T細胞受體(TCR)與抗原肽的特異性結合在調節和介導免疫的過程中發揮著關鍵作用,為腫瘤疫苗設計提供了必要的基礎。近年來的研究主要集中在主要組織相容性復合體(MHC)Ⅰ類抗原的TCR預測,針對MHC Ⅱ類抗原的TCR預測研究尚不充分,仍存在較大的提升空間。本研究利用ProtT5大模型進行MHC Ⅱ類抗原肽和TCR結合預測研究,探究其特征提取能力。此外,對模型進行微調,并構建前饋神經網絡結構進行融合從而實現預測模型。實驗結果顯示,本研究提出的方法較傳統方法表現更佳,預測準確度達到0.96,AUC達到0.93,驗證了本文提出模型的有效性。
引用本文: 許珉瑞, 張斯文, 魯曼曼, 高媛, 張夢歡, 林勇, 謝鷺. 基于大模型的MHC II抗原肽-T細胞受體結合預測. 生物醫學工程學雜志, 2024, 41(6): 1243-1249. doi: 10.7507/1001-5515.202405024 復制
版權信息: ?四川大學華西醫院華西期刊社《生物醫學工程學雜志》版權所有,未經授權不得轉載、改編
0 引言
腫瘤免疫治療被認為是癌癥治療領域的一次革命性突破,有效的靶向免疫療法需要準確預測可能觸發T細胞免疫應答的腫瘤特異性表位。抗原依據其特性,分為腫瘤相關抗原與腫瘤特異性抗原,它們通過主要組織相容性復合體(major histocompatibility complex,MHC)呈現在腫瘤細胞表面,隨后啟動T細胞介導的免疫應答,以消滅腫瘤細胞[1]。T細胞受體(T cell receptor,TCR)的激活依賴于它與肽-MHC(pMHC復合物)的識別過程[2]。TCR是由兩條不同肽鏈構成的二聚體,其胞外區由恒定區(C)和可變區(V)組成,V區又可分為三個互補決定區(CDR1-3),CDR1和CDR2負責識別MHC分子,而關鍵的CDR3區則是抗原的特異性識別部位[3]。CDR3的結構和功能在TCR與pMHC復合物的相互作用中至關重要,其特異性識別機制將影響免疫治療策略的精確設計。
高通量測序的發展推動了抗原肽與TCR結合計算方法的開發。基于傳統機器學習的方法,Gielis等[4]分析序列的物理化學性質,構建了基于隨機森林的模型評估并預測結合概率。De Neuter等[5]通過訓練隨機森林分類器以探究TCR識別抗原的機制。基于深度學習的方法,NetTCR-2.0[6]利用卷積網絡將肽和CDR3 β序列信息整合。ERGO[7]利用長短期記憶人工神經網絡來提取序列特征,用于預測兩者結合概率。本團隊利用自構建編碼方式AAPP,提出名為iTCep[8]的深度學習框架,預測MHC I類分子呈現的肽與TCR之間的相互作用。
盡管該研究領域展現出顯著的發展,但仍有潛力進一步提升和完善。傳統意義上認為CD8+ T細胞是主要的腫瘤殺傷細胞,但最近的臨床試驗表明CD4+ T細胞在腫瘤控制中也起到了重要作用。而現有研究主要集中于CD8+ T細胞與MHC Ⅰ類抗原結合預測,忽略了CD4+ T細胞與MHC Ⅱ類抗原的相互作用,模型性能仍有待提高。此外,現有模型多為單一模型,限制了從復雜數據中挖掘關聯性的能力,鑒于生物學數據通常呈現高維特征,開發能夠有效分析復雜數據集的模型變得尤為重要,因此本研究利用ProtT5模型對MHC Ⅱ類抗原肽與TCR的結合進行預測,希望能夠提供有益的探索經驗。
1 材料與方法
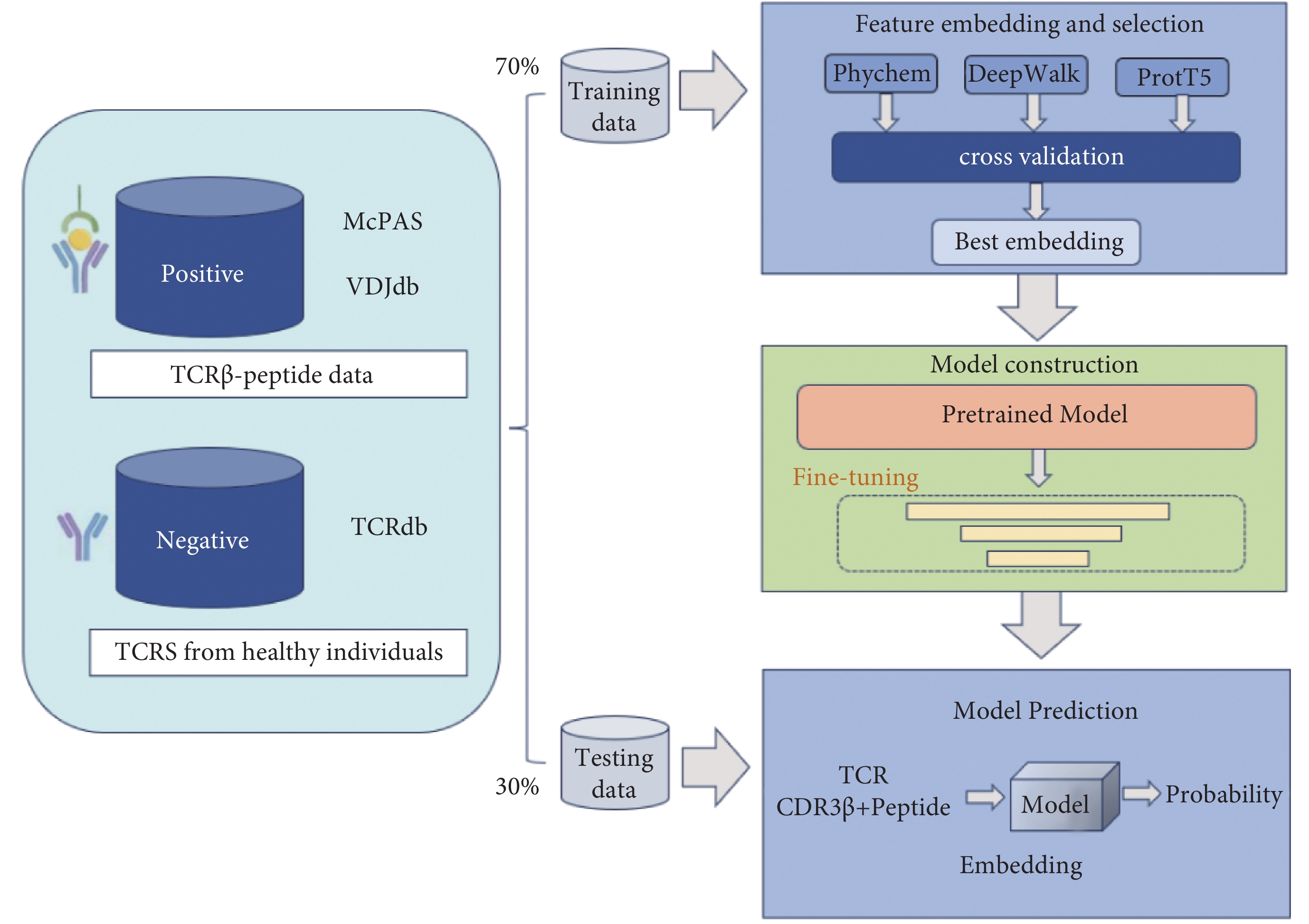
1.1 實驗總體流程
為提升MHC Ⅱ類抗原肽-TCR結合預測模型的性能,本研究在現有研究基礎上進行了改進。實驗框架設計如圖1所示,涵蓋數據收集、特征編碼與選擇、模型構建和模型預測四個關鍵步驟。
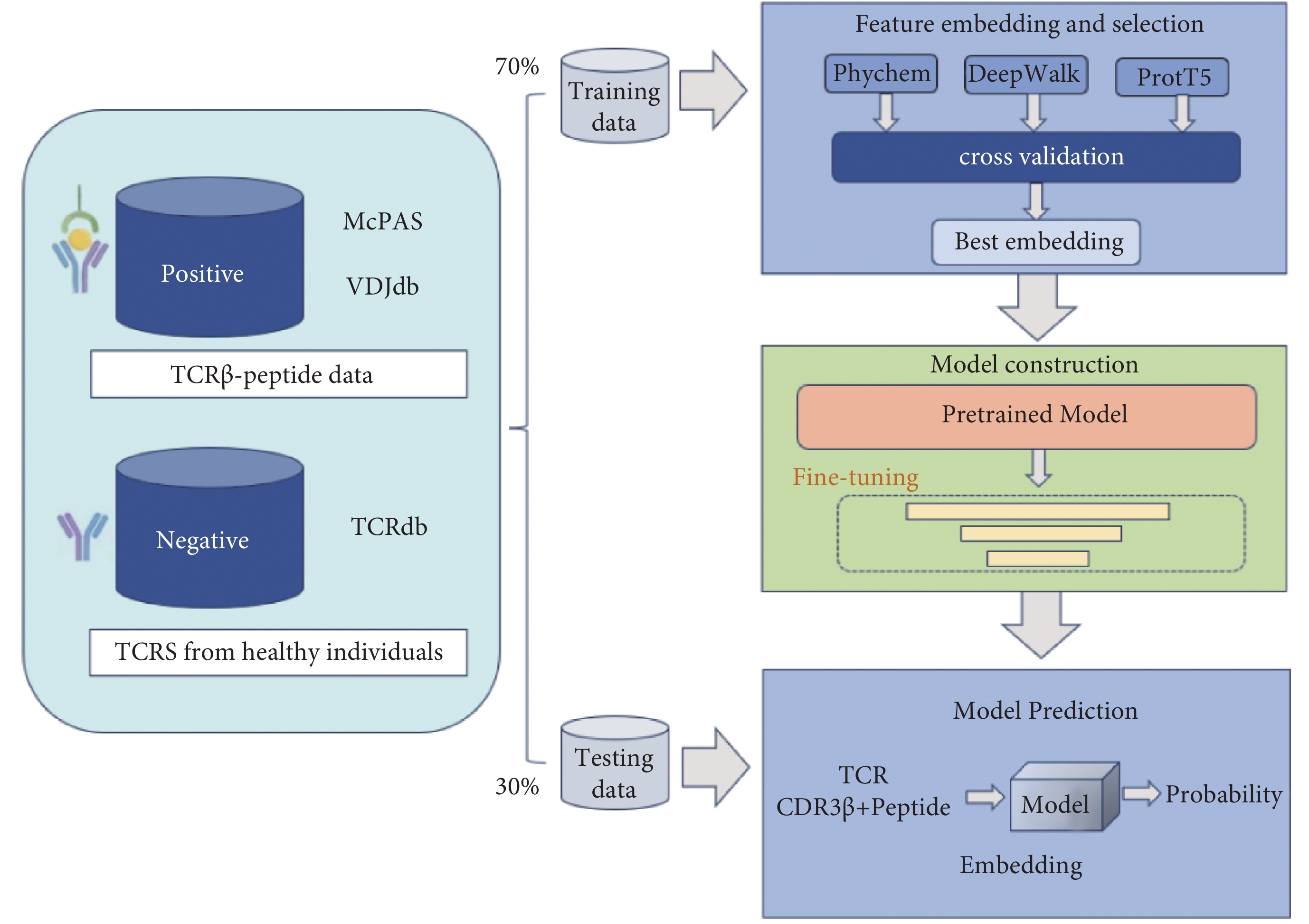 圖1
肽-TCR結合預測模型研究框架圖
Figure1.
Framework diagram for the study of peptide-TCR binding prediction model
圖1
肽-TCR結合預測模型研究框架圖
Figure1.
Framework diagram for the study of peptide-TCR binding prediction model
首先,本研究收集了公共數據庫中的TCR與II類MHC提呈抗原肽的數據,并進行了過濾和預處理。隨后,采用不同編碼策略將抗原肽和TCR序列轉換為相應的特征。為尋找最佳編碼方式,使用機器學習交叉驗證進行評估。在確定最優編碼策略后,對ProtT5模型進行了微調,以更好地適應研究需求。為全面評估模型性能,用多種指標綜合判斷。最后,將微調后的模型應用于測試集,以評估其泛化能力。
1.2 實驗數據
隨著測序技術的發展,越來越多的TCR序列及抗原肽數據被收集,現有數據庫提供了TCR與抗原對接的信息,為人工智能預測TCR與抗原的結合奠定了數據基礎。本研究使用的數據集源于公開的VDJdb[9]和McPAS-TCR[10]數據庫,從中提取MHC II類抗原肽和TCR信息,其中陽性數據分別為782條和898條。為了構建陰性數據并模擬真實世界TCR與抗原肽的情況,從TCRdb[11]數據庫中選取了來自健康供體的TCR β鏈序列共16 800條,使用的項目號為PRJNA390125(樣本編號為SRR5676649、SRR5676658)。經合并去重后得到一個完整的數據集,劃分訓練集、驗證集與測試集。
1.3 數據預處理
本研究剔除了MHC Ⅰ類抗原肽段,以及VDJdb數據庫中置信評分等于0的數據(0表示關鍵信息的缺失),著重研究MHC Ⅱ類抗原肽段;刪除序列中出現錯誤的氨基酸(B、J、O、U、X、Z);刪除不以半胱氨酸(C)開始或不以苯丙氨酸(F)結尾的CDR3序列。合并數據進行去重,最終得到有效陽性數據943條,陰性數據9 430條,共10 373條配對的抗原肽和TCR結合信息,其中MHC Ⅱ類分子呈遞的抗原肽大多是13和20個氨基酸數目的肽段。
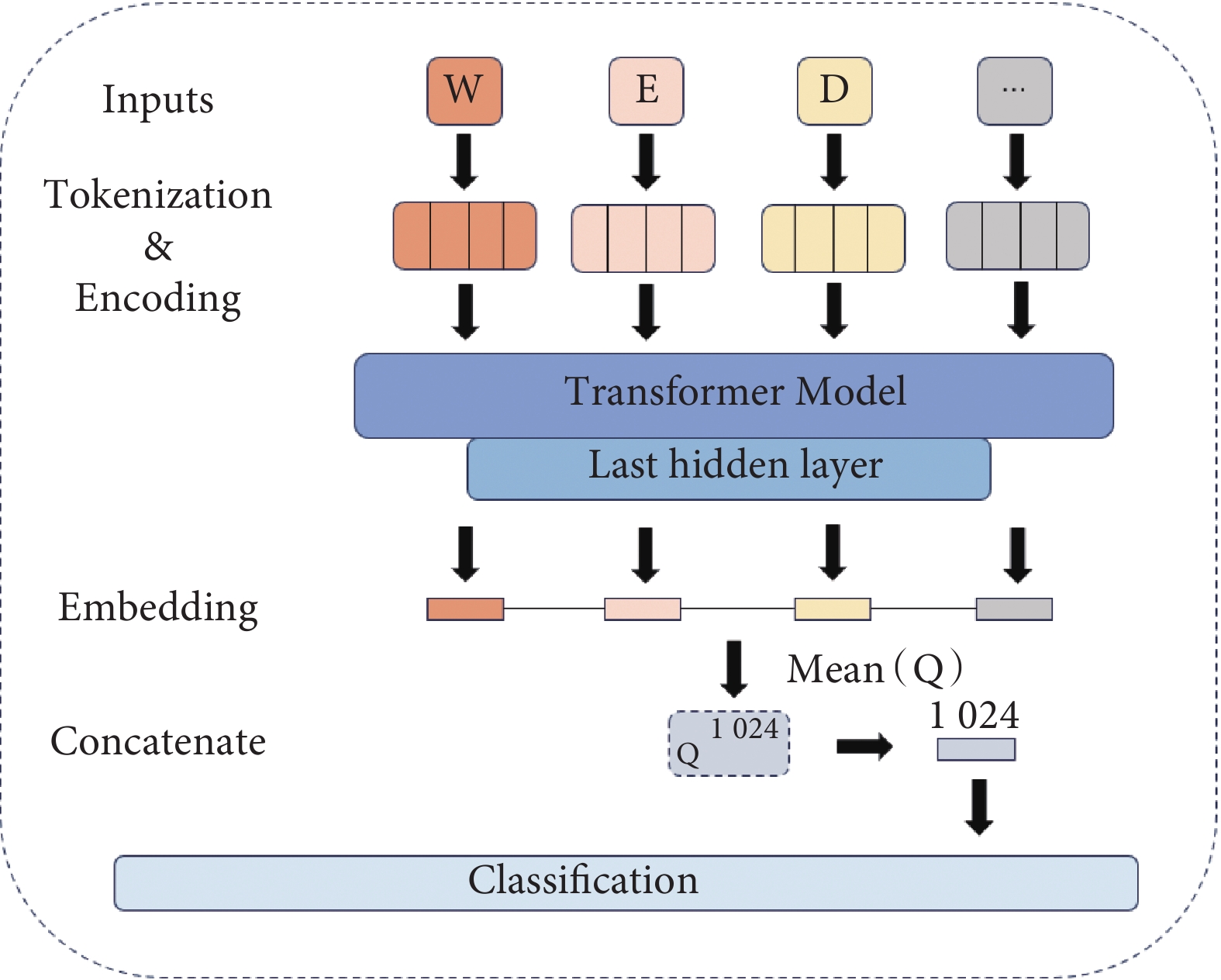
1.4 基于ProtT5模型特征提取分析
ProtT5模型[12]是一種基于Transformer架構的蛋白質預訓練模型,通過自監督方式在UniRef50數據集上(現包含6 600萬個蛋白質序列)進行訓練。模型從這些蛋白質序列中生成輸入和標簽,并隨機掩蓋輸入中的15%氨基酸,通過這種方式訓練得到的特征會捕捉到潛在的蛋白質信息。利用自注意力機制,ProtT5模型可以有效地識別和編碼蛋白質序列中的遠程交互依賴性。這使得ProtT5成為蛋白質相互作用預測等領域內一個強有力的工具。
在機器學習或深度學習模型的構建過程中,數據預處理環節涉及將原始數據編碼為數值化的向量或張量,這一步驟被稱為特征工程或特征表示學習。對于本研究中的TCR和Peptide序列數據,這一過程涉及到氨基酸殘基的向量化或嵌入(embedding),將生物序列轉換為機器學習算法可操作的數值形式,實現有效的模式識別和學習。
具體來說,在兩個序列之間添加一個分隔符,然后將它們作為一個整體輸入到模型中。ProtT5模型會處理這個長序列,并輸出它的隱藏狀態。提取出模型最后一層的隱藏嵌入,該層包含了一定數量的1 024維向量。這一處理過程旨在將抗原肽和TCR序列轉換為高維度的特征向量表示,這些特征向量不僅包含了整體的序列信息,還涵蓋了大量蛋白質功能等相關信息,具體流程如圖2所示。此外,對三種不同的編碼策略進行比較,分別是基于氨基酸理化性質的編碼(Phychem)[13]、隨機游走算法編碼(Deep Walk)[14]和基于ProtT5大模型的編碼,選取合適的特征提取方法是構建深度學習模型的重要一步。
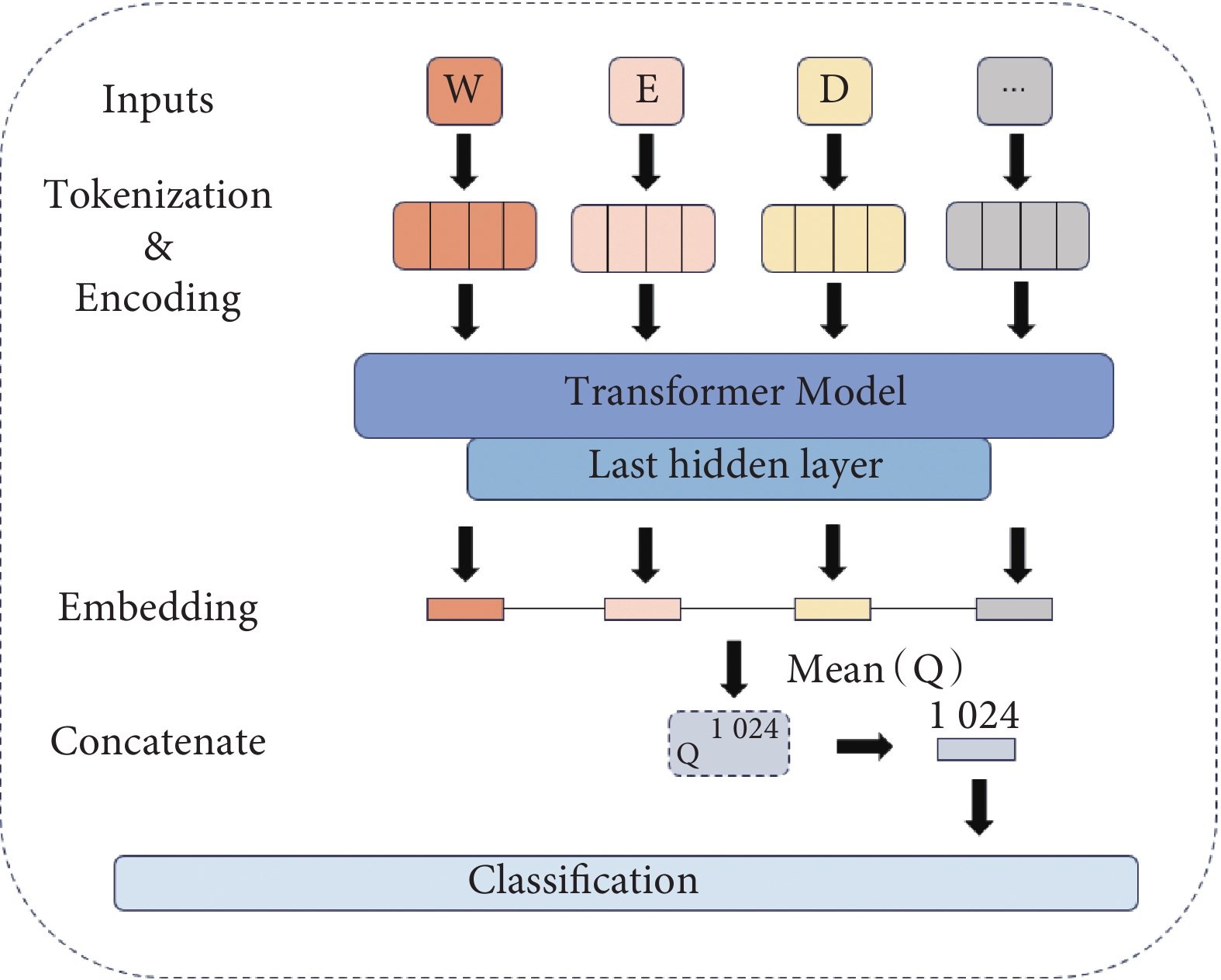 圖2
ProtT5模型特征提取流程圖
Figure2.
The feature extraction process diagram of the ProtT5 model
圖2
ProtT5模型特征提取流程圖
Figure2.
The feature extraction process diagram of the ProtT5 model
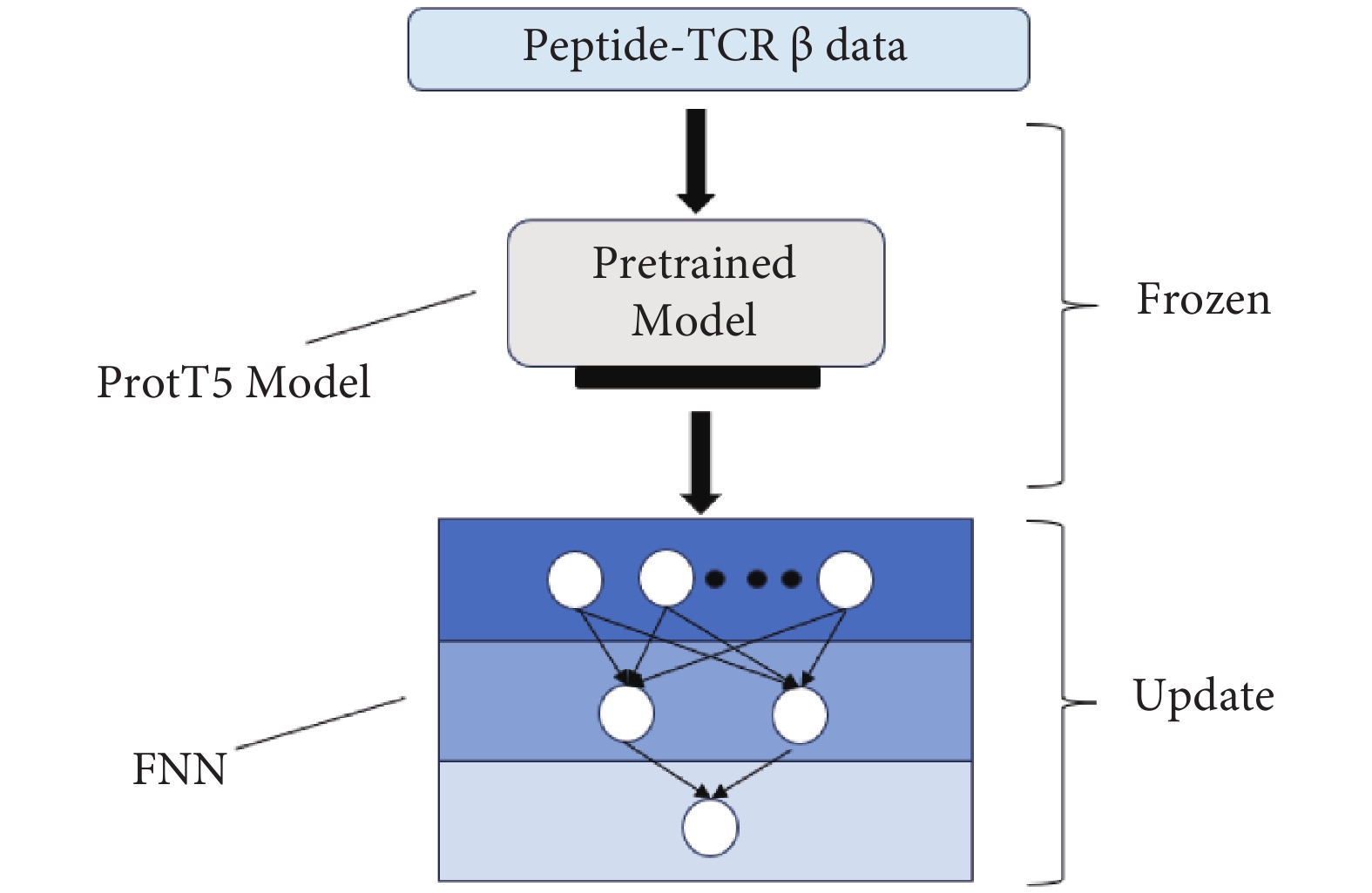
1.5 預測模型構建
盡管ProtT5模型經預訓練后積累了豐富的蛋白質信息,但在TCR與抗原肽的預測任務中并不完全適用。為更好地滿足研究需求,本研究對ProtT5模型進行了訓練和微調,顯著提升了其性能和適用性,能夠更有效地處理MHC II類抗原肽與TCR之間的復雜結合關系。
具體而言,本研究通過嵌入(Embedding)抗原肽和TCR序列以提取關鍵特征,獲取對當前數據的映射,并將嵌入后的向量輸入ProtT5模型進行訓練。由于ProtT5模型的前層網絡已能提供良好的底層特征,我們凍結了相對底層的權重,保留原有特征提取能力。為了更好地滿足任務需求,接入一個前饋神經網絡層,提取和組合底層特征,最終輸出預測結果。網絡模型結構示意圖見圖3。
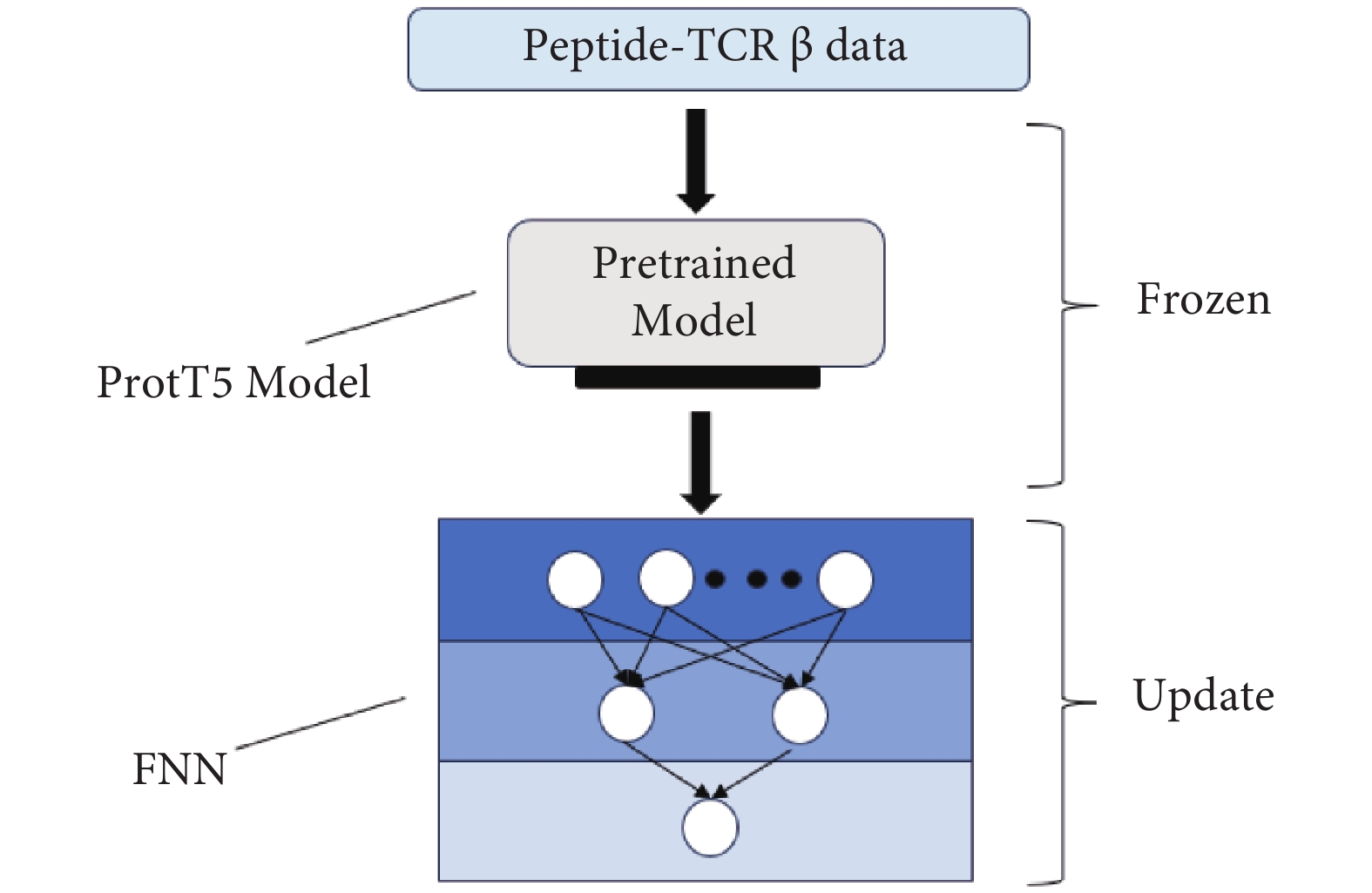 圖3
ProtT5模型微調結構圖
Figure3.
Structural diagram of the ProtT5 fine-tuned model
圖3
ProtT5模型微調結構圖
Figure3.
Structural diagram of the ProtT5 fine-tuned model
模型訓練過程中,采用學習率預熱(warm-up)的策略,優勢在于能夠有效避免模型在訓練初期出現的震蕩問題。在初始幾輪訓練中,采用較小的學習率讓模型逐漸穩定。當模型達到相對穩定狀態時,逐步增加學習率至預設值,以加速模型的收斂速度并提高效果。
1.6 針對不平衡數據集的訓練策略
由于數據集中正負樣本分布比例不平衡,分類器可能傾向于預測出現頻率較高的類別,進而忽略出現頻率較低的類別,導致訓練效果低下。為此,選取Focal Loss[15]作為損失函數,擴展評估指標范圍,而不僅僅局限于準確率。
Focal Loss(聚焦損失)是一種用于處理類別不平衡問題的損失函數,它具有調節正負樣本權重和易分類與難分類樣本權重的能力,通過增加困難樣本的權重,降低簡單樣本的權重,使模型能夠有針對性地學習難易樣本之間的差異。Focal Loss公式如式(1)所示:
 |
Pt表示預測概率,α表示控制正負樣本對loss的貢獻,本研究我們選取α為0.5、γ為2作為訓練的參數。
1.7 實驗環境和評價指標
本研究通過Python 3.9.7編寫實驗代碼,基于PyTorch框架實現,硬件環境為Intel(R) Core(TM) i9-12900K,顯卡為RTX
為了驗證不同編碼方式對模型性能的影響,我們采用準確率(Accuracy,ACC)、精準率(Precision)、召回率(Recall)、F1-score和受試者操作特征(receiver operating characteristic,ROC)曲線下面積(area under curve,AUC)五個測量指標進行評估。
準確率表示抗原肽與TCR預測正確的個數占預測所有數據個數的比例,計算公式如式(2)所示:
 |
精準率表示抗原肽與TCR預測為結合的對子中,實際上為真實數據的比例,公式如式(3)所示:
 |
召回率又叫做查全率,直觀地說是分類器找到所有正樣本的能力,公式如式(4)所示:
 |
F1-score是一個綜合評價分類模型性能的指標,適用于二分類問題,它同時考慮了模型的準確率和召回率,公式如式(5)所示:
 |
AUC表示ROC曲線下的面積,主要用于評估模型的泛化能力,即分類器效果的優劣。AUC作為一個數值指標,它比ROC更具可比性,能夠進行量化比較。其值范圍在0到1之間,越接近于1表示分類器性能越好。
2 實驗結果與分析
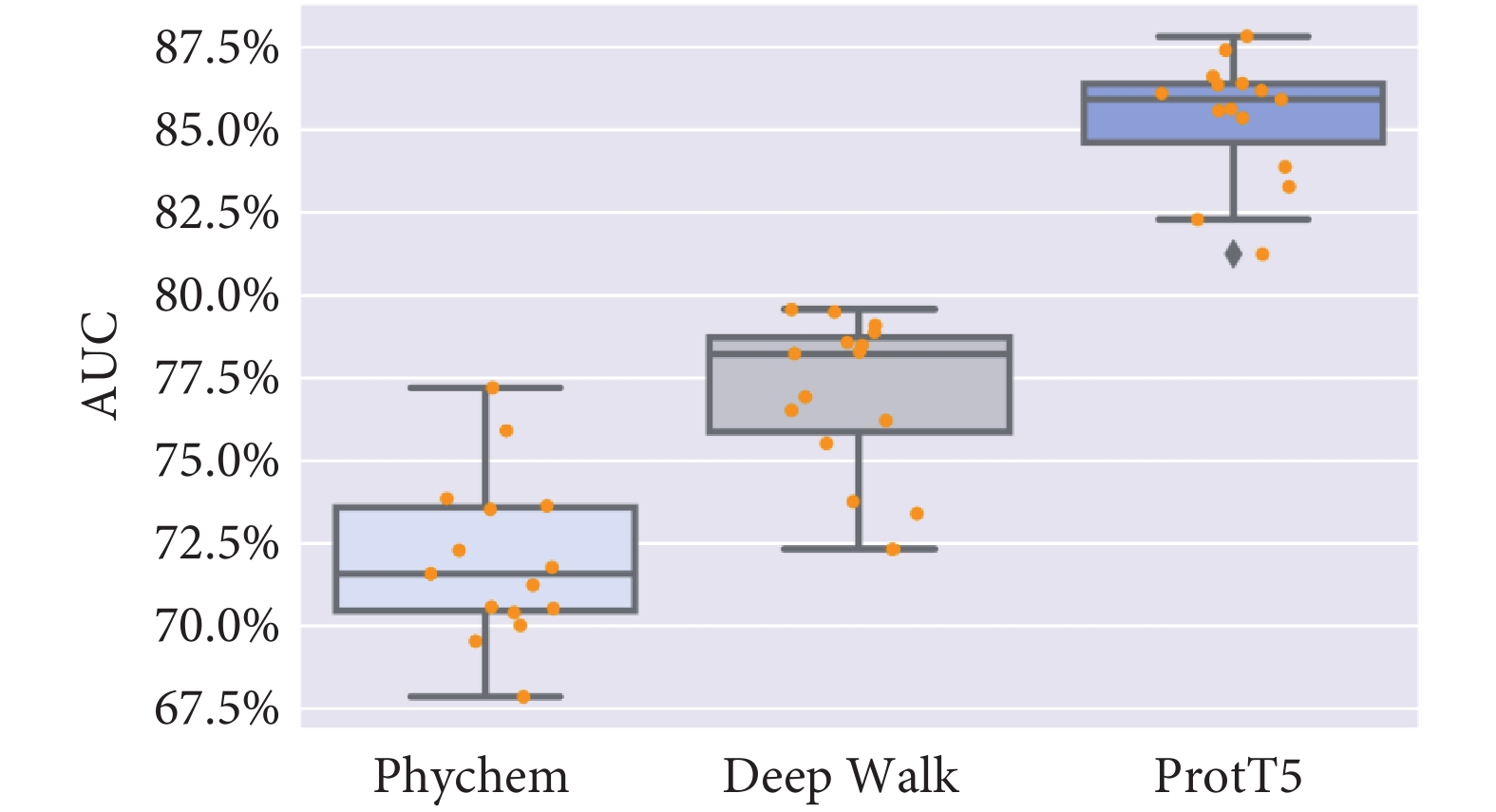
2.1 不同特征提取方法的比較
為研究不同特征提取方法對模型性能的影響,對三種編碼策略進行實驗測試,包括氨基酸理化性質編碼、隨機游走編碼和ProtT5模型生成的嵌入向量編碼。隨后,將原始數據集(訓練集、驗證集)分為五個相等大小的子集,結合不同編碼策略和SVM模型,采用三次獨立的五折交叉驗證評估各特征對模型性能的影響。在實驗中,將AUC作為主要評價指標,結果如圖4所示。結果說明,在TCR與MHC II類抗原肽結合預測任務中,ProtT5模型展現出顯著的優勢,其AUC的平均值為85.33%,高于氨基酸理化性質(71.99%)和隨機游走(77.06%)方法,表明了本文所提出模型的可靠性。
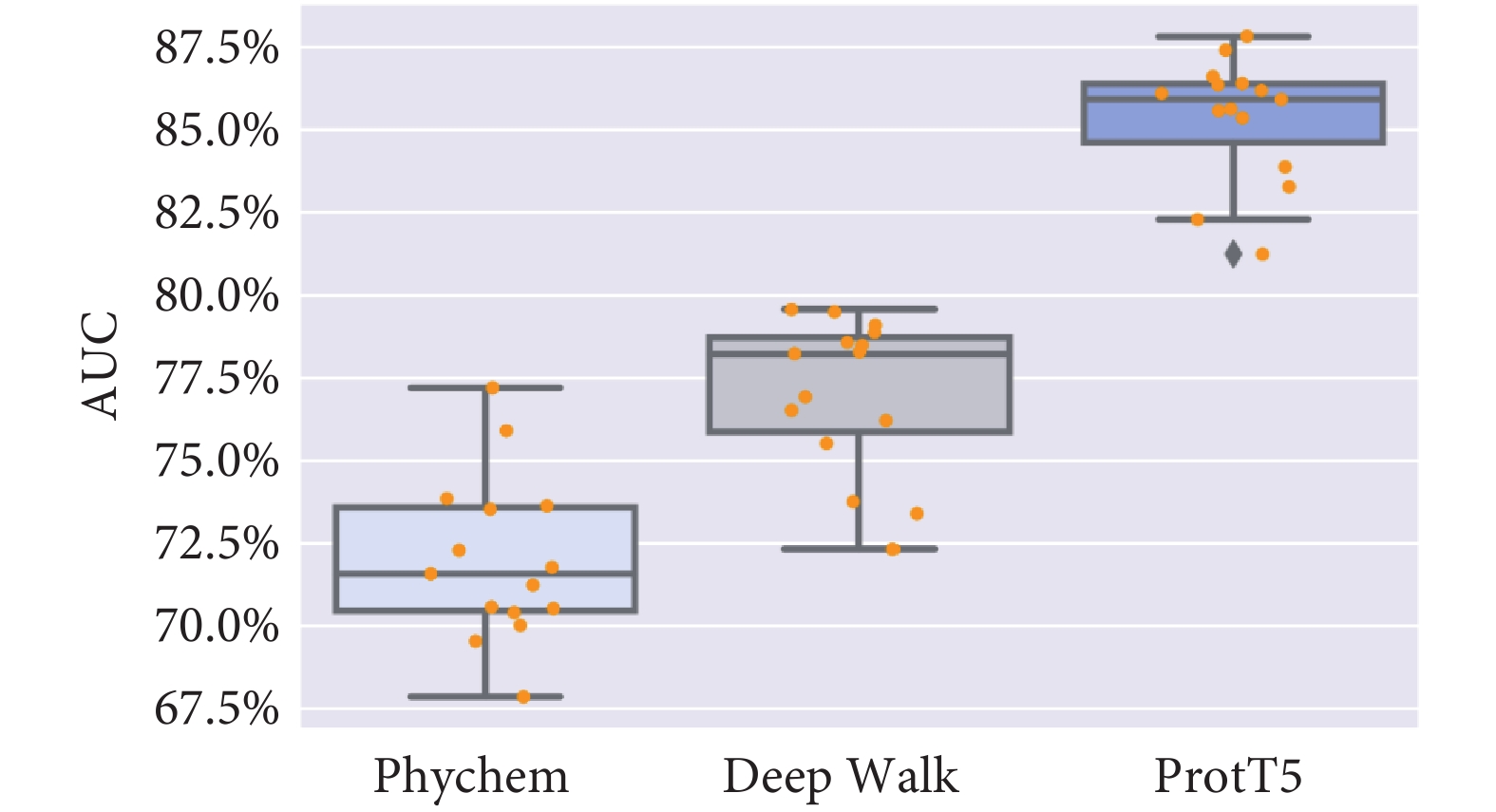 圖4
三種編碼特征的模型訓練五折交叉驗證的結果
Figure4.
Five-fold cross-validation results of model training with three encoding features
圖4
三種編碼特征的模型訓練五折交叉驗證的結果
Figure4.
Five-fold cross-validation results of model training with three encoding features
接下來,在完全獨立的測試集中評估模型的泛化能力,該測試集未參與原模型的訓練,用于在訓練結束后對原模型進行測試,以對比和分析不同編碼方法的性能表現,結果見表1。由表1結果可知,僅依賴氨基酸理化性質特征和隨機游走的方法并不能充分捕捉到抗原肽與TCR序列的信息,相比之下,基于ProtT5模型的編碼方法在不同特征中依然表現出顯著的優越性。
2.2 基于ProtT5的深度學習預測模型結果分析
為了對比研究不同的深度學習模型在TCR和MHC II類抗原肽結合預測任務中的表現,我們選用機器學習算法以及已發表的工具進行比較,包括K近鄰(KNN)、AttnTAP[16]和ATM-TCR[17]。為了保證公平性,使用相同的訓練集對模型進行訓練,通過多次實驗,綜合考察各模型在測試集上的準確度、召回率等性能指標,結果如表2所示。
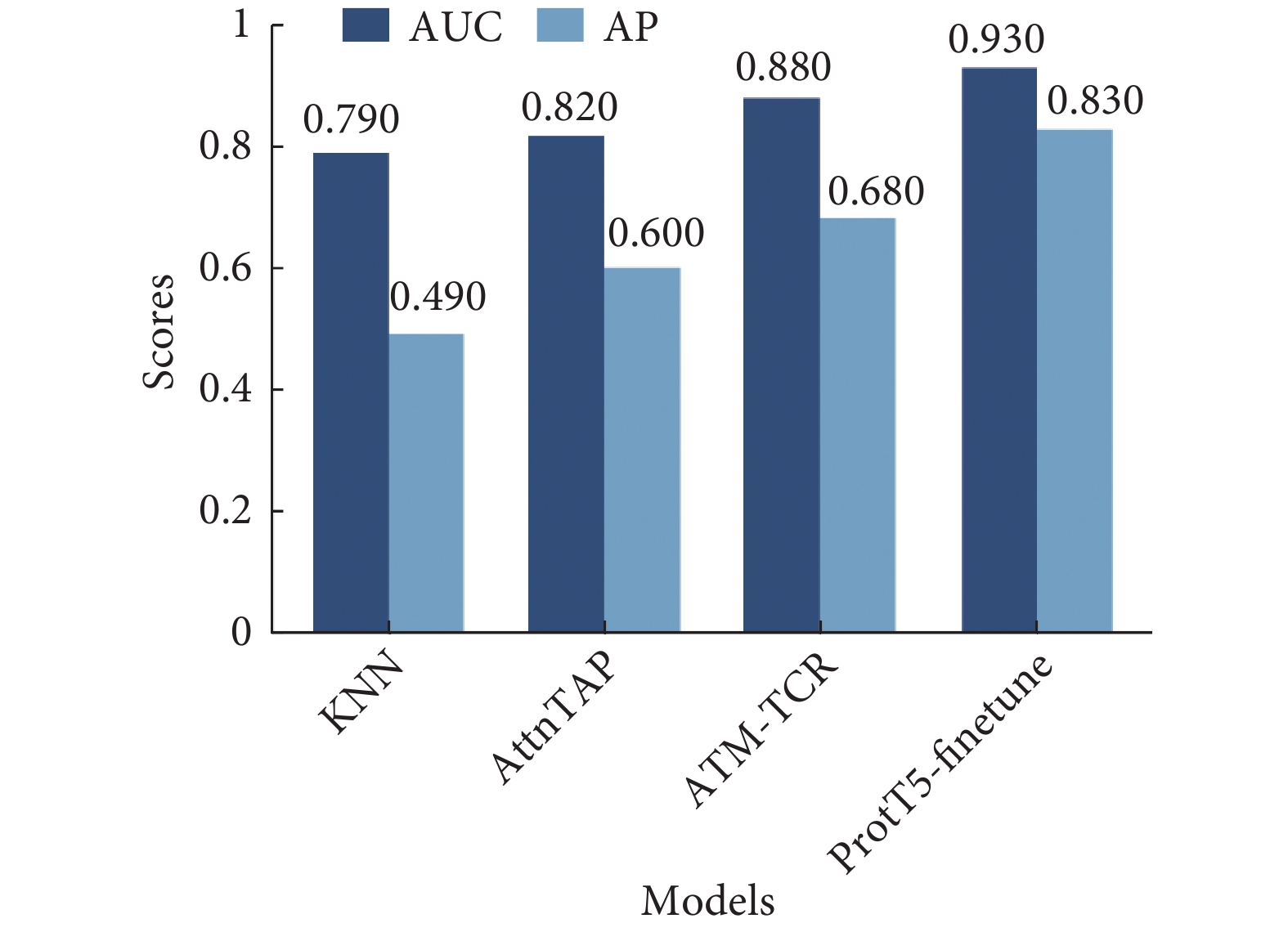
結果顯示,ATM-TCR和本研究的模型均表現良好。進一步分析比較兩者的AUC和平均精度(average precision,AP)(見圖5),可以觀察到本研究模型在區分正負樣本上具有明顯優勢(AUC = 0.93,AP = 0.83),優于其他分類器。高AP值還反映出模型在極度不平衡的肽-TCR陽性和陰性樣本中具備出色的適應性和魯棒性,能更有效地識別肽-TCR的真實相互作用,減小數據不平衡帶來的偏差。
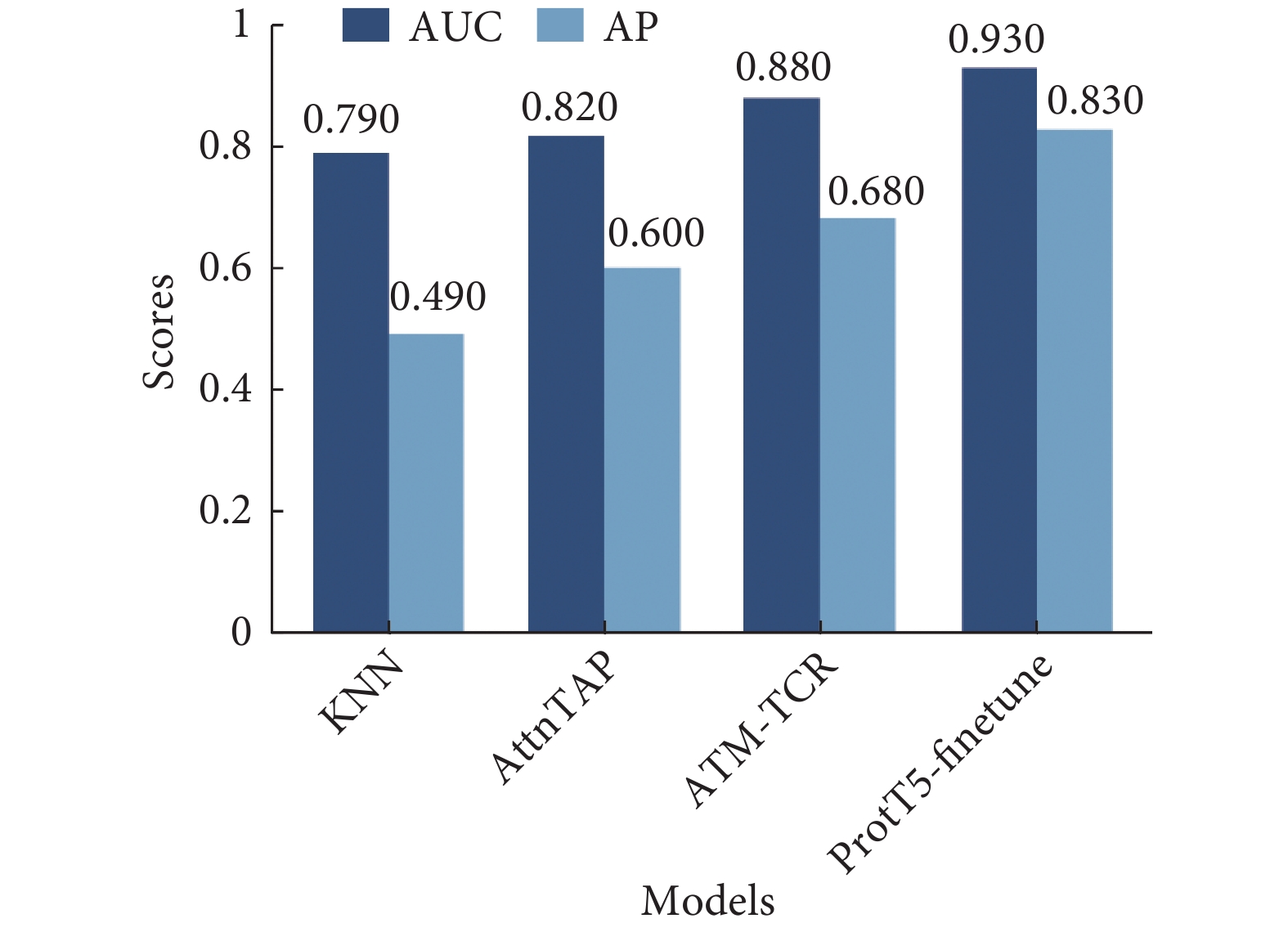 圖5
不同分類器在測試集預測MHC II類肽-TCR相互作用的AUC、AP值比較
Figure5.
Comparison of AUC and AP values for predicting MHC class II peptide TCR interactions using different classifiers on Testing set
圖5
不同分類器在測試集預測MHC II類肽-TCR相互作用的AUC、AP值比較
Figure5.
Comparison of AUC and AP values for predicting MHC class II peptide TCR interactions using different classifiers on Testing set
2.3 模型可解釋性
在當前的數據集中,篩選出與TCR匹配對數最多的肽以及相應的TCR序列,將兩段序列拼接后輸入ProtT5模型以提取對應嵌入特征。同樣地,選取另一種簡單的編碼方案——氨基酸理化特性作為比較,它將輸入序列中的每個氨基酸轉化為多個特征向量表示(有疏水性、β結構偏好、親水性等)。
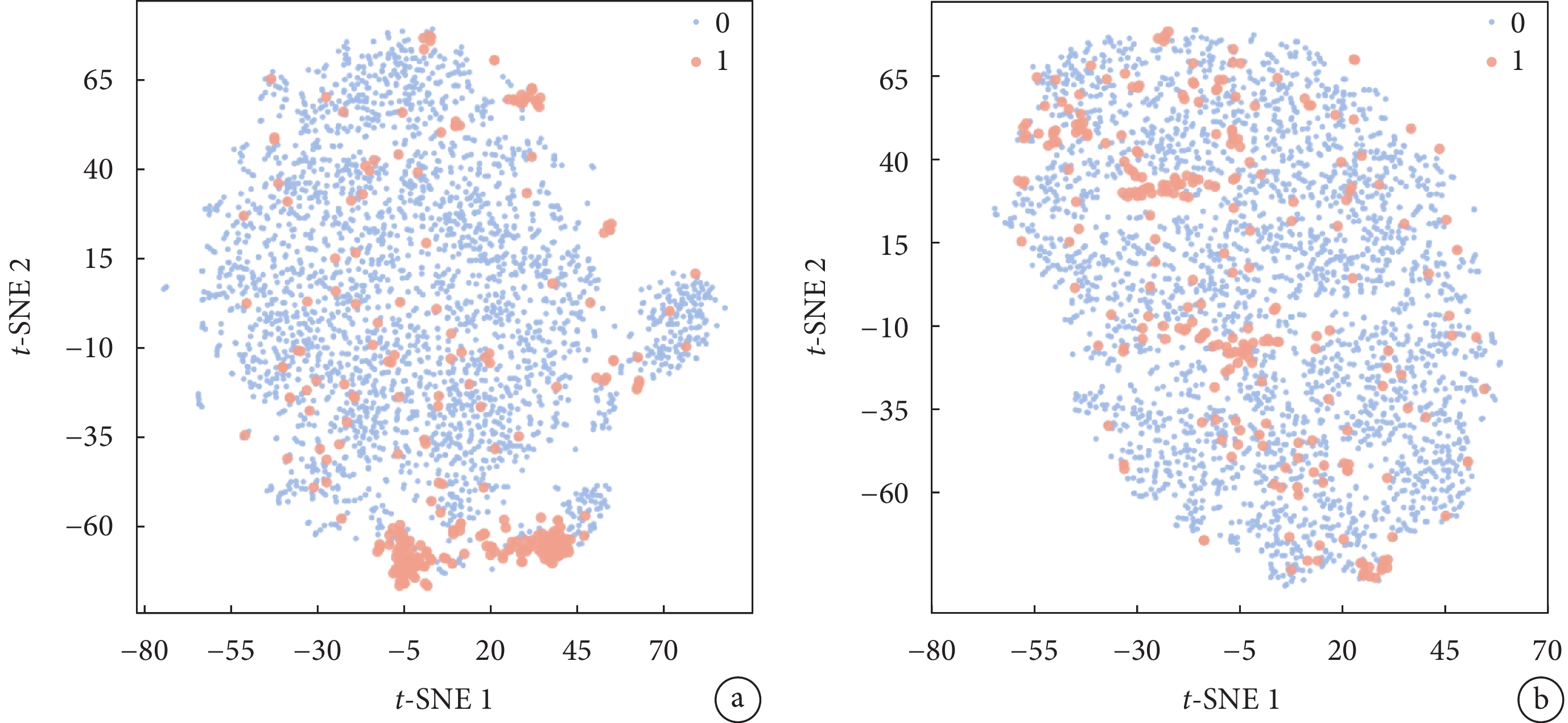
接下來,使用t分布隨機鄰居嵌入(t-distributed stochastic neighbor embedding,t-SNE)算法將這些高維向量降至二維,以便更直觀地展示它們之間的關系(見圖6a~b)。結果顯示,相較于氨基酸理化特性(見圖6b),經ProtT5嵌入后(見圖6a)的陽性和陰性肽-TCR顯著分離,尤其是在右下角形成了一個明顯的陽性肽-TCR富集區。這表明,經過ProtT5模型嵌入后,已經實現了良好的分類效果,這一結果也說明ProtT5預訓練模型在提取生物序列特征方面具有潛在優勢。
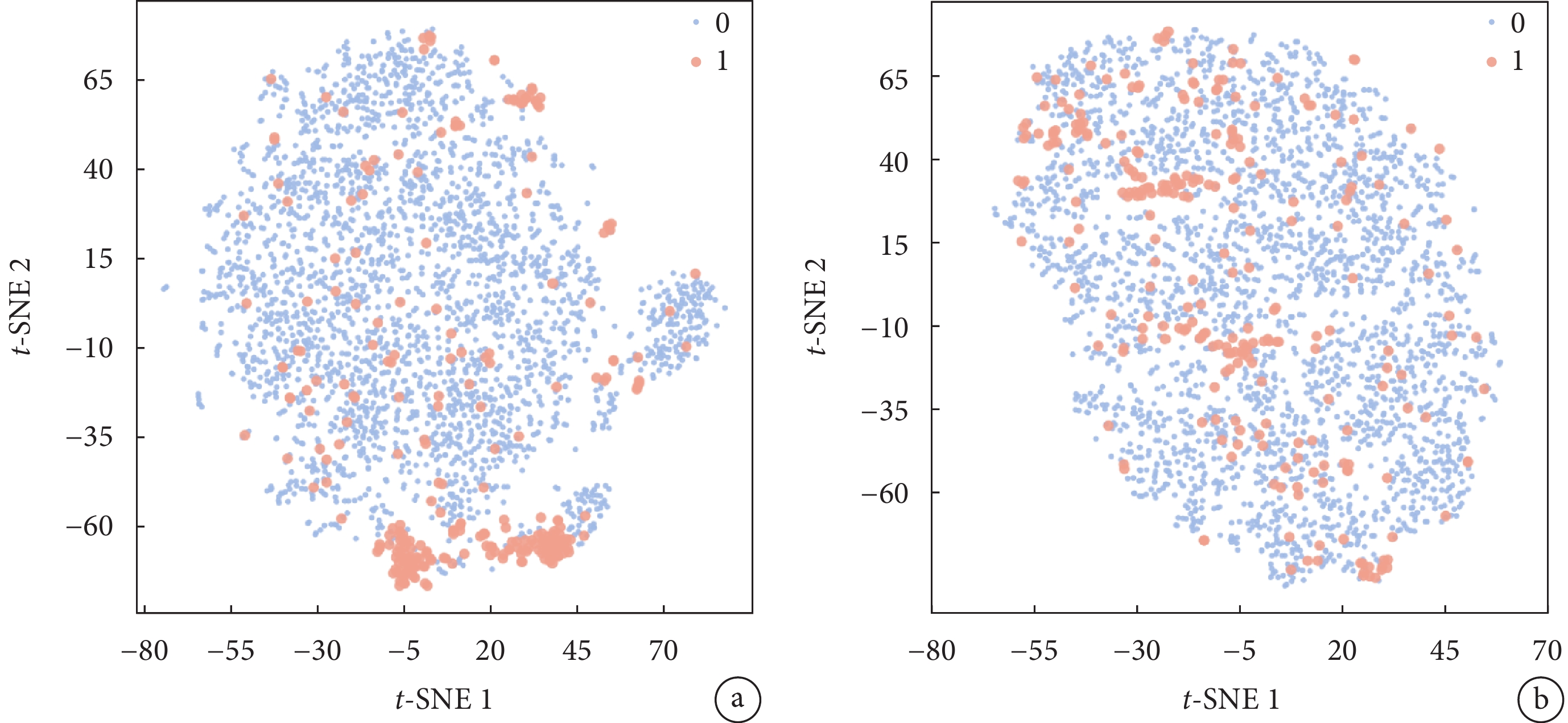 圖6
關于肽-TCR的t-SNE圖
圖6
關于肽-TCR的t-SNE圖
a. 經ProtT5嵌入后;b. 經氨基酸理化特性嵌入后。陽性TCR(標注為1)以橙色顯示,陰性TCR(標注為0)以淡藍色顯示
Figure6. t-SNE plot for the peptide-TCRa. after ProtT5 embedding; b. after embedding with amino acid physicochemical properties. Orange indicates positive TCR (labeled as 1), and light blue indicates negative TCR (labeled as 0)
我們猜測,ProtT5模型是通過大規模的蛋白質序列進行預訓練得到的,包括一級、二級結構以及氨基酸間的交互,這些都是傳統的人工篩選特征難以捕捉的,可能是ProtT5嵌入特征性能優異的一個重要原因。其次,ProtT5模型利用注意力機制的優勢來處理蛋白質序列的復雜性,能夠關注蛋白質序列中的關鍵部分,在不依賴于多序列比對的情況下可能實現準確的預測。
3 討論與結論
已有研究表明,CD4+ T細胞識別的MHC II分子對新發腫瘤的影響可能大于MHC I類,且它們也是CD8+ T細胞至關重要的輔助因子,在腫瘤發生中起著核心作用[18-19]。缺乏對MHC II類抗原免疫原性準確的預測,可能會導致很多患者對免疫治療無反應,即使這些患者腫瘤被預測含有免疫原性的MHC I抗原或具有有利的腫瘤突變負荷。
本研究構建了基于ProtT5模型的深度學習方法,利用預訓練蛋白質語言模型提取的特征作為輸入參數,并集成前饋全連接神經網絡模塊,以實現對TCR與MHC II類抗原肽結合的預測。通過多組實驗驗證,證明了模型在不同數據集上的穩定性和泛化能力,可以更準確地捕捉TCR與抗原肽序列的信息,有效地預測潛在的、可激活CD4+ T細胞的抗原靶點,縮小臨床上實驗驗證新抗原的范圍,提升靶點預測效率。
本研究雖然在TCR與MHC Ⅱ類呈遞抗原肽的免疫原性預測研究方面取得一定進展,但仍存在一些局限性。腫瘤的免疫反應是一個復雜的生物過程,其中涉及多肽的降解、蛋白酶的剪切加工、TAP的轉運以及MHC的結合呈遞等因素影響[20]。但由于訓練數據的有限性,這些影響抗原肽免疫原性的特征因素并未完全考慮到模型中。其次,大模型通常由數以億計的參數組成,使得解釋模型的決策過程變得更加困難。因此,準確地鑒定具有免疫原性的CD4+ T細胞表位仍然是一個技術瓶頸。但已有大量工作表明,深度學習方法可以用來解決這些問題,這在之前的工作中也得到了初步探索并驗證[8, 21]。我們相信隨著對腫瘤免疫行為的深入解析以及精準醫學時代下高質量數據的不斷累積,結合高通量的TCR免疫組庫測序技術[22],深度學習算法會不斷得到升級拓展,可有效幫助科研人員發現腫瘤治療的潛在通用靶點。
總體而言,本研究提出的模型準確預測出MHC Ⅱ類抗原肽和TCR的識別,有效地鑒定出高質量候選抗原肽,這一進展為開發更精確的TCR抗原預測模型奠定了基礎,并有望促進腫瘤免疫療法的優化和新治療策略的探索。本文模型訓練的數據集和代碼可在如下地址查看:
重要聲明
利益沖突聲明:本文全體作者均聲明不存在利益沖突。
作者貢獻說明:許珉瑞主要負責整體實驗的設計、算法修改及論文撰寫;張斯文、魯曼曼主要負責醫學背景指導及論文修訂;高媛、張夢歡主要負責數據處理方案設計及算法指導;林勇、謝鷺負責整體實驗的設計指導、算法技術指導與論文修訂。
0 引言
腫瘤免疫治療被認為是癌癥治療領域的一次革命性突破,有效的靶向免疫療法需要準確預測可能觸發T細胞免疫應答的腫瘤特異性表位。抗原依據其特性,分為腫瘤相關抗原與腫瘤特異性抗原,它們通過主要組織相容性復合體(major histocompatibility complex,MHC)呈現在腫瘤細胞表面,隨后啟動T細胞介導的免疫應答,以消滅腫瘤細胞[1]。T細胞受體(T cell receptor,TCR)的激活依賴于它與肽-MHC(pMHC復合物)的識別過程[2]。TCR是由兩條不同肽鏈構成的二聚體,其胞外區由恒定區(C)和可變區(V)組成,V區又可分為三個互補決定區(CDR1-3),CDR1和CDR2負責識別MHC分子,而關鍵的CDR3區則是抗原的特異性識別部位[3]。CDR3的結構和功能在TCR與pMHC復合物的相互作用中至關重要,其特異性識別機制將影響免疫治療策略的精確設計。
高通量測序的發展推動了抗原肽與TCR結合計算方法的開發。基于傳統機器學習的方法,Gielis等[4]分析序列的物理化學性質,構建了基于隨機森林的模型評估并預測結合概率。De Neuter等[5]通過訓練隨機森林分類器以探究TCR識別抗原的機制。基于深度學習的方法,NetTCR-2.0[6]利用卷積網絡將肽和CDR3 β序列信息整合。ERGO[7]利用長短期記憶人工神經網絡來提取序列特征,用于預測兩者結合概率。本團隊利用自構建編碼方式AAPP,提出名為iTCep[8]的深度學習框架,預測MHC I類分子呈現的肽與TCR之間的相互作用。
盡管該研究領域展現出顯著的發展,但仍有潛力進一步提升和完善。傳統意義上認為CD8+ T細胞是主要的腫瘤殺傷細胞,但最近的臨床試驗表明CD4+ T細胞在腫瘤控制中也起到了重要作用。而現有研究主要集中于CD8+ T細胞與MHC Ⅰ類抗原結合預測,忽略了CD4+ T細胞與MHC Ⅱ類抗原的相互作用,模型性能仍有待提高。此外,現有模型多為單一模型,限制了從復雜數據中挖掘關聯性的能力,鑒于生物學數據通常呈現高維特征,開發能夠有效分析復雜數據集的模型變得尤為重要,因此本研究利用ProtT5模型對MHC Ⅱ類抗原肽與TCR的結合進行預測,希望能夠提供有益的探索經驗。
1 材料與方法
1.1 實驗總體流程
為提升MHC Ⅱ類抗原肽-TCR結合預測模型的性能,本研究在現有研究基礎上進行了改進。實驗框架設計如圖1所示,涵蓋數據收集、特征編碼與選擇、模型構建和模型預測四個關鍵步驟。
圖1
肽-TCR結合預測模型研究框架圖
Figure1.
Framework diagram for the study of peptide-TCR binding prediction model
首先,本研究收集了公共數據庫中的TCR與II類MHC提呈抗原肽的數據,并進行了過濾和預處理。隨后,采用不同編碼策略將抗原肽和TCR序列轉換為相應的特征。為尋找最佳編碼方式,使用機器學習交叉驗證進行評估。在確定最優編碼策略后,對ProtT5模型進行了微調,以更好地適應研究需求。為全面評估模型性能,用多種指標綜合判斷。最后,將微調后的模型應用于測試集,以評估其泛化能力。
1.2 實驗數據
隨著測序技術的發展,越來越多的TCR序列及抗原肽數據被收集,現有數據庫提供了TCR與抗原對接的信息,為人工智能預測TCR與抗原的結合奠定了數據基礎。本研究使用的數據集源于公開的VDJdb[9]和McPAS-TCR[10]數據庫,從中提取MHC II類抗原肽和TCR信息,其中陽性數據分別為782條和898條。為了構建陰性數據并模擬真實世界TCR與抗原肽的情況,從TCRdb[11]數據庫中選取了來自健康供體的TCR β鏈序列共16 800條,使用的項目號為PRJNA390125(樣本編號為SRR5676649、SRR5676658)。經合并去重后得到一個完整的數據集,劃分訓練集、驗證集與測試集。
1.3 數據預處理
本研究剔除了MHC Ⅰ類抗原肽段,以及VDJdb數據庫中置信評分等于0的數據(0表示關鍵信息的缺失),著重研究MHC Ⅱ類抗原肽段;刪除序列中出現錯誤的氨基酸(B、J、O、U、X、Z);刪除不以半胱氨酸(C)開始或不以苯丙氨酸(F)結尾的CDR3序列。合并數據進行去重,最終得到有效陽性數據943條,陰性數據9 430條,共10 373條配對的抗原肽和TCR結合信息,其中MHC Ⅱ類分子呈遞的抗原肽大多是13和20個氨基酸數目的肽段。
1.4 基于ProtT5模型特征提取分析
ProtT5模型[12]是一種基于Transformer架構的蛋白質預訓練模型,通過自監督方式在UniRef50數據集上(現包含6 600萬個蛋白質序列)進行訓練。模型從這些蛋白質序列中生成輸入和標簽,并隨機掩蓋輸入中的15%氨基酸,通過這種方式訓練得到的特征會捕捉到潛在的蛋白質信息。利用自注意力機制,ProtT5模型可以有效地識別和編碼蛋白質序列中的遠程交互依賴性。這使得ProtT5成為蛋白質相互作用預測等領域內一個強有力的工具。
在機器學習或深度學習模型的構建過程中,數據預處理環節涉及將原始數據編碼為數值化的向量或張量,這一步驟被稱為特征工程或特征表示學習。對于本研究中的TCR和Peptide序列數據,這一過程涉及到氨基酸殘基的向量化或嵌入(embedding),將生物序列轉換為機器學習算法可操作的數值形式,實現有效的模式識別和學習。
具體來說,在兩個序列之間添加一個分隔符,然后將它們作為一個整體輸入到模型中。ProtT5模型會處理這個長序列,并輸出它的隱藏狀態。提取出模型最后一層的隱藏嵌入,該層包含了一定數量的1 024維向量。這一處理過程旨在將抗原肽和TCR序列轉換為高維度的特征向量表示,這些特征向量不僅包含了整體的序列信息,還涵蓋了大量蛋白質功能等相關信息,具體流程如圖2所示。此外,對三種不同的編碼策略進行比較,分別是基于氨基酸理化性質的編碼(Phychem)[13]、隨機游走算法編碼(Deep Walk)[14]和基于ProtT5大模型的編碼,選取合適的特征提取方法是構建深度學習模型的重要一步。
圖2
ProtT5模型特征提取流程圖
Figure2.
The feature extraction process diagram of the ProtT5 model
1.5 預測模型構建
盡管ProtT5模型經預訓練后積累了豐富的蛋白質信息,但在TCR與抗原肽的預測任務中并不完全適用。為更好地滿足研究需求,本研究對ProtT5模型進行了訓練和微調,顯著提升了其性能和適用性,能夠更有效地處理MHC II類抗原肽與TCR之間的復雜結合關系。
具體而言,本研究通過嵌入(Embedding)抗原肽和TCR序列以提取關鍵特征,獲取對當前數據的映射,并將嵌入后的向量輸入ProtT5模型進行訓練。由于ProtT5模型的前層網絡已能提供良好的底層特征,我們凍結了相對底層的權重,保留原有特征提取能力。為了更好地滿足任務需求,接入一個前饋神經網絡層,提取和組合底層特征,最終輸出預測結果。網絡模型結構示意圖見圖3。
圖3
ProtT5模型微調結構圖
Figure3.
Structural diagram of the ProtT5 fine-tuned model
模型訓練過程中,采用學習率預熱(warm-up)的策略,優勢在于能夠有效避免模型在訓練初期出現的震蕩問題。在初始幾輪訓練中,采用較小的學習率讓模型逐漸穩定。當模型達到相對穩定狀態時,逐步增加學習率至預設值,以加速模型的收斂速度并提高效果。
1.6 針對不平衡數據集的訓練策略
由于數據集中正負樣本分布比例不平衡,分類器可能傾向于預測出現頻率較高的類別,進而忽略出現頻率較低的類別,導致訓練效果低下。為此,選取Focal Loss[15]作為損失函數,擴展評估指標范圍,而不僅僅局限于準確率。
Focal Loss(聚焦損失)是一種用于處理類別不平衡問題的損失函數,它具有調節正負樣本權重和易分類與難分類樣本權重的能力,通過增加困難樣本的權重,降低簡單樣本的權重,使模型能夠有針對性地學習難易樣本之間的差異。Focal Loss公式如式(1)所示:
|
Pt表示預測概率,α表示控制正負樣本對loss的貢獻,本研究我們選取α為0.5、γ為2作為訓練的參數。
1.7 實驗環境和評價指標
本研究通過Python 3.9.7編寫實驗代碼,基于PyTorch框架實現,硬件環境為Intel(R) Core(TM) i9-12900K,顯卡為RTX
為了驗證不同編碼方式對模型性能的影響,我們采用準確率(Accuracy,ACC)、精準率(Precision)、召回率(Recall)、F1-score和受試者操作特征(receiver operating characteristic,ROC)曲線下面積(area under curve,AUC)五個測量指標進行評估。
準確率表示抗原肽與TCR預測正確的個數占預測所有數據個數的比例,計算公式如式(2)所示:
|
精準率表示抗原肽與TCR預測為結合的對子中,實際上為真實數據的比例,公式如式(3)所示:
|
召回率又叫做查全率,直觀地說是分類器找到所有正樣本的能力,公式如式(4)所示:
|
F1-score是一個綜合評價分類模型性能的指標,適用于二分類問題,它同時考慮了模型的準確率和召回率,公式如式(5)所示:
|
AUC表示ROC曲線下的面積,主要用于評估模型的泛化能力,即分類器效果的優劣。AUC作為一個數值指標,它比ROC更具可比性,能夠進行量化比較。其值范圍在0到1之間,越接近于1表示分類器性能越好。
2 實驗結果與分析
2.1 不同特征提取方法的比較
為研究不同特征提取方法對模型性能的影響,對三種編碼策略進行實驗測試,包括氨基酸理化性質編碼、隨機游走編碼和ProtT5模型生成的嵌入向量編碼。隨后,將原始數據集(訓練集、驗證集)分為五個相等大小的子集,結合不同編碼策略和SVM模型,采用三次獨立的五折交叉驗證評估各特征對模型性能的影響。在實驗中,將AUC作為主要評價指標,結果如圖4所示。結果說明,在TCR與MHC II類抗原肽結合預測任務中,ProtT5模型展現出顯著的優勢,其AUC的平均值為85.33%,高于氨基酸理化性質(71.99%)和隨機游走(77.06%)方法,表明了本文所提出模型的可靠性。
圖4
三種編碼特征的模型訓練五折交叉驗證的結果
Figure4.
Five-fold cross-validation results of model training with three encoding features
接下來,在完全獨立的測試集中評估模型的泛化能力,該測試集未參與原模型的訓練,用于在訓練結束后對原模型進行測試,以對比和分析不同編碼方法的性能表現,結果見表1。由表1結果可知,僅依賴氨基酸理化性質特征和隨機游走的方法并不能充分捕捉到抗原肽與TCR序列的信息,相比之下,基于ProtT5模型的編碼方法在不同特征中依然表現出顯著的優越性。
2.2 基于ProtT5的深度學習預測模型結果分析
為了對比研究不同的深度學習模型在TCR和MHC II類抗原肽結合預測任務中的表現,我們選用機器學習算法以及已發表的工具進行比較,包括K近鄰(KNN)、AttnTAP[16]和ATM-TCR[17]。為了保證公平性,使用相同的訓練集對模型進行訓練,通過多次實驗,綜合考察各模型在測試集上的準確度、召回率等性能指標,結果如表2所示。
結果顯示,ATM-TCR和本研究的模型均表現良好。進一步分析比較兩者的AUC和平均精度(average precision,AP)(見圖5),可以觀察到本研究模型在區分正負樣本上具有明顯優勢(AUC = 0.93,AP = 0.83),優于其他分類器。高AP值還反映出模型在極度不平衡的肽-TCR陽性和陰性樣本中具備出色的適應性和魯棒性,能更有效地識別肽-TCR的真實相互作用,減小數據不平衡帶來的偏差。
圖5
不同分類器在測試集預測MHC II類肽-TCR相互作用的AUC、AP值比較
Figure5.
Comparison of AUC and AP values for predicting MHC class II peptide TCR interactions using different classifiers on Testing set
2.3 模型可解釋性
在當前的數據集中,篩選出與TCR匹配對數最多的肽以及相應的TCR序列,將兩段序列拼接后輸入ProtT5模型以提取對應嵌入特征。同樣地,選取另一種簡單的編碼方案——氨基酸理化特性作為比較,它將輸入序列中的每個氨基酸轉化為多個特征向量表示(有疏水性、β結構偏好、親水性等)。
接下來,使用t分布隨機鄰居嵌入(t-distributed stochastic neighbor embedding,t-SNE)算法將這些高維向量降至二維,以便更直觀地展示它們之間的關系(見圖6a~b)。結果顯示,相較于氨基酸理化特性(見圖6b),經ProtT5嵌入后(見圖6a)的陽性和陰性肽-TCR顯著分離,尤其是在右下角形成了一個明顯的陽性肽-TCR富集區。這表明,經過ProtT5模型嵌入后,已經實現了良好的分類效果,這一結果也說明ProtT5預訓練模型在提取生物序列特征方面具有潛在優勢。
圖6
關于肽-TCR的t-SNE圖
a. 經ProtT5嵌入后;b. 經氨基酸理化特性嵌入后。陽性TCR(標注為1)以橙色顯示,陰性TCR(標注為0)以淡藍色顯示
Figure6. t-SNE plot for the peptide-TCRa. after ProtT5 embedding; b. after embedding with amino acid physicochemical properties. Orange indicates positive TCR (labeled as 1), and light blue indicates negative TCR (labeled as 0)
我們猜測,ProtT5模型是通過大規模的蛋白質序列進行預訓練得到的,包括一級、二級結構以及氨基酸間的交互,這些都是傳統的人工篩選特征難以捕捉的,可能是ProtT5嵌入特征性能優異的一個重要原因。其次,ProtT5模型利用注意力機制的優勢來處理蛋白質序列的復雜性,能夠關注蛋白質序列中的關鍵部分,在不依賴于多序列比對的情況下可能實現準確的預測。
3 討論與結論
已有研究表明,CD4+ T細胞識別的MHC II分子對新發腫瘤的影響可能大于MHC I類,且它們也是CD8+ T細胞至關重要的輔助因子,在腫瘤發生中起著核心作用[18-19]。缺乏對MHC II類抗原免疫原性準確的預測,可能會導致很多患者對免疫治療無反應,即使這些患者腫瘤被預測含有免疫原性的MHC I抗原或具有有利的腫瘤突變負荷。
本研究構建了基于ProtT5模型的深度學習方法,利用預訓練蛋白質語言模型提取的特征作為輸入參數,并集成前饋全連接神經網絡模塊,以實現對TCR與MHC II類抗原肽結合的預測。通過多組實驗驗證,證明了模型在不同數據集上的穩定性和泛化能力,可以更準確地捕捉TCR與抗原肽序列的信息,有效地預測潛在的、可激活CD4+ T細胞的抗原靶點,縮小臨床上實驗驗證新抗原的范圍,提升靶點預測效率。
本研究雖然在TCR與MHC Ⅱ類呈遞抗原肽的免疫原性預測研究方面取得一定進展,但仍存在一些局限性。腫瘤的免疫反應是一個復雜的生物過程,其中涉及多肽的降解、蛋白酶的剪切加工、TAP的轉運以及MHC的結合呈遞等因素影響[20]。但由于訓練數據的有限性,這些影響抗原肽免疫原性的特征因素并未完全考慮到模型中。其次,大模型通常由數以億計的參數組成,使得解釋模型的決策過程變得更加困難。因此,準確地鑒定具有免疫原性的CD4+ T細胞表位仍然是一個技術瓶頸。但已有大量工作表明,深度學習方法可以用來解決這些問題,這在之前的工作中也得到了初步探索并驗證[8, 21]。我們相信隨著對腫瘤免疫行為的深入解析以及精準醫學時代下高質量數據的不斷累積,結合高通量的TCR免疫組庫測序技術[22],深度學習算法會不斷得到升級拓展,可有效幫助科研人員發現腫瘤治療的潛在通用靶點。
總體而言,本研究提出的模型準確預測出MHC Ⅱ類抗原肽和TCR的識別,有效地鑒定出高質量候選抗原肽,這一進展為開發更精確的TCR抗原預測模型奠定了基礎,并有望促進腫瘤免疫療法的優化和新治療策略的探索。本文模型訓練的數據集和代碼可在如下地址查看:
重要聲明
利益沖突聲明:本文全體作者均聲明不存在利益沖突。
作者貢獻說明:許珉瑞主要負責整體實驗的設計、算法修改及論文撰寫;張斯文、魯曼曼主要負責醫學背景指導及論文修訂;高媛、張夢歡主要負責數據處理方案設計及算法指導;林勇、謝鷺負責整體實驗的設計指導、算法技術指導與論文修訂。