引用本文: 王源, 耿佳, 熊文羽, 盧宇, 袁慧軍. 一例白化病合并尋常型魚鱗病患者 TYR 和 FLG 基因變異分析. 華西醫學, 2025, 40(1): 153-156. doi: 10.7507/1002-0179.202404082 復制
版權信息: ?四川大學華西醫院華西期刊社《華西醫學》版權所有,未經授權不得轉載、改編
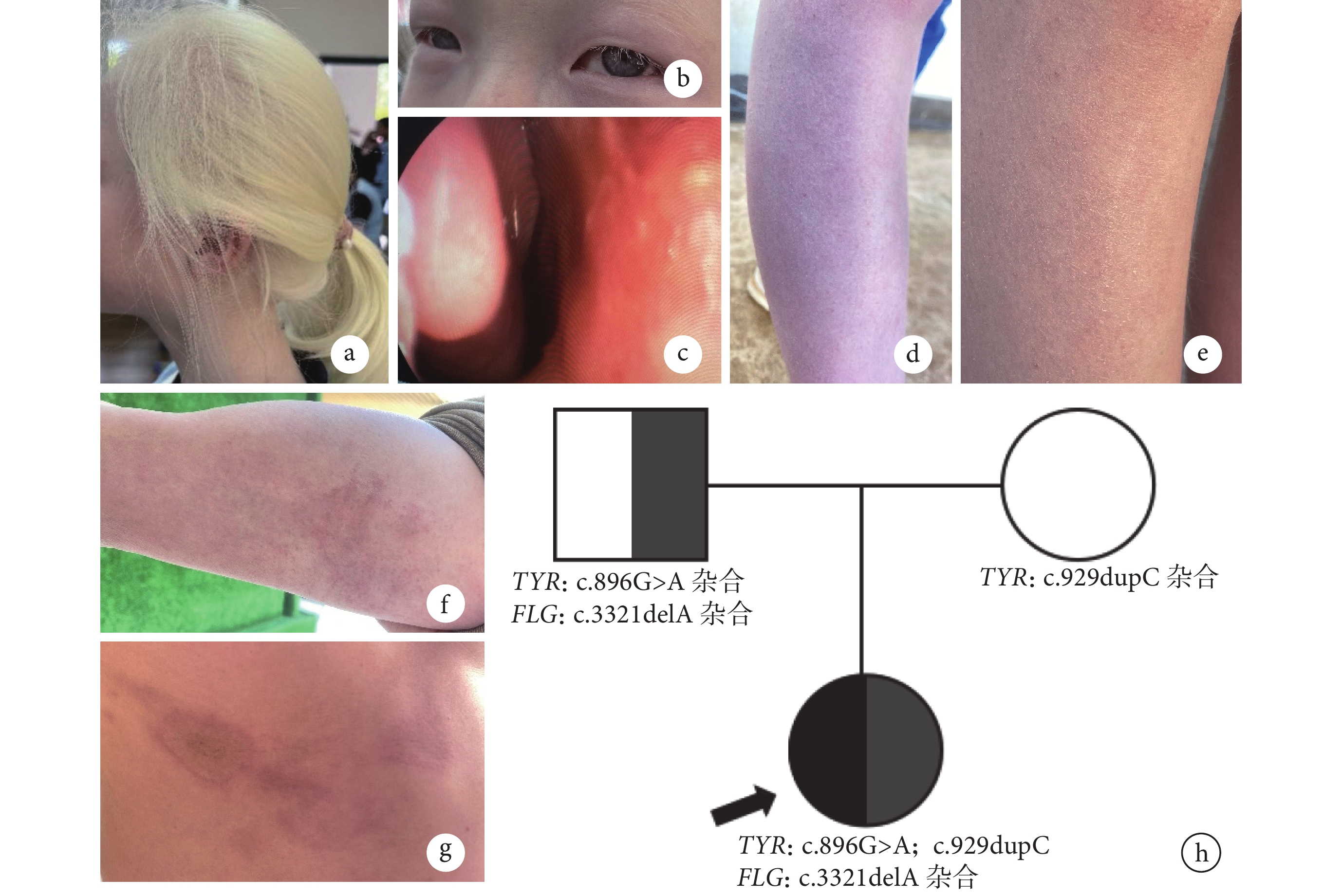
病例介紹 先證者(Ⅱ-1),女,12 歲,為 2023 年 6 月于四川省涼山彝族自治州采集的眼皮膚白化病(oculocutaneous albinism, OCA)患者。先證者足月順產,出生時全身皮膚毛發缺乏黑色素,隨著年齡增長,皮膚毛發顏色仍未改變(圖1a)。先證者先天性雙眼視力差,6 歲確診參與本研究時輔助檢查發現雙眼近視,目前
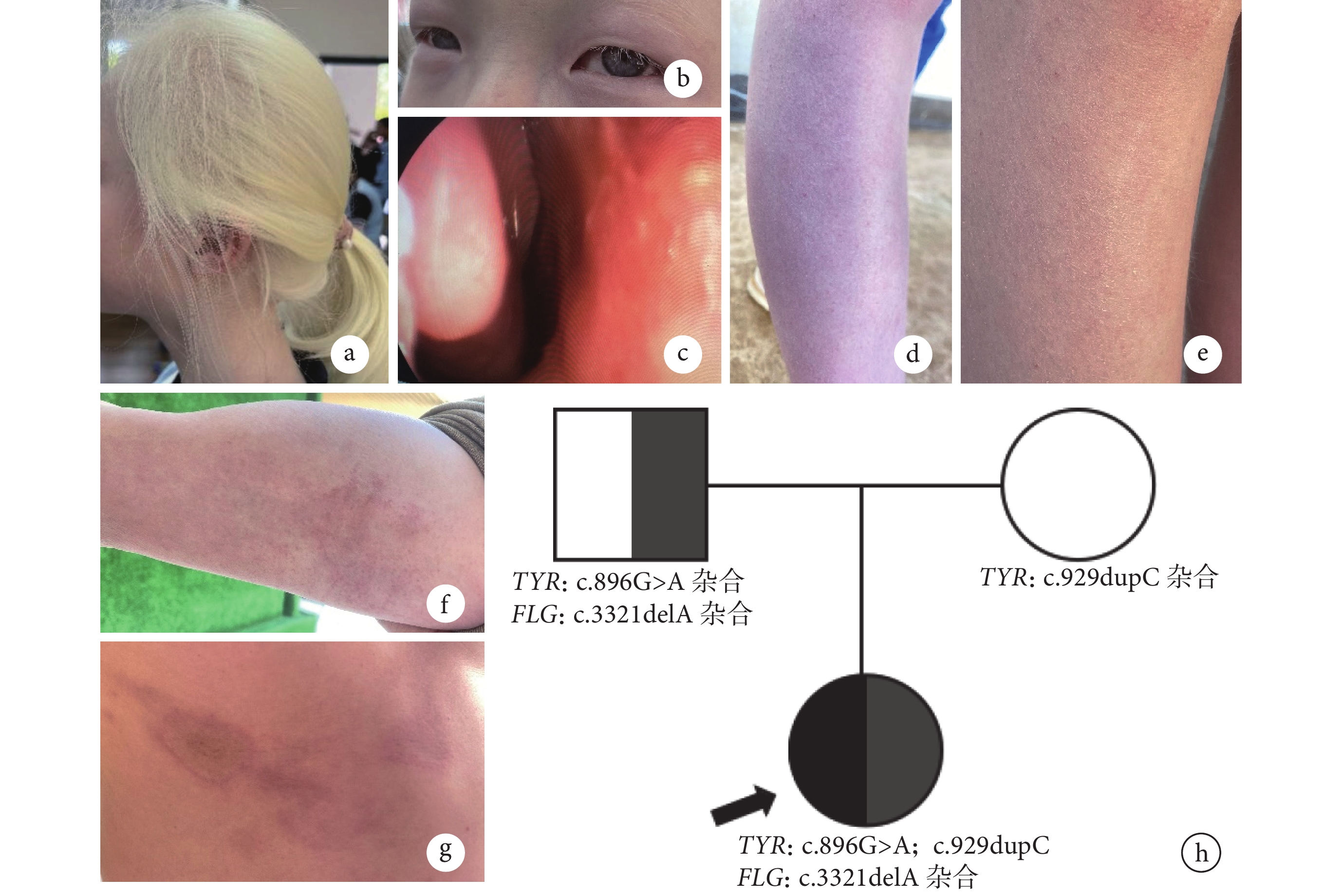 圖1
患者表型、患者父親表型及患者家系圖
圖1
患者表型、患者父親表型及患者家系圖
a、b. 患者眼皮膚白化病表型,全身毛發及皮膚缺乏黑色素,雙眼虹膜異色;c. 患者過敏性鼻炎表型,鼻腔黏膜水腫、充血,下鼻甲肥大;d、e. 患者尋常型魚鱗病表型,皮膚干燥、脫屑,可見毛囊性小丘疹毛周角化癥;f、g. 患者父親特應性皮炎表型,右臂肘部、上腹部及右季肋部可見紅斑、硬紅斑,邊緣堤狀隆起;h. 患者家系圖,黑箭所指為先證者,其中黑色代表
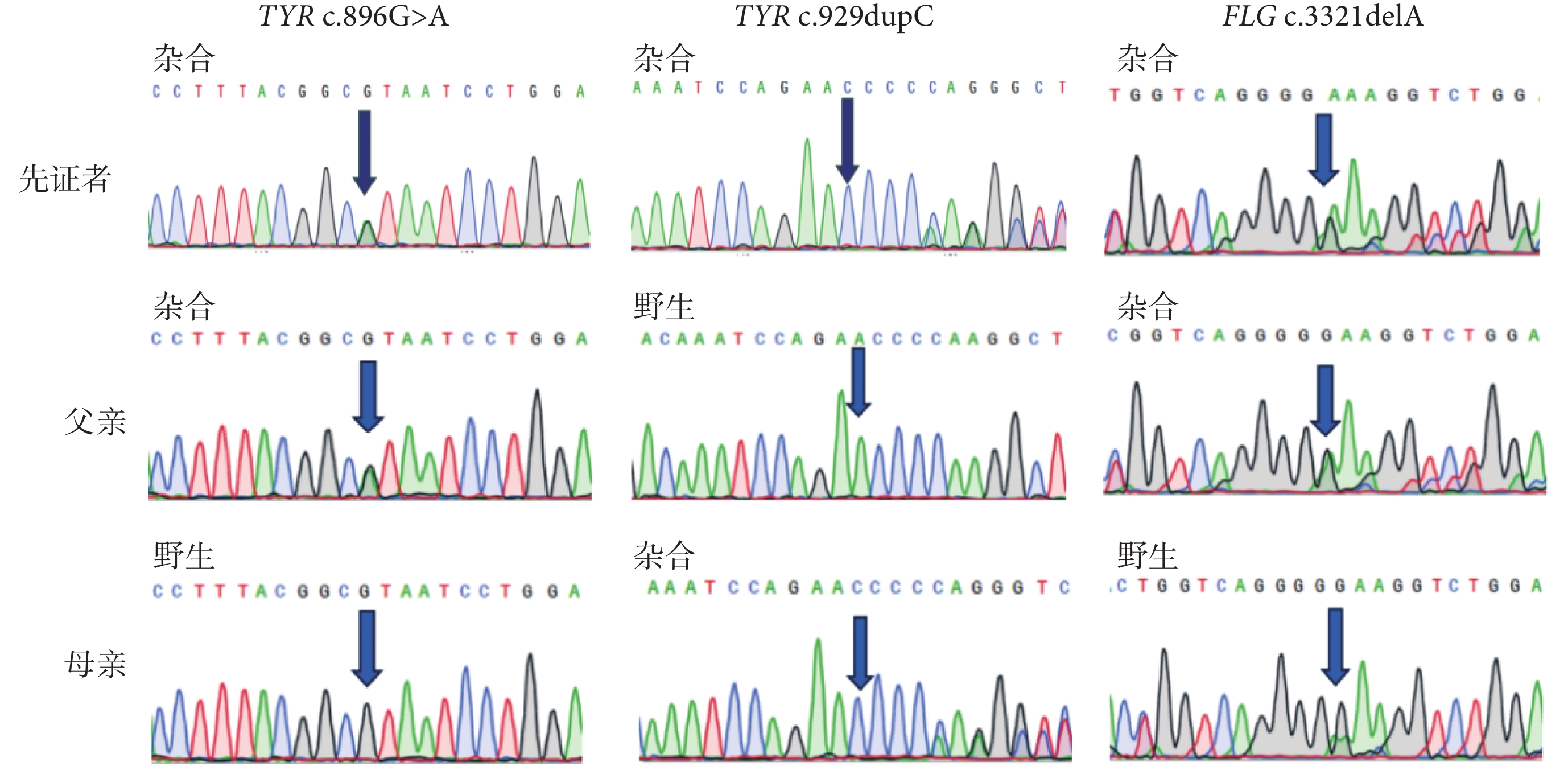
遺傳分析經過及結果:收集該先證者相關臨床資料,包括先證者病史與家族遺傳史、體格檢查與實驗室檢查。采集該先證者的外周靜脈血樣 5 mL,提取基因組 DNA。使用華大智造 DNBSEQ-T7 測序平臺對先證者血液提取到的 DNA 進行全基因組測序(whole genome sequencing, WGS),篩選位于編碼區和/或剪切位點區域的變異,結合臨床表型參照美國醫學遺傳學與基因組學學會相關標準與指南對候選變異進行致病性分析。根據 WGS 檢出的候選致病基因變異,在變異兩端設計聚合酶鏈反應引物,對患者進行聚合酶鏈反應實驗和 Sanger 測序驗證。測序數據通過質量控制后,應用 Mutation-Surveyor 軟件進行序列比對,判讀基因型。WGS 結果提示,先證者在 OCA 致病候選基因列表中檢出 TYR 基因存在 NM_000372.5:c.896G>A(p.Arg299His)和 c.929dupC(p.Arg311Lysfs*7)復合雜合變異,在尋常型魚鱗病的候選基因列表中檢出 FLG 基因 NM_002016.2:c.3321delA 雜合變異。經 Sanger 測序驗證,先證者基因型表型共分離,其中 TYR 基因 c.896G>A 及 FLG 基因 c.3321delA 來自父親,TYR 基因 c.929dupC 來自母親(圖2)。TYR 基因的兩個變異在 ClinVar 數據庫中已分類為致病變異,且在多個 OCA 患者中被報道[1-9],為明確的致病性變異(c.896G>A:PM3_Strong+PM5+PM2_Supporting+PP3+PP4;c.929dupC:PVS1+PM3_Strong+PM2_Supporting+PP4)。先證者攜帶的 FLG 基因的變異既往在日本尋常型魚鱗病家系和特應性皮炎患者中檢出[10],在漢族人群中與特應性皮炎具有相關性[11],考慮該變異的雜合是導致先證者尋常型魚鱗病輕型反復發生的遺傳因素。
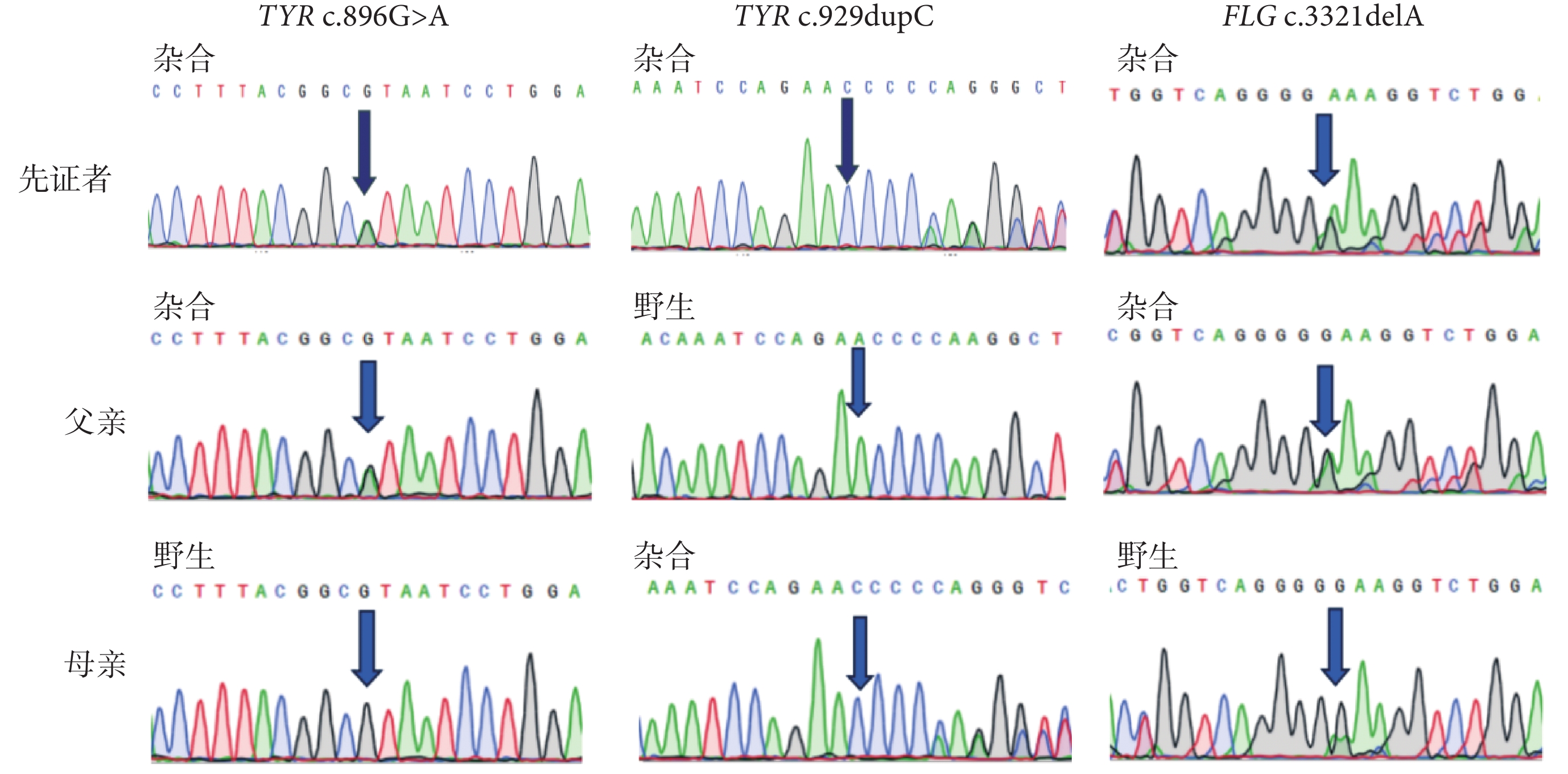 圖2
先證者及雙親 TYR 與 FLG 基因突變位點 Sanger 測序結果
圖2
先證者及雙親 TYR 與 FLG 基因突變位點 Sanger 測序結果
先證者
討論 白化病是由于酪氨酸酶缺乏或功能減退引起的一種皮膚及附屬器官黑色素缺乏或合成障礙所導致的遺傳性白斑病,全球總體發病率約為 1∶
尋常型魚鱗病是一種常染色體遺傳病,致病模式為半顯性,是遺傳性魚鱗病最常見的一種類型,其致病基因主要是由絲聚蛋白編碼基因(FLG)及相關序列純合或雜合型變異引起[19]。FLG 基因定位于染色體 1q21.3,有 3 個外顯子。該基因共編碼
此外,在本報道中,白化病合并其他基因變異導致了先證者臨床表型的復雜性。由中華遺傳學會發布的 2020 版《白化病的臨床實踐指南》重點說明了白化病的常見鑒別診斷[15],但還未說明有關多基因致病的臨床實踐診療方式。因此我們篩選并整理了白化病基因合并其他基因變異導致的白化病合并其他系統疾病,通過文獻聯網檢索 PubMed、Medline、中國知網、中華醫學會數據庫、Web of Science 及 OMIM 數據庫等,對 2013 年—2023 年間的國內外相關文獻進行檢索,中文檢索詞包括“TYR 基因變異”“白化病基因變異”,英文檢索詞包括“TYR gene”“Albinism gene”“variant or mutant”“variation or mutation”。文獻納入標準:① 原始研究報告;② 研究白化病基因與其他基因變異的關系;③ 提供完善的原始數據:例數、基因變異位點信息和相關臨床表現;④ 報道的變異位點為致病性的或可能致病性的,并與患者表型關聯。文獻排除標準:① 重復發表;② 只包含白化病基因變異具體信息,但合并的其他基因變異信息不全;③ 會議文獻。經篩選最終納入 4 篇文獻,共報道了 6 例先證者:1 例 OCA2 基因合并 NTRK1 基因變異,臨床表型為 OCAⅡ型合并先天性無痛無汗癥[29];1 例 OCA2 基因合并 HBB 基因及 a 珠蛋白基因變異,臨床表型為 OCAⅡ型合并輕度鐮狀細胞病 1 例[30];2 例 TYR 基因合并 CNV 變異,其中 1 例臨床表型為 OCAⅠ型合并智力障礙,1 例臨床表型為 OCAⅠ型合并智力障礙及注意力缺陷多動障礙[31];2 例 GPR143 基因合并 ABCA4 基因變異,臨床表型均為 X 連鎖眼白化病合并 Stargardt 病[32]。在臨床診療情景中,當白化病合并其他系統病變時,臨床上容易忽略其他基因變異導致的多種復雜交錯的疾病表型,特別是皮膚相關表型。以本研究為例,第一印象診斷中,我們往往忽略患者皮膚表型的病因多樣化。同時,白化病目前由于缺乏有效的治療,通過產前診斷預防患兒出生尤為重要,特別是 Hermansky-Pudlak 綜合征和 Chediak-Higashi 綜合征危害嚴重的綜合征型白化病[20]。在基因檢測的基礎上開展產前診斷、遺傳咨詢和婚育指導,可以有效地預防重型患兒的出生。因此,建議對罕見病患者進行全面的基因檢測和分析,以提高診斷的質量和效率,為個體化醫療和疾病控制提供更多機會。
總之,本研究通過對 1 例臨床診斷為 OCA 的先證者進行 WGS,明確了 TYR 基因 NM_000372.5:c.896G>A(p.Arg299His)和 c.929dupC(p.Arg311Lysfs*7)復合雜合變異是該先證者 OCA 的致病原因,同時發現先證者及父親 FLG 基因 NM_002016.2:c.3321delA 雜合變異,是導致尋常型魚鱗病或特應性皮炎的致病性變異,明確先證者是由不同基因變異分別導致白化病與尋常型魚鱗病,為患者提供了更全面精準的臨床診斷和遺傳咨詢。
利益沖突:所有作者聲明不存在利益沖突。
病例介紹 先證者(Ⅱ-1),女,12 歲,為 2023 年 6 月于四川省涼山彝族自治州采集的眼皮膚白化病(oculocutaneous albinism, OCA)患者。先證者足月順產,出生時全身皮膚毛發缺乏黑色素,隨著年齡增長,皮膚毛發顏色仍未改變(圖1a)。先證者先天性雙眼視力差,6 歲確診參與本研究時輔助檢查發現雙眼近視,目前
圖1
患者表型、患者父親表型及患者家系圖
a、b. 患者眼皮膚白化病表型,全身毛發及皮膚缺乏黑色素,雙眼虹膜異色;c. 患者過敏性鼻炎表型,鼻腔黏膜水腫、充血,下鼻甲肥大;d、e. 患者尋常型魚鱗病表型,皮膚干燥、脫屑,可見毛囊性小丘疹毛周角化癥;f、g. 患者父親特應性皮炎表型,右臂肘部、上腹部及右季肋部可見紅斑、硬紅斑,邊緣堤狀隆起;h. 患者家系圖,黑箭所指為先證者,其中黑色代表
遺傳分析經過及結果:收集該先證者相關臨床資料,包括先證者病史與家族遺傳史、體格檢查與實驗室檢查。采集該先證者的外周靜脈血樣 5 mL,提取基因組 DNA。使用華大智造 DNBSEQ-T7 測序平臺對先證者血液提取到的 DNA 進行全基因組測序(whole genome sequencing, WGS),篩選位于編碼區和/或剪切位點區域的變異,結合臨床表型參照美國醫學遺傳學與基因組學學會相關標準與指南對候選變異進行致病性分析。根據 WGS 檢出的候選致病基因變異,在變異兩端設計聚合酶鏈反應引物,對患者進行聚合酶鏈反應實驗和 Sanger 測序驗證。測序數據通過質量控制后,應用 Mutation-Surveyor 軟件進行序列比對,判讀基因型。WGS 結果提示,先證者在 OCA 致病候選基因列表中檢出 TYR 基因存在 NM_000372.5:c.896G>A(p.Arg299His)和 c.929dupC(p.Arg311Lysfs*7)復合雜合變異,在尋常型魚鱗病的候選基因列表中檢出 FLG 基因 NM_002016.2:c.3321delA 雜合變異。經 Sanger 測序驗證,先證者基因型表型共分離,其中 TYR 基因 c.896G>A 及 FLG 基因 c.3321delA 來自父親,TYR 基因 c.929dupC 來自母親(圖2)。TYR 基因的兩個變異在 ClinVar 數據庫中已分類為致病變異,且在多個 OCA 患者中被報道[1-9],為明確的致病性變異(c.896G>A:PM3_Strong+PM5+PM2_Supporting+PP3+PP4;c.929dupC:PVS1+PM3_Strong+PM2_Supporting+PP4)。先證者攜帶的 FLG 基因的變異既往在日本尋常型魚鱗病家系和特應性皮炎患者中檢出[10],在漢族人群中與特應性皮炎具有相關性[11],考慮該變異的雜合是導致先證者尋常型魚鱗病輕型反復發生的遺傳因素。
圖2
先證者及雙親 TYR 與 FLG 基因突變位點 Sanger 測序結果
先證者
討論 白化病是由于酪氨酸酶缺乏或功能減退引起的一種皮膚及附屬器官黑色素缺乏或合成障礙所導致的遺傳性白斑病,全球總體發病率約為 1∶
尋常型魚鱗病是一種常染色體遺傳病,致病模式為半顯性,是遺傳性魚鱗病最常見的一種類型,其致病基因主要是由絲聚蛋白編碼基因(FLG)及相關序列純合或雜合型變異引起[19]。FLG 基因定位于染色體 1q21.3,有 3 個外顯子。該基因共編碼
此外,在本報道中,白化病合并其他基因變異導致了先證者臨床表型的復雜性。由中華遺傳學會發布的 2020 版《白化病的臨床實踐指南》重點說明了白化病的常見鑒別診斷[15],但還未說明有關多基因致病的臨床實踐診療方式。因此我們篩選并整理了白化病基因合并其他基因變異導致的白化病合并其他系統疾病,通過文獻聯網檢索 PubMed、Medline、中國知網、中華醫學會數據庫、Web of Science 及 OMIM 數據庫等,對 2013 年—2023 年間的國內外相關文獻進行檢索,中文檢索詞包括“TYR 基因變異”“白化病基因變異”,英文檢索詞包括“TYR gene”“Albinism gene”“variant or mutant”“variation or mutation”。文獻納入標準:① 原始研究報告;② 研究白化病基因與其他基因變異的關系;③ 提供完善的原始數據:例數、基因變異位點信息和相關臨床表現;④ 報道的變異位點為致病性的或可能致病性的,并與患者表型關聯。文獻排除標準:① 重復發表;② 只包含白化病基因變異具體信息,但合并的其他基因變異信息不全;③ 會議文獻。經篩選最終納入 4 篇文獻,共報道了 6 例先證者:1 例 OCA2 基因合并 NTRK1 基因變異,臨床表型為 OCAⅡ型合并先天性無痛無汗癥[29];1 例 OCA2 基因合并 HBB 基因及 a 珠蛋白基因變異,臨床表型為 OCAⅡ型合并輕度鐮狀細胞病 1 例[30];2 例 TYR 基因合并 CNV 變異,其中 1 例臨床表型為 OCAⅠ型合并智力障礙,1 例臨床表型為 OCAⅠ型合并智力障礙及注意力缺陷多動障礙[31];2 例 GPR143 基因合并 ABCA4 基因變異,臨床表型均為 X 連鎖眼白化病合并 Stargardt 病[32]。在臨床診療情景中,當白化病合并其他系統病變時,臨床上容易忽略其他基因變異導致的多種復雜交錯的疾病表型,特別是皮膚相關表型。以本研究為例,第一印象診斷中,我們往往忽略患者皮膚表型的病因多樣化。同時,白化病目前由于缺乏有效的治療,通過產前診斷預防患兒出生尤為重要,特別是 Hermansky-Pudlak 綜合征和 Chediak-Higashi 綜合征危害嚴重的綜合征型白化病[20]。在基因檢測的基礎上開展產前診斷、遺傳咨詢和婚育指導,可以有效地預防重型患兒的出生。因此,建議對罕見病患者進行全面的基因檢測和分析,以提高診斷的質量和效率,為個體化醫療和疾病控制提供更多機會。
總之,本研究通過對 1 例臨床診斷為 OCA 的先證者進行 WGS,明確了 TYR 基因 NM_000372.5:c.896G>A(p.Arg299His)和 c.929dupC(p.Arg311Lysfs*7)復合雜合變異是該先證者 OCA 的致病原因,同時發現先證者及父親 FLG 基因 NM_002016.2:c.3321delA 雜合變異,是導致尋常型魚鱗病或特應性皮炎的致病性變異,明確先證者是由不同基因變異分別導致白化病與尋常型魚鱗病,為患者提供了更全面精準的臨床診斷和遺傳咨詢。
利益沖突:所有作者聲明不存在利益沖突。