所示ARI值達到了0.620,優于DeepST算法的ARI值0.615。
圖3
DLPFC數據集151673切片上ARI值的對比
Figure3.
The comparison of ARI on section 151507 in the DLPFC dataset
對于DLPFC數據集的151676切片,本文通過STCAGAE算法進行了空間域的聚類。通過ARI值計算識別空間域的精度,如圖4所示ARI值達到了0.638。然后,本文在該數據集上通過其他空間算法進行實驗,并比較各種聚類算法的性能,發現STCAGAE效果優于軟件包Scanpy 1.9.1(Theis Lab實驗室,德國)(ARI = 0.29)、SEDR(ARI = 0.42)、SpaGCN(ARI = 0.44)、BayesSpace(ARI = 0.44)和用于空間解析轉錄組學的自適應圖注意力自動編碼器(deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder,STAGATE)方法[20](ARI = 0.60)。
圖4
DLPFC數據集上151676切片的人工注釋以及STCAGAE聚類結果
Figure4.
Human annotation and clustering result of section 151676 in the DLPFC dataset
本文在DLPFC數據集上測試STCAGAE的性能,發現STCAGAE可以有效地將數據劃分為6個皮質層和1個白質層[21-22]。其中,皮質層1主要由神經膠質細胞、軸突、樹突和少量神經元組成,對神經元活動的調節非常敏感,功能涉及到信息處理、信號傳遞和空間感知等方面;皮質層2是DLPFC的主要輸入層,具有調節和控制信息傳遞的功能,在決策制定、工作記憶和靈活的認知控制等高級認知過程中扮演著重要的角色;皮質層3主要包含錐形神經元,在視覺加工和感知處理中扮演重要的角色;皮質層4主要包含大錐形神經元,參與計劃、決策和行動控制等許多高級認知過程;皮質層5主要由大錐形神經元和星形神經元等神經元組成,對調節神經元活動起到重要的作用,同時也與情緒調節、行動控制和認知過程等方面有關;皮質層6主要由星形神經元和橫向神經元等組成,在感知處理、計劃和動作控制等方面具有重要的作用;白質層是DLPFC的最內層,主要由神經元軸突組成。白質對于DLPFC與其他腦區的信息交流具有重要作用,同時也與情緒調節、決策制定和計劃等各種高級認知過程密切相關。與其他聚類方法相比,STCAGAE實現了顯著的改善。STCAGAE清晰地勾畫出了層的邊界,并獲得了最好的聚類精度。而通過聚類得到的空間域中的各種細胞類型的有機結合與相互作用,能夠行使特定的生物學功能。
3 結論
本文通過整合空間位置、組織學和基因表達來建模空間嵌入表示,提出了一種基于圖注意力網絡的深度學習方法,用于空間轉錄組數據的聚類分析。實驗結果表明,本文提出的方法可以有效地聚類空間轉錄組數據,并且與其他的聚類方法相比具有更好的性能。總之,本文所提方法可以幫助研究人員更好地理解細胞在組織中的分布和表達方式,并為相關領域的研究提供新的工具和思路。
本文通過圖注意力網絡對空間轉錄組數據進行了特征提取,并通過Leiden算法進行聚類。值得注意的是,圖注意力網絡中的注意力是靜態的,也就是說對于某個節點,它的相鄰點的注意力系數是固定不變的,這一點后續會加以改進。另外,進行特征提取之后,聚類分析的方法有很多種,包括基于傳統聚類算法的方法、基于自監督學習的方法、基于端對端訓練的方法等。不同聚類方法的效果不同,什么樣的方法能在不同的空間轉錄組數據上得到最好的效果,這一點還有待進一步探索。
重要聲明
利益沖突聲明:本文全體作者均聲明不存在利益沖突。
作者貢獻聲明:吳瀚文主要負責算法程序設計、數據記錄與分析、論文編寫;高潔主要負責項目主持、數據分析指導、論文審閱修訂。