引用本文: 何喜寧, 陸庭斌, 侯新芳, 李紅霞, 劉俐. 黏脂貯積癥Ⅲα/β 型一例. 華西醫學, 2024, 39(8): 1337-1340. doi: 10.7507/1002-0179.202403298 復制
版權信息: ?四川大學華西醫院華西期刊社《華西醫學》版權所有,未經授權不得轉載、改編
病例介紹 患兒,男,8 歲 4 個月,因肢體活動受限伴身材矮小于 2022 年 8 月 16 日入陜西省康復醫院。患兒系第 1 胎第 1 產,母孕時 24 周歲,孕期體健,足月試產失敗后行剖宮產(具體不詳),出生體重
入院體格檢查:神志清楚,精神好,情緒穩定,交流態度好,言語清晰流利;身高 118 cm,面容粗陋,全身皮膚黝黑、粗糙,眉毛、頭發濃黑,頸短,四肢、手指粗短,爪形手,言語、認知能力基本正常。徒手肌力:雙側上肢肱二頭肌、肱三頭肌 3 級,腹內外斜肌 3 級,豎脊肌 3 級,臀大肌、臀中肌 3 級,雙側股四頭肌 4 級,雙側脛前肌 4 級。肌張力 Ashworth 評定:雙側肱三頭肌Ⅰ級,雙側小腿三頭肌Ⅰ級;雙手指伸肌Ⅱ級。關節活動度:肩關節前屈 0~100°,后伸 0~30°,外展 0~90°;肘關節屈曲 0~110°,伸展 5°;前臂旋前 0~30°,旋后 0~40°;腕關節掌屈 0~60°,背伸 0~15°;髖關節屈曲 0~90°,伸展 0~15°,外展 0~35°;膝關節屈曲 0~130°,伸展 0~150°;踝關節主動背屈 0~20°,跖屈 0~–25°。肱二頭肌腱反射正常,肱三頭肌腱反射稍亢進,雙側膝腱反射亢進,雙側跟腱反射亢進,雙側髕陣攣、踝陣攣未引出,雙側巴賓斯基征陰性。患兒可獨站、獨行,獨行時脊椎伸展不充分,快走時雙膝關節屈曲,可獨自上下樓梯;跑步動作不協調,緩慢,易摔倒;雙手遠端指間關節伸展不充分,呈屈曲狀,可滿把抓握,可擰蓋瓶蓋,能握筆書寫,會畫圈、三角形等,可一頁一頁翻書,動作笨拙,不能完成拇食指捏小物;能使用筷子進食,動作笨拙,獨自脫簡單寬松開衫,穿上衣需家長輔助,大小便可以自理,日常生活基本獨立。韋氏智力量表評估:智商 91 分;能力低下兒童評定量表(Pediatric Evaluation of Disability Inventory, PEDI)評分:移動能力 59 分(正常),交流能力 65 分(正常),日常活動 62 分(低下);日常生活能力評定(Activity of Daily Living, ADL)評分:80 分。
輔助檢查:① 脊柱及四肢關節 X 線片:頸胸腰椎椎體發育異常,胸腰椎生理曲度變直;雙膝關節骨質發育延遲;雙手腕骨齡符合 4.5 歲男孩特征;雙肘關節骨化中心延遲;雙肩關節肩胛盂較扁平(圖1)。② 四肢肌電圖檢查:右側正中神經重度-完全病損,感覺、運動均累及,軸索改變為主;左側正中神經重度病損,感覺運動均累及,感覺重于運動,脫髓鞘改變為主;雙側正中神經腕部病變可能。③ 溶酶體酶檢測:β-半乳糖苷酶 132 nmol/(g·min),β-葡萄糖醛酸苷酶 140.9 nmol/(g·min),血漿總氨基己糖苷酶 908.9 nmol/(L·min),血漿 β-葡萄糖醛酸苷酶
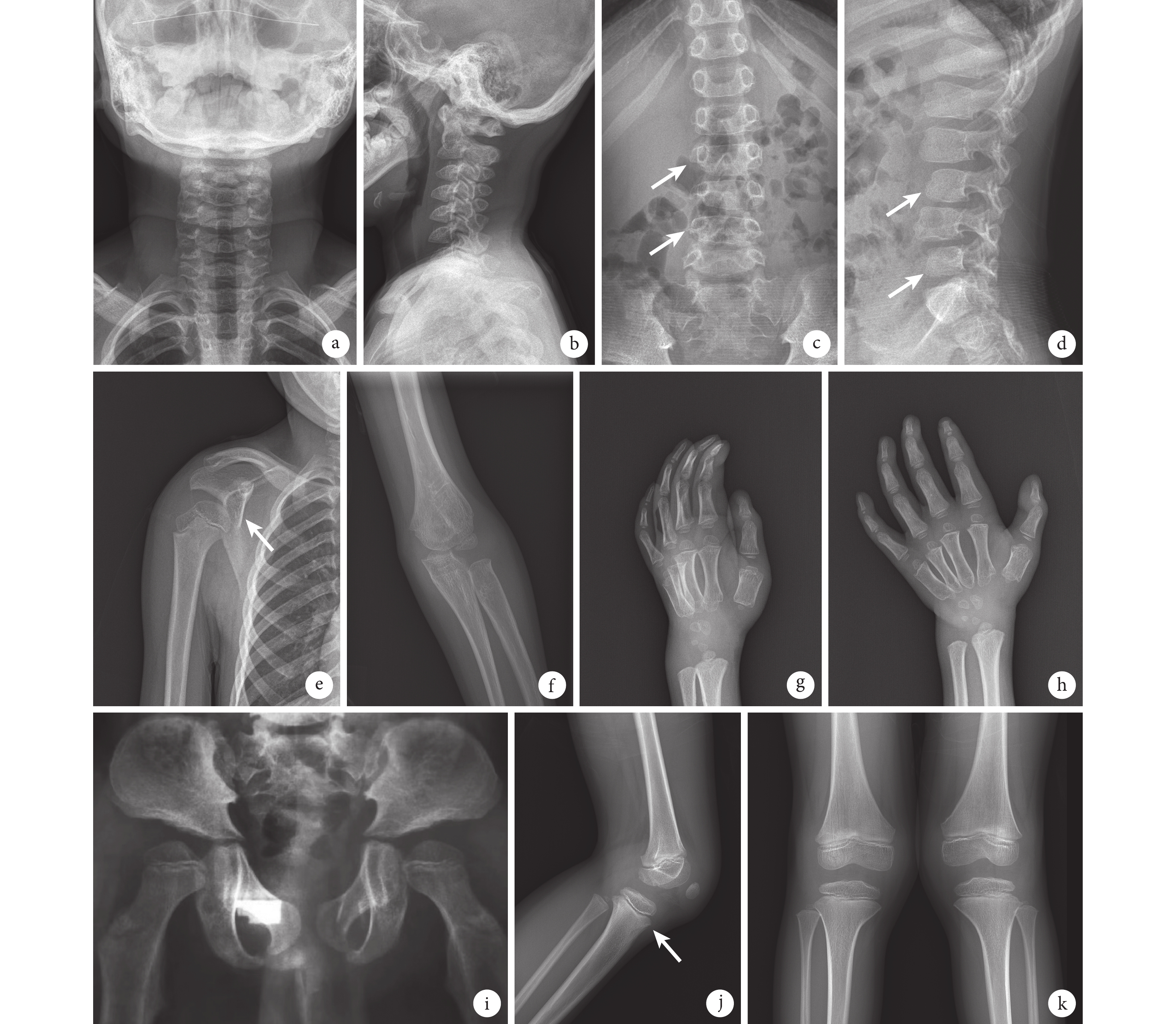
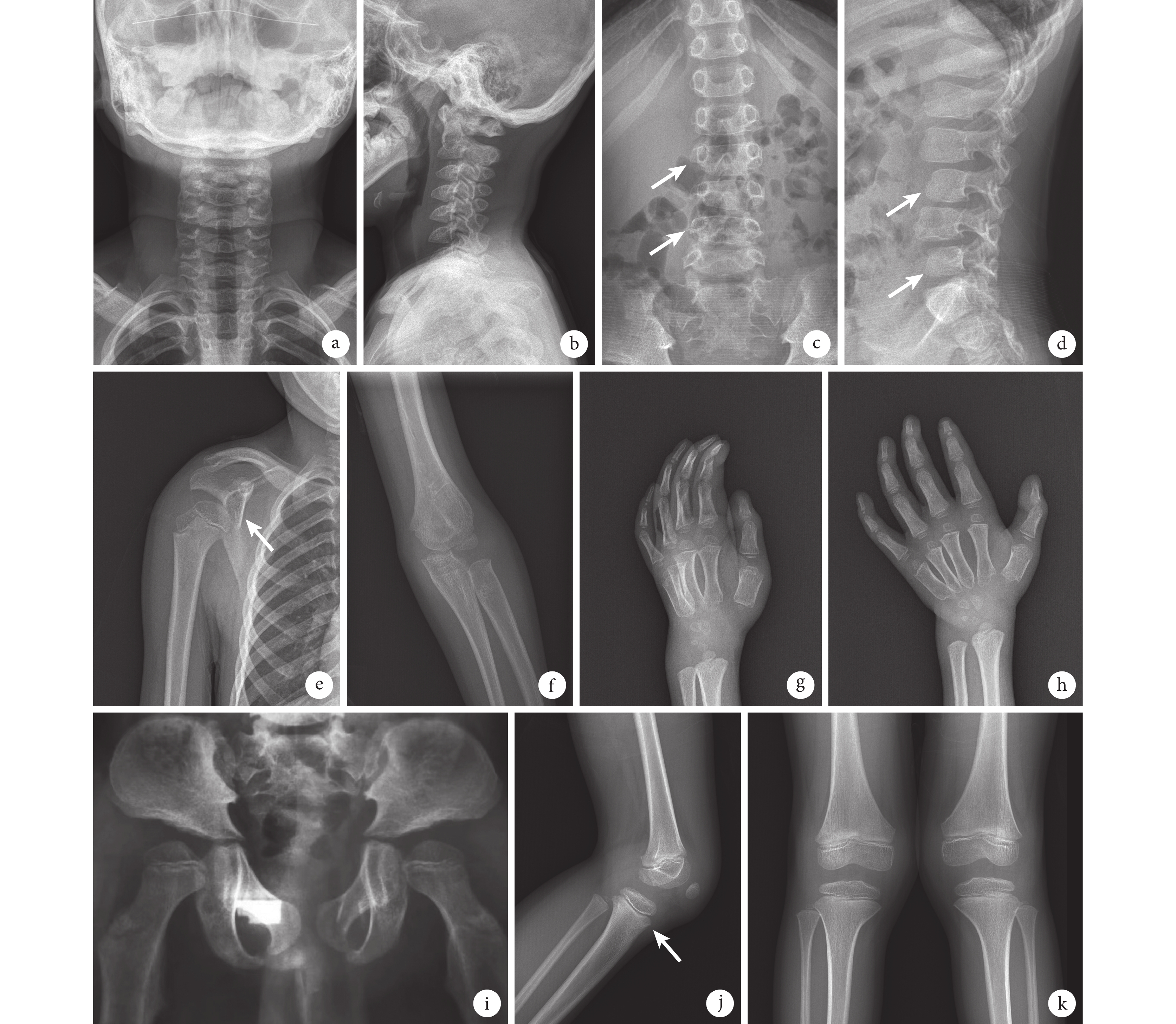 圖1
患兒入院時脊柱及四肢關節 X 線片
圖1
患兒入院時脊柱及四肢關節 X 線片
a、b. 頸椎發育異常,頸椎椎體呈三角形改變;c、d. 腰椎椎體發育異常,腰 2、腰 4 椎體較小(白箭);e. 右肩關節肩胛盂扁平(白箭);f. 左肘關節骨化中心延遲出現;g、h. 左手發育遲緩,骨齡相當于 4.5 歲男孩特征;i. 雙側髖外翻;j、k. 膝關節骨骺發育遲緩,脛骨前結節骨骺未萌出(白箭)
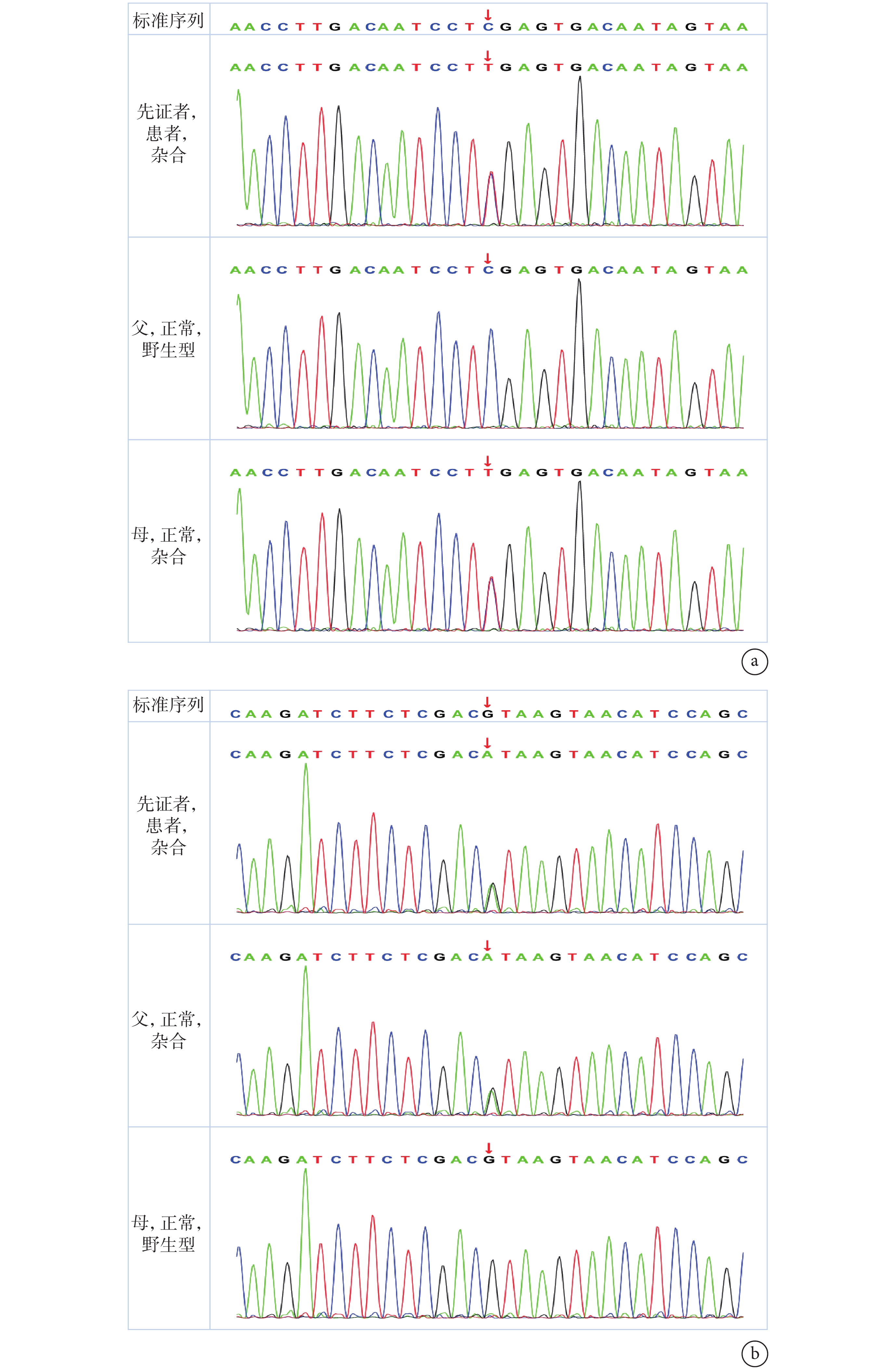
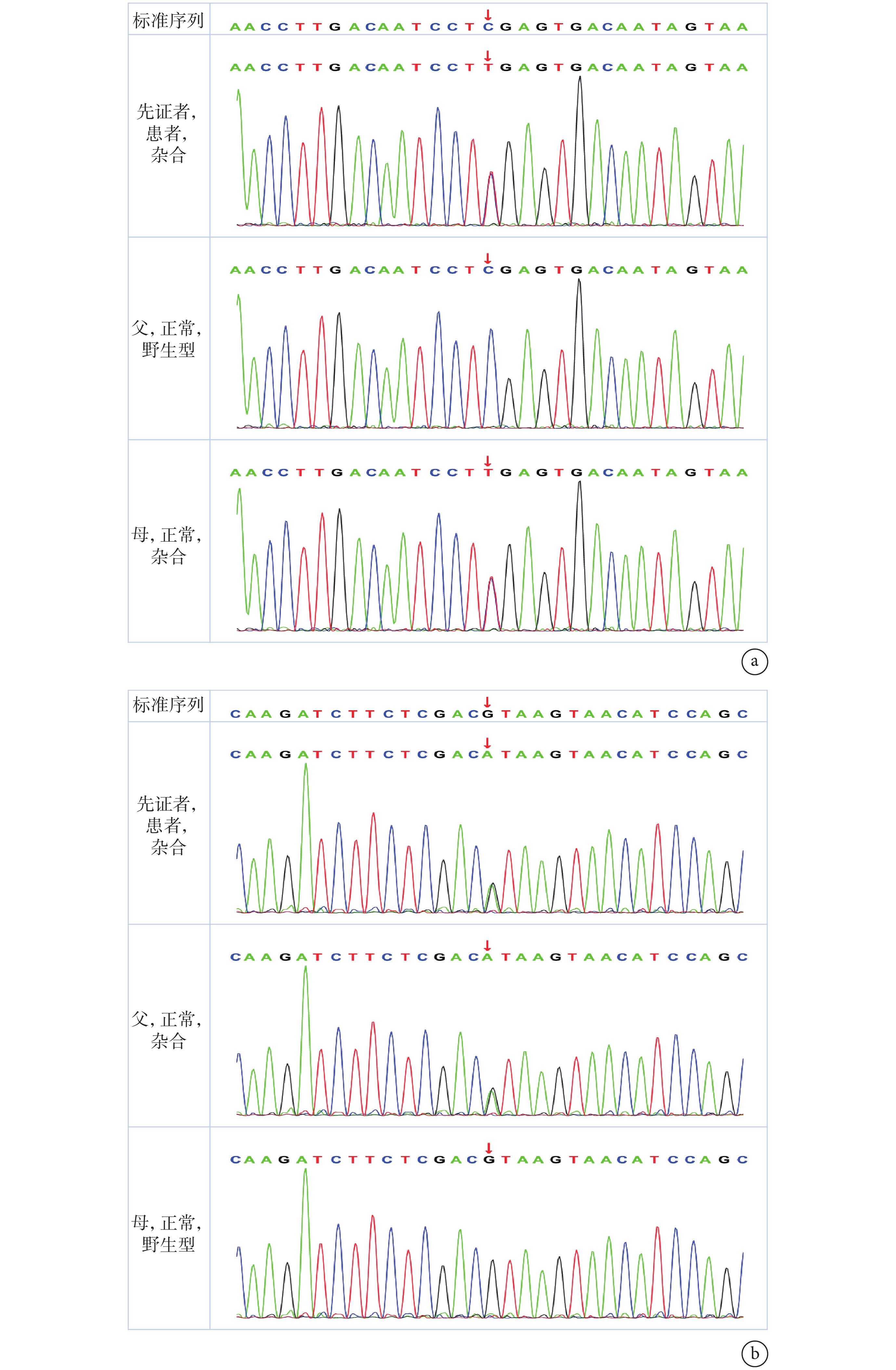 圖2
患兒 GNPTAB 基因 2 個雜合變異
圖2
患兒 GNPTAB 基因 2 個雜合變異
a. 先證者(患兒)獲得
診斷:綜合體格檢查、臨床癥狀、X 線、基因檢測,患兒診斷為黏脂貯積癥(mucolipidosis, ML)Ⅲα/β 型。
康復治療情況:入院由康復醫生組織團隊評估,康復醫生、康復護士、物理治療師、作業治療師、心理治療師、針灸醫師、推拿醫師、患兒母親參加。主要問題:① 雙側髖關節、膝關節、踝關節活動范圍明顯受限,蹲起、跑步等困難,活動多時會出現疼痛;② 雙側肩關節、腕、手指關節僵硬,握拳困難,間斷橈側三指麻木;③ 日常洗漱、穿衣、書寫等表現不良,活動需要部分輔助;④ 學校生活適應困難,兒童和家庭很困擾。結合家庭期望制訂康復目標,康復治療以運動、作業、物理因子、傳統中醫等綜合治療。物理因子治療、針灸緩解關節僵硬及損傷性炎癥;以目標導向設計游戲活動,主、被動活動關節,放松關節周圍肌肉、韌帶;手操作能力訓練,如書寫、操作玩具、日常生活活動訓練;親子同訓;每周 1 次 1 h 的家長課堂進行疾病及相關康復知識宣教、指導;每周 2 次對兒童和家長在訓練結束時進行心理疏導。1 個月后評估多關節活動范圍輕微改善,如,雙手握拳、拇他指對指、雙上肢上舉改善,扶物完成下蹲動作改善。家屬自述兒童日常生活活動表現改善,自信心增強。量表評估:PEDI 測評日常活動 67 分(穿脫開衫、開合拉鏈、插上和取下拉鏈能力改善),ADL 評分 90 分。
隨訪:患兒返回校園,普通小學就讀,每月進行電話、視頻指導,在家庭開展適宜訓練,學習成績良好。經過 20 個月,患兒身高未再增加,關節活動受限無明顯進展,仍間斷出現雙側膝關節疼痛及手指麻木,跑、跳動作質量輕微倒退。
討論 ML 為常染色體隱性遺傳性疾病,臨床分為 4 型,即 ML Ⅰ型、Ⅱ型、Ⅲ型、Ⅳ型,是一種罕見的遺傳代謝病。其中Ⅲ型分為 ML Ⅲα/β 和 ML Ⅲγ 兩種,臨床癥狀相對輕,病情進展緩慢,臨床關注不多。以往關于輕癥病例的自然發展史報道少,但該病因骨代謝異常引起肢體運動障礙對兒童的影響不容忽視。ML 是溶酶體貯積病中的一類,合并發病率約為 1∶
ML Ⅲ型的骨營養不良顯著導致骨骼和關節癥狀以及進行性和破壞性骨病,這比黏多糖病帶來的虛弱、疼痛和殘疾負擔更重。這類骨疾病可能由成骨細胞和破骨細胞之間失衡引起,破骨細胞的骨吸收區中破骨酶的存在增加導致軟骨和骨發育異常,多關節受累,表現關節炎、骨痛癥狀,患者絕大多數會表現骨關節發育障礙,易與風濕類疾病、骨關節病相混淆,早期診斷比較困難,常常出現誤診、漏診[3]。在臨床上出現以下特征有助于 ML Ⅲα/β 型的診斷[2]:發病年齡在嬰兒期晚期到童年晚期;臨床和放射學上表現明顯異常;生長緩慢;關節僵硬最初出現在肩膀、臀部和手指,因劇烈運動或物理治療而加重的關節疼痛;漸進性的面部粗陋特征;輕至中度的脊柱側凸;正常至輕度認知發育缺陷,伴隨疼痛的骨質疏松。本例患兒臨床表現典型,生長緩慢,身材矮小,骨、關節發育畸形,多關節活動受限及至疼痛。自 8 歲 4 個月隨訪至 10 歲,患兒身高未再增長,全身多關節活動受限進展緩慢,四肢遠端癥狀相對重,踝關節、腕關節僵硬明顯,活動后膝關節疼痛明顯。
目前針對該病仍沒有公認有效的治療方法,骨關節畸形、關節周圍組織攣縮、僵硬等引起關節功能逐漸退化、機械性損傷,會明顯影響患者的生活質量,最終會失去行走能力。一個多中心回顧性研究描述了 13 例成人型 ML Ⅲ型的臨床過程,大約一半患者有明顯身體活動限制,或依賴輪椅,或需要日常生活輔助;所有患者均出現疼痛,3/4 的患者成人期可以參加工作;所有患者均有廣泛的骨骼損害,需要在 20 歲或 30 歲時進行骨科手術干預[6]。骨關節矯形術在疾病發展到關節活動明顯限制步行能力時實施,期望改善患者日常生活活動。常見的手術有髖關節置換術、腕管綜合征松解術等。近年來有臍帶血干細胞、造血干細胞、骨髓移植等方法應用,僅見于發達國家應用于極少數的 ML Ⅱ型重癥病例,但并未發現確切證據支持這些方法能改善患者預后。故而對于該類患者的日常康復管理可能是有益的,適度、科學的日常家庭康復方法在一定程度上幫助患者緩解痛苦,改善日常生活質量,延緩疾病進展。
綜上所述,由骨骼發育異常和嚴重進行性骨關節炎導致的嚴重骨骼異常是 ML Ⅲ型的常見癥狀,鑒別診斷應排除相關骨、軟骨發育異常疾病,未來對該疾病隨訪應關注改善軟骨和骨質量,通過適當的康復干預預防骨骼并發癥和改善關節活動度。本例患兒經過 1 個月余機構康復治療及親子同訓指導后出院。指導開展家庭康復,使患兒學會日常訓練方法、知曉如何盡量預防繼發損傷,可較好地幫助患兒融入學校,融入社會生活。該病例隨訪時間短,骨關節功能退化不明顯,另外也缺少定期溶酶體代謝相關指標檢測,所以遠期以及多病例隨訪可進一步幫助了解 ML 疾病自然史,為長期的綜合康復管理提供更多有益的建議。
利益沖突:所有作者聲明不存在利益沖突。
病例介紹 患兒,男,8 歲 4 個月,因肢體活動受限伴身材矮小于 2022 年 8 月 16 日入陜西省康復醫院。患兒系第 1 胎第 1 產,母孕時 24 周歲,孕期體健,足月試產失敗后行剖宮產(具體不詳),出生體重
入院體格檢查:神志清楚,精神好,情緒穩定,交流態度好,言語清晰流利;身高 118 cm,面容粗陋,全身皮膚黝黑、粗糙,眉毛、頭發濃黑,頸短,四肢、手指粗短,爪形手,言語、認知能力基本正常。徒手肌力:雙側上肢肱二頭肌、肱三頭肌 3 級,腹內外斜肌 3 級,豎脊肌 3 級,臀大肌、臀中肌 3 級,雙側股四頭肌 4 級,雙側脛前肌 4 級。肌張力 Ashworth 評定:雙側肱三頭肌Ⅰ級,雙側小腿三頭肌Ⅰ級;雙手指伸肌Ⅱ級。關節活動度:肩關節前屈 0~100°,后伸 0~30°,外展 0~90°;肘關節屈曲 0~110°,伸展 5°;前臂旋前 0~30°,旋后 0~40°;腕關節掌屈 0~60°,背伸 0~15°;髖關節屈曲 0~90°,伸展 0~15°,外展 0~35°;膝關節屈曲 0~130°,伸展 0~150°;踝關節主動背屈 0~20°,跖屈 0~–25°。肱二頭肌腱反射正常,肱三頭肌腱反射稍亢進,雙側膝腱反射亢進,雙側跟腱反射亢進,雙側髕陣攣、踝陣攣未引出,雙側巴賓斯基征陰性。患兒可獨站、獨行,獨行時脊椎伸展不充分,快走時雙膝關節屈曲,可獨自上下樓梯;跑步動作不協調,緩慢,易摔倒;雙手遠端指間關節伸展不充分,呈屈曲狀,可滿把抓握,可擰蓋瓶蓋,能握筆書寫,會畫圈、三角形等,可一頁一頁翻書,動作笨拙,不能完成拇食指捏小物;能使用筷子進食,動作笨拙,獨自脫簡單寬松開衫,穿上衣需家長輔助,大小便可以自理,日常生活基本獨立。韋氏智力量表評估:智商 91 分;能力低下兒童評定量表(Pediatric Evaluation of Disability Inventory, PEDI)評分:移動能力 59 分(正常),交流能力 65 分(正常),日常活動 62 分(低下);日常生活能力評定(Activity of Daily Living, ADL)評分:80 分。
輔助檢查:① 脊柱及四肢關節 X 線片:頸胸腰椎椎體發育異常,胸腰椎生理曲度變直;雙膝關節骨質發育延遲;雙手腕骨齡符合 4.5 歲男孩特征;雙肘關節骨化中心延遲;雙肩關節肩胛盂較扁平(圖1)。② 四肢肌電圖檢查:右側正中神經重度-完全病損,感覺、運動均累及,軸索改變為主;左側正中神經重度病損,感覺運動均累及,感覺重于運動,脫髓鞘改變為主;雙側正中神經腕部病變可能。③ 溶酶體酶檢測:β-半乳糖苷酶 132 nmol/(g·min),β-葡萄糖醛酸苷酶 140.9 nmol/(g·min),血漿總氨基己糖苷酶 908.9 nmol/(L·min),血漿 β-葡萄糖醛酸苷酶
圖1
患兒入院時脊柱及四肢關節 X 線片
a、b. 頸椎發育異常,頸椎椎體呈三角形改變;c、d. 腰椎椎體發育異常,腰 2、腰 4 椎體較小(白箭);e. 右肩關節肩胛盂扁平(白箭);f. 左肘關節骨化中心延遲出現;g、h. 左手發育遲緩,骨齡相當于 4.5 歲男孩特征;i. 雙側髖外翻;j、k. 膝關節骨骺發育遲緩,脛骨前結節骨骺未萌出(白箭)
圖2
患兒 GNPTAB 基因 2 個雜合變異
a. 先證者(患兒)獲得
診斷:綜合體格檢查、臨床癥狀、X 線、基因檢測,患兒診斷為黏脂貯積癥(mucolipidosis, ML)Ⅲα/β 型。
康復治療情況:入院由康復醫生組織團隊評估,康復醫生、康復護士、物理治療師、作業治療師、心理治療師、針灸醫師、推拿醫師、患兒母親參加。主要問題:① 雙側髖關節、膝關節、踝關節活動范圍明顯受限,蹲起、跑步等困難,活動多時會出現疼痛;② 雙側肩關節、腕、手指關節僵硬,握拳困難,間斷橈側三指麻木;③ 日常洗漱、穿衣、書寫等表現不良,活動需要部分輔助;④ 學校生活適應困難,兒童和家庭很困擾。結合家庭期望制訂康復目標,康復治療以運動、作業、物理因子、傳統中醫等綜合治療。物理因子治療、針灸緩解關節僵硬及損傷性炎癥;以目標導向設計游戲活動,主、被動活動關節,放松關節周圍肌肉、韌帶;手操作能力訓練,如書寫、操作玩具、日常生活活動訓練;親子同訓;每周 1 次 1 h 的家長課堂進行疾病及相關康復知識宣教、指導;每周 2 次對兒童和家長在訓練結束時進行心理疏導。1 個月后評估多關節活動范圍輕微改善,如,雙手握拳、拇他指對指、雙上肢上舉改善,扶物完成下蹲動作改善。家屬自述兒童日常生活活動表現改善,自信心增強。量表評估:PEDI 測評日常活動 67 分(穿脫開衫、開合拉鏈、插上和取下拉鏈能力改善),ADL 評分 90 分。
隨訪:患兒返回校園,普通小學就讀,每月進行電話、視頻指導,在家庭開展適宜訓練,學習成績良好。經過 20 個月,患兒身高未再增加,關節活動受限無明顯進展,仍間斷出現雙側膝關節疼痛及手指麻木,跑、跳動作質量輕微倒退。
討論 ML 為常染色體隱性遺傳性疾病,臨床分為 4 型,即 ML Ⅰ型、Ⅱ型、Ⅲ型、Ⅳ型,是一種罕見的遺傳代謝病。其中Ⅲ型分為 ML Ⅲα/β 和 ML Ⅲγ 兩種,臨床癥狀相對輕,病情進展緩慢,臨床關注不多。以往關于輕癥病例的自然發展史報道少,但該病因骨代謝異常引起肢體運動障礙對兒童的影響不容忽視。ML 是溶酶體貯積病中的一類,合并發病率約為 1∶
ML Ⅲ型的骨營養不良顯著導致骨骼和關節癥狀以及進行性和破壞性骨病,這比黏多糖病帶來的虛弱、疼痛和殘疾負擔更重。這類骨疾病可能由成骨細胞和破骨細胞之間失衡引起,破骨細胞的骨吸收區中破骨酶的存在增加導致軟骨和骨發育異常,多關節受累,表現關節炎、骨痛癥狀,患者絕大多數會表現骨關節發育障礙,易與風濕類疾病、骨關節病相混淆,早期診斷比較困難,常常出現誤診、漏診[3]。在臨床上出現以下特征有助于 ML Ⅲα/β 型的診斷[2]:發病年齡在嬰兒期晚期到童年晚期;臨床和放射學上表現明顯異常;生長緩慢;關節僵硬最初出現在肩膀、臀部和手指,因劇烈運動或物理治療而加重的關節疼痛;漸進性的面部粗陋特征;輕至中度的脊柱側凸;正常至輕度認知發育缺陷,伴隨疼痛的骨質疏松。本例患兒臨床表現典型,生長緩慢,身材矮小,骨、關節發育畸形,多關節活動受限及至疼痛。自 8 歲 4 個月隨訪至 10 歲,患兒身高未再增長,全身多關節活動受限進展緩慢,四肢遠端癥狀相對重,踝關節、腕關節僵硬明顯,活動后膝關節疼痛明顯。
目前針對該病仍沒有公認有效的治療方法,骨關節畸形、關節周圍組織攣縮、僵硬等引起關節功能逐漸退化、機械性損傷,會明顯影響患者的生活質量,最終會失去行走能力。一個多中心回顧性研究描述了 13 例成人型 ML Ⅲ型的臨床過程,大約一半患者有明顯身體活動限制,或依賴輪椅,或需要日常生活輔助;所有患者均出現疼痛,3/4 的患者成人期可以參加工作;所有患者均有廣泛的骨骼損害,需要在 20 歲或 30 歲時進行骨科手術干預[6]。骨關節矯形術在疾病發展到關節活動明顯限制步行能力時實施,期望改善患者日常生活活動。常見的手術有髖關節置換術、腕管綜合征松解術等。近年來有臍帶血干細胞、造血干細胞、骨髓移植等方法應用,僅見于發達國家應用于極少數的 ML Ⅱ型重癥病例,但并未發現確切證據支持這些方法能改善患者預后。故而對于該類患者的日常康復管理可能是有益的,適度、科學的日常家庭康復方法在一定程度上幫助患者緩解痛苦,改善日常生活質量,延緩疾病進展。
綜上所述,由骨骼發育異常和嚴重進行性骨關節炎導致的嚴重骨骼異常是 ML Ⅲ型的常見癥狀,鑒別診斷應排除相關骨、軟骨發育異常疾病,未來對該疾病隨訪應關注改善軟骨和骨質量,通過適當的康復干預預防骨骼并發癥和改善關節活動度。本例患兒經過 1 個月余機構康復治療及親子同訓指導后出院。指導開展家庭康復,使患兒學會日常訓練方法、知曉如何盡量預防繼發損傷,可較好地幫助患兒融入學校,融入社會生活。該病例隨訪時間短,骨關節功能退化不明顯,另外也缺少定期溶酶體代謝相關指標檢測,所以遠期以及多病例隨訪可進一步幫助了解 ML 疾病自然史,為長期的綜合康復管理提供更多有益的建議。
利益沖突:所有作者聲明不存在利益沖突。