引用本文: 翟麗穎, 叢金鵬, 糜麗云, 鞏海紅, 于文成. PD-1/PD-L1軸在慢性阻塞性肺疾病中的研究進展. 中國呼吸與危重監護雜志, 2024, 23(1): 69-75. doi: 10.7507/1671-6205.202211038 復制
版權信息: ?四川大學華西醫院華西期刊社《中國呼吸與危重監護雜志》版權所有,未經授權不得轉載、改編
由于人口增長和老齡化,慢性阻塞性肺疾病(簡稱慢阻肺)的患病率和發病率不斷上升。近十幾年來,全球慢阻肺死亡人數增加了17.5%。預計2040年慢阻肺將成為全球第四大死因[1]。其發病機制目前尚無定論,治療方法也較局限。近年來越來越多的研究關注到免疫調節及紊亂在其中發揮著重要作用。吸入刺激物會導致呼吸道內的結構細胞和炎癥細胞激活并釋放出大量的趨化因子,從而進一步招募和活化大量免疫細胞,觸發級聯免疫反應。導致組織破壞和粘液過度分泌,最終導致不可逆的呼吸道結構改變和氣流受限[2]。程序性細胞死亡蛋白-1(programmed cell death protein 1,PD-1)/程序性細胞死亡配體1(programmed cell death ligand 1,PD-L1)軸是機體免疫中不可或缺的信號通路。然而目前關于PD-1/PD-L1信號通路的研究大多集中于腫瘤領域,與慢阻肺相關的研究相對較少。因此,我們對PD-1/PD-L1軸在慢阻肺中的研究進展作一綜述。
1 PD-1/PD-L1的概念及意義
1.1 PD-1/PD-L1的發現、結構及表達
1992年,Ishida等[3]發現了免疫球蛋白基因超家族新成員PD-1。PD-1又稱CD279,由三部分組成,包括含有IgV樣結構域的胞外區,跨膜結構域以及帶有兩個酪氨酸信號基序的胞內區[4]。PD-1表達于包括T細胞在內的多種免疫細胞[5-7],但它在靜息T細胞上不表達,而是在刺激后24 h內在活化的成熟T細胞上誘導表達[8]。2000年,Freeman等[9]發現了一種類似B7的分子,并證明它是PD-1的配體,后來被命名為PD-L1。Dong等[10]報道了與PD-L1相同的分子。PD-L1又稱B7-H1或CD274,由三部分組成,包括含有IgV和IgC樣結構域的胞外區、跨膜結構域和不含典型信號基序的短胞質尾[11-12]。PD-L1不僅表達于T細胞等免疫細胞,還表達于一些健康組織細胞,例如血管、皮膚、胰腺、胎盤、角膜、肝臟和肺的一些細胞[12-13]。
1.2 PD-1/PD-L1軸對T細胞免疫應答的作用
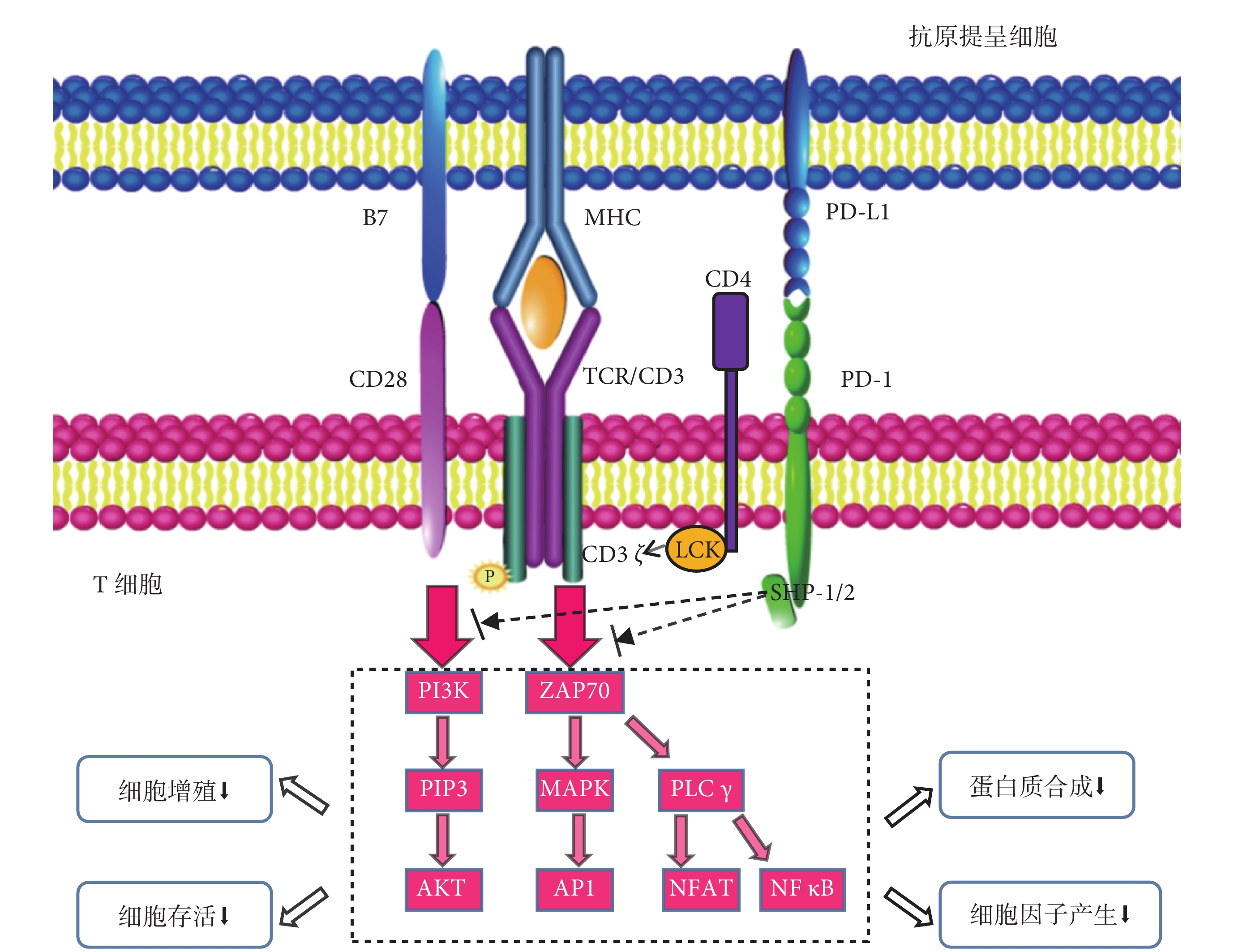
免疫應答發生時,T細胞與抗原提呈細胞之間形成免疫連接,由T細胞受體(T cell receptor,TCR)發出第一特異性免疫信號[14]。TCR信號可以激活其對應的多條信號通路,觸發級聯免疫反應[15]。然而激活初始T細胞還需CD28發出第二協同刺激信號。在CD28信號激活其相應的信號通路后,T細胞才能有效激活并開始擴增[16]。而PD-1與PD-L1結合后,通過TCR信號募集的淋巴細胞特異性蛋白酪氨酸激酶可以快速磷酸化PD-1胞質區信號基序的酪氨酸,招募并激活下游信號分子[8, 17-18],從而抑制TCR和CD28介導的信號傳導[12, 19],最終對T細胞免疫應答產生負性調控作用[20-21],見圖1。
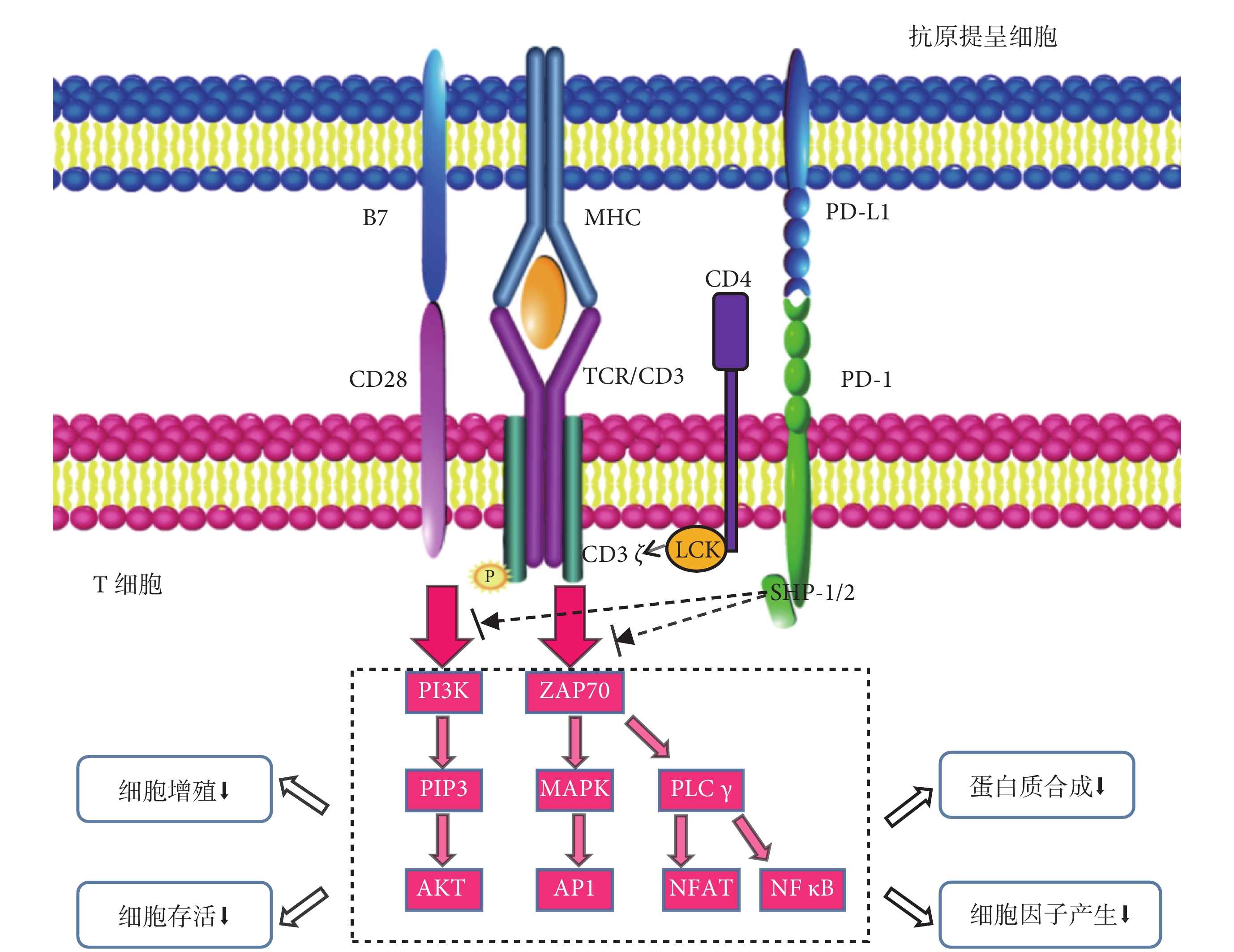 圖1
PD-1/PD-L1信號通路對T細胞免疫應答的作用
圖1
PD-1/PD-L1信號通路對T細胞免疫應答的作用
免疫應答發生時,由TCR和APC上載有抗原肽的MHC接觸發出第一信號,后CD4分子以TCR/CD3復合物為中心在T細胞表面多聚成簇,連于CD4受體胞內段的LCK也由此被招募至TCR/CD3受體,LCK被激活后會磷酸化CD3ζ鏈胞內段的ITAM,進而使ZAP70分子被募集至CD3ζ鏈并被激活,活化的ZAP70介導PLCγ的活化,PLCγ被活化后進一步激活NFAT和NF-κB信號通路。另外,活化的ZAP70還通過活化GRB-2 進一步激活Ras-MAPK信號通路,最后NFAT、NF-κB 以及AP-1等轉錄因子被活化并進入核內調控相關靶基因的轉錄。然而激活初始T細胞還需協同刺激信號的輔助,CD28與B7結合后激活細胞內的PI3K,形成PIP3并進一步激活AKT,使T細胞有效激活并開始擴增。PD-1與PD-L1相互識別并結合,其胞質區的ITSM酪氨酸被LCK磷酸化并招募和激活SHP-2/SHP-1,從而使通過PI3K途徑和通過ZAP70途徑的信號去磷酸化,最終導致T細胞活化、增殖、存活、細胞因子產生減少以及分化和代謝特征的改變。PD-1,程序性細胞死亡蛋白-1;PD-L1,程序性細胞死亡配體1;TCR,T細胞受體;APC,抗原提呈細胞;MHC,主要組織相容性復合體;LCK,淋巴細胞特異性蛋白酪氨酸激酶;ITAM,免疫受體酪氨酸活化基序;ZAP70,Zeta鏈相關蛋白激酶70;PLC,磷脂酶C;NFAT,T細胞核因子;NF-κB,核因子κB;GRB-2,生長因子受體結合蛋白-2;MAPK,絲裂原活化蛋白激酶;AP-1,激活蛋白1;PI3K,磷脂酰肌醇3激酶;PIP3,磷脂酰肌醇-3,4,5-三磷酸;AKT,蛋白激酶B;ITSM,免疫受體酪氨酸開關基序;SHP-2,含Src同源區2結構域的酪氨酸磷酸酶2;SHP-1,含Src同源區2結構域的酪氨酸磷酸酶1。
2 慢阻肺的T細胞免疫紊亂
慢阻肺通常與香煙煙霧以及環境中的有害顆粒或氣體有關,免疫功能紊亂在其中具有重要作用,部分學者提出慢阻肺是一種吸煙引起的自身免疫性疾病的觀點[22]。
2.1 慢阻肺中的CD8+ T細胞(慢阻肺中的CD8+/CD4+ T細胞失衡)
Williams等[23]發現慢阻肺患者肺中的CD8+ T細胞數量增加,并且顆粒酶和/或穿孔素的表達以及Fas/FasL途徑增強。研究還發現其凋亡率也較低[24]。動物實驗研究亦發現慢阻肺小鼠模型中出現肺CD8+ T細胞寡克隆擴增,并且這一現象在戒煙后仍持續存在[25]。另一方面,慢阻肺患者CD4+ T淋巴細胞未發現差異或減少[26-28],CD8+/CD4+ T細胞失衡。CD8+ T細胞數量增多不僅可以通過細胞毒性作用導致肺破壞[29],還可以誘導基質金屬蛋白酶產生從而降解肺彈性蛋白[30]。研究證實慢阻肺患者CD8+ T細胞數量與氣道阻塞程度及肺氣腫的嚴重程度呈正比[31-32]。并且CD8+ T淋巴細胞數量與第1秒用力呼氣容積(forced expiratory volume in the first second,FEV1)呈負相關[33]。
2.2 慢阻肺中的CD4+ T細胞
初始CD4+ T細胞在激活后可以分化為不同的亞群效應細胞(表1)。這些亞群細胞在某些條件下可以相互轉化,共同維持機體的免疫平衡。在慢阻肺中,不同的CD4+ T細胞亞群平衡發生了不同程度的紊亂。
2.2.1 慢阻肺中的Th1/Th2失衡
研究發現慢阻肺患者外周血中γ干擾素(interferon-γ,IFN-γ)水平升高而IL-4水平降低[34-35],說明慢阻肺患者出現Th1/Th2失衡,并且傾向于Th1細胞。近來一項研究表明慢阻肺患者Th1細胞分泌的IL-2與氣流受限程度呈正相關,IL-8與外周血氧飽和度呈負相關[36]。亦有研究報道Th1反應與肺氣腫的嚴重程度相關[37]。Th1細胞可以分泌IFN-γ,誘導產生金屬蛋白酶降解肺基質彈性蛋白,使肺組織失去彈力從而形成肺氣腫。同時IFN-γ還可以趨化炎癥細胞,導致炎癥浸潤和組織破壞從而降低患者的肺功能[38]。
2.2.2 慢阻肺中的TH17/Treg失衡
Louren?o等[39]總結穩定型慢阻肺相關研究,發現與對照組相比,慢阻肺中Th17細胞及其相關細胞因子呈增加趨勢,而Treg及其相關細胞因子呈減少趨勢。并且研究表明,Th17/Treg比率的增加與用力肺活量(forced vital capacity,FVC)、FEV1以及FEV1/FVC呈負相關[40]。還有研究報道慢阻肺患者的CD4+IL-10+ T淋巴細胞與下肢肌肉力量呈正相關[36]。
2.3 慢阻肺急性加重中的免疫紊亂
慢阻肺自然病程中會出現呼吸道癥狀反復惡化,稱為慢阻肺急性加重。感染是其主要原因之一,急性加重將加劇并進一步導致穩定型慢阻肺患者肺實質的病理表現、肺組織結構破壞和肺功能下降[41]。
2.3.1 慢阻肺急性加重中的CD4+和CD8+ T細胞
研究發現與穩定型慢阻肺相比,慢阻肺急性加重患者中性粒細胞和嗜酸性粒細胞數量增加[28,42-43],而在淋巴細胞亞群方面研究結果各不相同。但是我們在穩定型慢阻肺中發現的CD8+/CD4+ T細胞失衡在慢阻肺急性加重中依然存在。研究表明慢阻肺急性加重患者與健康對照組相比外周血中CD4+/CD8+ T細胞比率降低,CD8+ T細胞數量增加而CD4+ T細胞數量減少,并且CD4+ T細胞與肺功能指標呈顯著正相關[44]。
2.3.2 慢阻肺急性加重中的Th1/Th2以及TH17/Treg
盡管關于急性加重與穩定型慢阻肺患者Th1/Th2細胞比率的研究得出了不同的結論,但當與健康對照組相比,急性加重患者中出現了類似于穩定型慢阻肺的發現,即Th1顯著升高,而Th2顯著降低,Th1/Th2細胞比例增加[44]。Louren?o等[39]匯總相關研究發現慢阻肺急性加重與穩定型慢阻肺相比,Th17細胞數量增加,其相應細胞因子水平也增加,而Treg細胞數量增加,細胞因子減少。
包括香煙煙霧等環境因素在內的復雜多機制導致慢阻肺患者的慢性炎癥,肺臟中T細胞免疫紊亂,具有防御作用的CD8+ T細胞、Th1、Th17及相關細胞因子異常增多,導致肺的破壞。然而,當病原體侵犯時,這些免疫細胞卻不能產生正常的免疫應答,使慢阻肺患者發生反復的急性加重,從而進一步導致肺的破壞。研究表明慢阻肺患者的免疫紊亂與PD-1/PD-L1軸有關。
3 PD-1/PD-L1在慢阻肺中的作用
3.1 PD-1/PD-L1在穩定型慢阻肺中的表達
慢阻肺患者與健康對照組相比,樹突狀細胞(dendritic cells,DCs)上抗炎共刺激分子PD-L1的表達減少[45],而在香煙煙霧或機動車尾氣暴露誘導慢阻肺模型大鼠肺上皮細胞上PD-L1上調[46]。穩定型慢阻肺患者與對照組相比外周血PD-1+T細胞數量增加,并且在阻斷PD-1后,CD4+和CD8+效應T細胞的增殖增強,IFN-γ的產生也增加[47]。還有研究發現慢阻肺中表達PD-1的CD8+ T細胞比例大于對照組[48]。
3.2 PD-1/PD-L1在慢阻肺急性加重中的表達
研究表明慢阻肺急性加重患者外周血中PD-1+CD4+ T細胞的頻率高于穩定型慢阻肺患者和對照組。而CD8+ T細胞表達PD-1的頻率比較低并且各組之間相似。此外,慢阻肺急性加重患者PD-1+ Treg細胞的比例也明顯高于對照組。PD-1阻斷后,IL-10的產生增加,表明Treg、Th2和Th1細胞亞群的功能增加。因此可以得出結論,在慢阻肺急性加重中,PD-1+CD4+ T細胞比例升高,T細胞細胞因子反應受損,阻斷PD-1可以逆轉T細胞衰竭并增強細胞因子反應[49]。慢阻肺的一個主要復雜因素是氣道感染在急性加重以及疾病進展中的重要作用。研究發現感染使PD-1在慢阻肺和對照組T細胞上的表達上調。與對照組相比,慢阻肺巨噬細胞上病毒誘導的配體PD-L1表達降低,感染導致慢阻肺的IFN-γ釋放量相應增加。同時,流感顯著上調了對照組CD8+ T細胞上的細胞毒性脫顆粒(CD107a)標志物,但對慢阻肺患者的細胞毒性脫顆粒物(CD107a)沒有上調。說明慢阻肺急性加重患者CD8+ T細胞功能障礙[48]。另外一項研究發現與對照組相比,慢阻肺患者抗原刺激的IFN-γ產生減少,PD-1表達增加[50]。動物實驗證實,香煙煙霧誘導的和細菌誘導的炎癥與表達PD-1和PD-L1的肺CD4+、CD8+ T細胞數量增加有關。阻斷PD-1可以減少肺損傷和中性粒細胞炎癥。并且慢阻肺急性加重的血清中PD-L1濃度增加。PD-L1濃度與血清和肺泡灌洗液中PD-1濃度呈正相關[51]。還有研究表明PD-1抑制劑治療可通過降低PD-1表達和增加PD-L1表達來減輕慢阻肺的肺部炎癥[52]。
3.3 PD-1/PD-L1介導慢阻肺中T細胞衰竭
T細胞衰竭是T細胞功能障礙的一種獨特的分化狀態。在衰竭過程中,功能的喪失以分層的方式發生。通常從細胞增殖能力、細胞因子的產生和體外殺傷能力的喪失開始,然后是脫顆粒等細胞毒性功能的喪失,最后是T細胞數量的減少[53]。T細胞衰竭模式已在人類的許多慢性感染和癌癥期間出現[54-56]。免疫調節是T細胞衰竭的核心,而PD-1及其配體的軸似乎是參與T細胞衰竭的主要抑制性受體途徑。然而衰竭的T細胞并不是完全終結的狀態,通過阻斷PD-1通路可以逆轉T細胞衰竭[57]。
研究表明與健康對照組相比,慢阻肺患者的初始T細胞數量顯著減少,而表達衰竭標記物(PD-1)的初始和記憶T細胞比例增加[58]。另外一項研究發現與健康志愿者相比,CD8+初始T細胞的頻率在慢阻肺中顯著降低。慢阻肺和慢阻肺急性加重患者活化的CD4+ T細胞的比例降低,效應記憶CD8+ T細胞(effector memory CD8+ T cells,CD8+ TEM)的頻率顯著增加,并且PD1+的CD8+ TEM的頻率顯著高于健康組。與健康組相比,慢阻肺急性加重中PD1+的CD8+ TEM中CD38的表達顯著增加。CD38是獲得性抵抗PD-1/PD-L1阻斷的主要機制,導致CD8+ T細胞抗病毒活性的保留[59]。此外,研究證明PD-1在呼吸道病毒再感染過程中損害次級效應肺CD8+ T細胞。他們發現肺部CD8+ T細胞損傷類似于在慢性病毒感染期間觀察到的衰竭表型,其中CD8+ T細胞上調PD-1,并且當被病毒抗原再次刺激時沒有反應。CD8+ T細胞衰竭發生在感染后幾周,產生IFN-γ和TNF-α的能力首先喪失,隨后是脫顆粒和細胞毒性能力受損[60-61]。而阻斷PD-1可以逆轉T細胞衰竭并恢復細胞因子反應[49,57],從而減少慢阻肺的肺損傷和肺部炎癥[51-52]。
綜上,慢阻肺中存在T細胞衰竭,PD-1誘導慢阻肺中T細胞衰竭,阻斷PD-1可以逆轉T細胞衰竭。
3.4 慢阻肺、PD-1/PD-L1和免疫代謝
免疫反應高度依賴于代謝過程中的主要變化。不同的免疫細胞利用不同的代謝程序來實現其效應功能。正常生理情況下,大多數靜息免疫細胞消耗能量以三磷酸腺苷(adenosine triphosphate,ATP)的形式,主要通過糖的有氧氧化產生。記憶T細胞、Treg細胞和未成熟骨髓來源的DCs也可以利用脂肪酸氧化。抑制糖酵解可以促進記憶T細胞的形成,而抑制脂肪酸氧化和氧化磷酸化可減少記憶T細胞的形成。免疫細胞被激活后將從靜止狀態轉化為效應模式,進入細胞的葡萄糖轉運的增加驅動糖酵解活性的提高,導致糖酵解中間分子作為生物合成過程的前體分子的過度利用,例如磷酸戊糖途徑,脂質合成以及乳酸生成的增加[62]。
3.4.1 慢阻肺中的免疫代謝變化
慢阻肺患者與健康對照組相比,外周血單核細胞(peripheral blood mononuclear cells,PBMCs)在利用葡萄糖時細胞外酸化率(extracellular acidification rate,ECAR)和棕櫚酸或丙酮酸的線粒體耗氧率(oxygen consumption rate,OCR)顯著降低。在葡萄糖或棕櫚酸存在下,細胞體外實驗顯示暴露于香煙煙霧后產生類似的代謝變化和細胞吞噬能力的降低。并且觀察到健康吸煙者中PBMCs的棕櫚酸OCR與FEV1和FVC呈正相關。健康吸煙者向脂肪酸代謝的轉變促進了炎性細胞因子反應[63]。而另一項研究表明非吸煙者對照組、吸煙組和慢阻肺組的肺泡巨噬細胞(alveolar macrophages,AMs)基礎氧化應激反應和ECAR相似,但也觀察到慢阻肺患者有較低的線粒體呼吸和有缺陷的代償性糖酵解,并且與較低的FEV1%pred相關[64]。還有研究發現慢阻肺患者的中性粒細胞糖原循環、糖異生和糖酵解減少,導致功能受損[65]。動物實驗表明,香煙煙霧暴露改變了肺葡萄糖代謝,導致糖酵解速率降低和戊糖磷酸途徑增加,這分別由甘油醛-3-磷酸脫氫酶和葡萄糖-6-磷酸脫氫酶的表達和活性改變所致[66]。香煙煙霧暴露使Ⅱ型肺泡細胞代謝葡萄糖時細胞外酸化率降低,而棕櫚酸代謝增加,并且肉毒堿-棕櫚酰轉移酶1A(carnitine palmitoyl-transferase 1A,CPT1A)在Ⅱ型肺泡細胞中的表達增加[67]。慢阻肺患者AMs的轉錄組和脂質組譜顯示了GOLD等級依賴的變化,如膽固醇代謝和干擾素-α和γ反應。據推測,膽固醇的細胞內積聚是細胞炎癥反應的觸發因素。而香煙煙霧可以通過降低ATP結合盒轉運蛋白A1的轉運活性導致膽固醇在細胞內積聚[68]。
3.4.2 PD-1/PD-L1在免疫代謝中的作用
兩個主要的靶通路包括Ras-絲裂原活化蛋白激酶和磷脂酰肌醇3激酶/蛋白激酶B,它們是T細胞代謝的關鍵調節因子,主要整合來自TCR、共刺激因子和細胞因子受體信號來促進糖酵解表型。而PD-1與PD-L1結合可以抑制這些途徑,從而抑制T細胞的糖酵解以及氨基酸轉運和代謝,最終導致T細胞發揮效應功能所需的能量和物質合成減少。但同時也減少效應T細胞由糖酵解引起的快速死亡和細胞終末分化,使T細胞壽命得以延長。這與前面我們提到的慢阻肺患者效應記憶T細胞的比例增加但功能受損相符合。同時,PD-1/PD-L1信號通路可以通過增加CPT1A的表達促進內源性脂肪酸氧化,并通過增加脂肪酶的表達誘導脂肪分解,而使代謝平衡向脂肪酸代謝模式傾斜[69-70]。這與前文提到在健康吸煙者和香煙煙霧暴露的肺泡細胞中觀察到的向脂肪酸代謝的轉變一致,而在慢阻肺中仍缺乏相關研究。
炎癥性T細胞反應依賴于有氧糖酵解代謝。葡萄糖代謝的調節對免疫細胞的激活和功能具有關鍵作用,葡萄糖的缺乏直接阻礙了細胞因子的產生。研究發現與對照組相比,慢阻肺急性加重表現出代謝活性減弱,包括糖酵解、膽固醇代謝、脂肪生成等,慢阻肺急性加重中的CD4+或CD8+ T細胞攝取葡萄糖的能力降低。此外,慢阻肺急性加重中的T細胞表現出線粒體質量的損失和糖酵解代謝的依賴性,使它們產生更少的ATP和在激活中執行關鍵細胞功能的能力受損。并且,與健康對照組相比,PD-1在慢阻肺急性加重中過度表達。為了證實糖酵解調節慢阻肺急性加重的適應性免疫系統,他們用mTOR激活劑和抗PD-1抑制劑處理慢阻肺急性加重的T細胞,發現T細胞產生IFN-γ、葡萄糖轉運蛋白1表達和葡萄糖攝取增加[71]。
綜上,目前有限的研究表明慢阻肺患者存在免疫代謝重編程,其中葡萄糖代謝方面均提示糖酵解減少,而脂肪酸代謝方面研究較少而不能得出結論。糖酵解的減少將導致慢阻肺患者免疫細胞的能量和物質合成不足,尤其是慢阻肺急性加重時T細胞顯示出對糖酵解代謝的依賴性。而PD-1/PD-L1信號通路在抑制T細胞的糖酵解方面發揮重要作用。因此,我們假設PD-1在慢阻肺的上調導致免疫代謝紊亂,不能為T細胞發揮效應功能提供足夠的能量和物質基礎,從而在慢阻肺的發病機制中發揮作用。
4 抗PD-1/PD-L1在慢阻肺中的治療前景
PD-1/PD-L1抑制劑治療主要集中在腫瘤領域,目前各種PD-1和(或)PD-L1單抗藥物已經獲批應用于PD-L1高表達(腫瘤細胞≥1%~50%或免疫細胞≥1%~10%)的患者[72]。穩定型慢阻肺患者肺中CD8+ T細胞中PD-1表達的百分比高達16.2%,CD4+ T細胞中PD-1表達的百分比高達12.5%[48,50],急性加重時PD-1+ T細胞的頻率要高于穩定型慢阻肺患者[49]。并且合并慢阻肺的肺癌患者PD-L1陽性表達要高于非慢阻肺肺癌患者[73]。肺癌合并慢阻肺患者可能從免疫治療中獲益更多,具有更好的預后和肺功能改善[74]。此外,PD-1/PD-L1抑制劑可增強T細胞反應,提供針對多種感染的免疫防御[75]。在2019年新型冠狀病毒大流行期間,抗PD-1/PD-L1治療顯示其收益和風險并存。一方面通過抗PD-1/PD-L1干擾抑制信號的轉導,可提高COVID -19患者體內T細胞的數量和功能,進而提高T細胞對病毒的清除率。另一方面衰竭T細胞的再激活導致炎癥細胞因子釋放加劇,引起免疫相關不良事件[76]。
慢阻肺中存在的免疫紊亂和T細胞功能障礙為靶向PD-1/PD-L1治療提供了理論依據,但同時仍然有許多問題需要思考。首先是治療時機的選擇,PD-1或PD-L1表達的百分比高達多少時需要治療,是否需要對穩定期慢阻肺患者進行治療,還是僅僅在急性加重時給予治療。其次是安全性如何,免疫相關不良事件的危害不容忽視。
5 總結與展望
慢阻肺的發病機制迄今尚未完全闡明,大量的研究結果表明慢阻肺中存在免疫紊亂,包括免疫細胞數量變化、比例失衡、功能障礙以及代謝改變。PD-1是一個關鍵的免疫檢查點抑制受體,越來越多的研究發現慢阻肺患者中PD-1的表達上調。PD-1/PD-L1軸可以誘導T細胞衰竭,使T細胞的增殖、活化和效應功能降低。不管是在穩定型慢阻肺的慢性炎癥環境還是在慢阻肺急性加重中,幾乎所有研究均發現了PD-1+ T細胞的數量增加并且功能受損。阻斷PD-1后T細胞的功能得到恢復,肺部炎癥和損傷減輕。此外,PD-1/PD-L1信號通路還可以介導慢阻肺中的免疫代謝紊亂,然而相關研究較少,這將是未來的研究方向之一。總的來說,PD-1/PD-L1在慢阻肺慢性炎癥環境和急性加重中均發揮一定的作用。但是PD-1/PD-L1信號通路對慢阻肺炎癥、免疫和代謝的調控具有復雜性,其對慢阻肺的確切作用與機制仍有待進一步研究。
利益沖突:本文不涉及任何利益沖突
由于人口增長和老齡化,慢性阻塞性肺疾病(簡稱慢阻肺)的患病率和發病率不斷上升。近十幾年來,全球慢阻肺死亡人數增加了17.5%。預計2040年慢阻肺將成為全球第四大死因[1]。其發病機制目前尚無定論,治療方法也較局限。近年來越來越多的研究關注到免疫調節及紊亂在其中發揮著重要作用。吸入刺激物會導致呼吸道內的結構細胞和炎癥細胞激活并釋放出大量的趨化因子,從而進一步招募和活化大量免疫細胞,觸發級聯免疫反應。導致組織破壞和粘液過度分泌,最終導致不可逆的呼吸道結構改變和氣流受限[2]。程序性細胞死亡蛋白-1(programmed cell death protein 1,PD-1)/程序性細胞死亡配體1(programmed cell death ligand 1,PD-L1)軸是機體免疫中不可或缺的信號通路。然而目前關于PD-1/PD-L1信號通路的研究大多集中于腫瘤領域,與慢阻肺相關的研究相對較少。因此,我們對PD-1/PD-L1軸在慢阻肺中的研究進展作一綜述。
1 PD-1/PD-L1的概念及意義
1.1 PD-1/PD-L1的發現、結構及表達
1992年,Ishida等[3]發現了免疫球蛋白基因超家族新成員PD-1。PD-1又稱CD279,由三部分組成,包括含有IgV樣結構域的胞外區,跨膜結構域以及帶有兩個酪氨酸信號基序的胞內區[4]。PD-1表達于包括T細胞在內的多種免疫細胞[5-7],但它在靜息T細胞上不表達,而是在刺激后24 h內在活化的成熟T細胞上誘導表達[8]。2000年,Freeman等[9]發現了一種類似B7的分子,并證明它是PD-1的配體,后來被命名為PD-L1。Dong等[10]報道了與PD-L1相同的分子。PD-L1又稱B7-H1或CD274,由三部分組成,包括含有IgV和IgC樣結構域的胞外區、跨膜結構域和不含典型信號基序的短胞質尾[11-12]。PD-L1不僅表達于T細胞等免疫細胞,還表達于一些健康組織細胞,例如血管、皮膚、胰腺、胎盤、角膜、肝臟和肺的一些細胞[12-13]。
1.2 PD-1/PD-L1軸對T細胞免疫應答的作用
免疫應答發生時,T細胞與抗原提呈細胞之間形成免疫連接,由T細胞受體(T cell receptor,TCR)發出第一特異性免疫信號[14]。TCR信號可以激活其對應的多條信號通路,觸發級聯免疫反應[15]。然而激活初始T細胞還需CD28發出第二協同刺激信號。在CD28信號激活其相應的信號通路后,T細胞才能有效激活并開始擴增[16]。而PD-1與PD-L1結合后,通過TCR信號募集的淋巴細胞特異性蛋白酪氨酸激酶可以快速磷酸化PD-1胞質區信號基序的酪氨酸,招募并激活下游信號分子[8, 17-18],從而抑制TCR和CD28介導的信號傳導[12, 19],最終對T細胞免疫應答產生負性調控作用[20-21],見圖1。
圖1
PD-1/PD-L1信號通路對T細胞免疫應答的作用
免疫應答發生時,由TCR和APC上載有抗原肽的MHC接觸發出第一信號,后CD4分子以TCR/CD3復合物為中心在T細胞表面多聚成簇,連于CD4受體胞內段的LCK也由此被招募至TCR/CD3受體,LCK被激活后會磷酸化CD3ζ鏈胞內段的ITAM,進而使ZAP70分子被募集至CD3ζ鏈并被激活,活化的ZAP70介導PLCγ的活化,PLCγ被活化后進一步激活NFAT和NF-κB信號通路。另外,活化的ZAP70還通過活化GRB-2 進一步激活Ras-MAPK信號通路,最后NFAT、NF-κB 以及AP-1等轉錄因子被活化并進入核內調控相關靶基因的轉錄。然而激活初始T細胞還需協同刺激信號的輔助,CD28與B7結合后激活細胞內的PI3K,形成PIP3并進一步激活AKT,使T細胞有效激活并開始擴增。PD-1與PD-L1相互識別并結合,其胞質區的ITSM酪氨酸被LCK磷酸化并招募和激活SHP-2/SHP-1,從而使通過PI3K途徑和通過ZAP70途徑的信號去磷酸化,最終導致T細胞活化、增殖、存活、細胞因子產生減少以及分化和代謝特征的改變。PD-1,程序性細胞死亡蛋白-1;PD-L1,程序性細胞死亡配體1;TCR,T細胞受體;APC,抗原提呈細胞;MHC,主要組織相容性復合體;LCK,淋巴細胞特異性蛋白酪氨酸激酶;ITAM,免疫受體酪氨酸活化基序;ZAP70,Zeta鏈相關蛋白激酶70;PLC,磷脂酶C;NFAT,T細胞核因子;NF-κB,核因子κB;GRB-2,生長因子受體結合蛋白-2;MAPK,絲裂原活化蛋白激酶;AP-1,激活蛋白1;PI3K,磷脂酰肌醇3激酶;PIP3,磷脂酰肌醇-3,4,5-三磷酸;AKT,蛋白激酶B;ITSM,免疫受體酪氨酸開關基序;SHP-2,含Src同源區2結構域的酪氨酸磷酸酶2;SHP-1,含Src同源區2結構域的酪氨酸磷酸酶1。
2 慢阻肺的T細胞免疫紊亂
慢阻肺通常與香煙煙霧以及環境中的有害顆粒或氣體有關,免疫功能紊亂在其中具有重要作用,部分學者提出慢阻肺是一種吸煙引起的自身免疫性疾病的觀點[22]。
2.1 慢阻肺中的CD8+ T細胞(慢阻肺中的CD8+/CD4+ T細胞失衡)
Williams等[23]發現慢阻肺患者肺中的CD8+ T細胞數量增加,并且顆粒酶和/或穿孔素的表達以及Fas/FasL途徑增強。研究還發現其凋亡率也較低[24]。動物實驗研究亦發現慢阻肺小鼠模型中出現肺CD8+ T細胞寡克隆擴增,并且這一現象在戒煙后仍持續存在[25]。另一方面,慢阻肺患者CD4+ T淋巴細胞未發現差異或減少[26-28],CD8+/CD4+ T細胞失衡。CD8+ T細胞數量增多不僅可以通過細胞毒性作用導致肺破壞[29],還可以誘導基質金屬蛋白酶產生從而降解肺彈性蛋白[30]。研究證實慢阻肺患者CD8+ T細胞數量與氣道阻塞程度及肺氣腫的嚴重程度呈正比[31-32]。并且CD8+ T淋巴細胞數量與第1秒用力呼氣容積(forced expiratory volume in the first second,FEV1)呈負相關[33]。
2.2 慢阻肺中的CD4+ T細胞
初始CD4+ T細胞在激活后可以分化為不同的亞群效應細胞(表1)。這些亞群細胞在某些條件下可以相互轉化,共同維持機體的免疫平衡。在慢阻肺中,不同的CD4+ T細胞亞群平衡發生了不同程度的紊亂。
2.2.1 慢阻肺中的Th1/Th2失衡
研究發現慢阻肺患者外周血中γ干擾素(interferon-γ,IFN-γ)水平升高而IL-4水平降低[34-35],說明慢阻肺患者出現Th1/Th2失衡,并且傾向于Th1細胞。近來一項研究表明慢阻肺患者Th1細胞分泌的IL-2與氣流受限程度呈正相關,IL-8與外周血氧飽和度呈負相關[36]。亦有研究報道Th1反應與肺氣腫的嚴重程度相關[37]。Th1細胞可以分泌IFN-γ,誘導產生金屬蛋白酶降解肺基質彈性蛋白,使肺組織失去彈力從而形成肺氣腫。同時IFN-γ還可以趨化炎癥細胞,導致炎癥浸潤和組織破壞從而降低患者的肺功能[38]。
2.2.2 慢阻肺中的TH17/Treg失衡
Louren?o等[39]總結穩定型慢阻肺相關研究,發現與對照組相比,慢阻肺中Th17細胞及其相關細胞因子呈增加趨勢,而Treg及其相關細胞因子呈減少趨勢。并且研究表明,Th17/Treg比率的增加與用力肺活量(forced vital capacity,FVC)、FEV1以及FEV1/FVC呈負相關[40]。還有研究報道慢阻肺患者的CD4+IL-10+ T淋巴細胞與下肢肌肉力量呈正相關[36]。
2.3 慢阻肺急性加重中的免疫紊亂
慢阻肺自然病程中會出現呼吸道癥狀反復惡化,稱為慢阻肺急性加重。感染是其主要原因之一,急性加重將加劇并進一步導致穩定型慢阻肺患者肺實質的病理表現、肺組織結構破壞和肺功能下降[41]。
2.3.1 慢阻肺急性加重中的CD4+和CD8+ T細胞
研究發現與穩定型慢阻肺相比,慢阻肺急性加重患者中性粒細胞和嗜酸性粒細胞數量增加[28,42-43],而在淋巴細胞亞群方面研究結果各不相同。但是我們在穩定型慢阻肺中發現的CD8+/CD4+ T細胞失衡在慢阻肺急性加重中依然存在。研究表明慢阻肺急性加重患者與健康對照組相比外周血中CD4+/CD8+ T細胞比率降低,CD8+ T細胞數量增加而CD4+ T細胞數量減少,并且CD4+ T細胞與肺功能指標呈顯著正相關[44]。
2.3.2 慢阻肺急性加重中的Th1/Th2以及TH17/Treg
盡管關于急性加重與穩定型慢阻肺患者Th1/Th2細胞比率的研究得出了不同的結論,但當與健康對照組相比,急性加重患者中出現了類似于穩定型慢阻肺的發現,即Th1顯著升高,而Th2顯著降低,Th1/Th2細胞比例增加[44]。Louren?o等[39]匯總相關研究發現慢阻肺急性加重與穩定型慢阻肺相比,Th17細胞數量增加,其相應細胞因子水平也增加,而Treg細胞數量增加,細胞因子減少。
包括香煙煙霧等環境因素在內的復雜多機制導致慢阻肺患者的慢性炎癥,肺臟中T細胞免疫紊亂,具有防御作用的CD8+ T細胞、Th1、Th17及相關細胞因子異常增多,導致肺的破壞。然而,當病原體侵犯時,這些免疫細胞卻不能產生正常的免疫應答,使慢阻肺患者發生反復的急性加重,從而進一步導致肺的破壞。研究表明慢阻肺患者的免疫紊亂與PD-1/PD-L1軸有關。
3 PD-1/PD-L1在慢阻肺中的作用
3.1 PD-1/PD-L1在穩定型慢阻肺中的表達
慢阻肺患者與健康對照組相比,樹突狀細胞(dendritic cells,DCs)上抗炎共刺激分子PD-L1的表達減少[45],而在香煙煙霧或機動車尾氣暴露誘導慢阻肺模型大鼠肺上皮細胞上PD-L1上調[46]。穩定型慢阻肺患者與對照組相比外周血PD-1+T細胞數量增加,并且在阻斷PD-1后,CD4+和CD8+效應T細胞的增殖增強,IFN-γ的產生也增加[47]。還有研究發現慢阻肺中表達PD-1的CD8+ T細胞比例大于對照組[48]。
3.2 PD-1/PD-L1在慢阻肺急性加重中的表達
研究表明慢阻肺急性加重患者外周血中PD-1+CD4+ T細胞的頻率高于穩定型慢阻肺患者和對照組。而CD8+ T細胞表達PD-1的頻率比較低并且各組之間相似。此外,慢阻肺急性加重患者PD-1+ Treg細胞的比例也明顯高于對照組。PD-1阻斷后,IL-10的產生增加,表明Treg、Th2和Th1細胞亞群的功能增加。因此可以得出結論,在慢阻肺急性加重中,PD-1+CD4+ T細胞比例升高,T細胞細胞因子反應受損,阻斷PD-1可以逆轉T細胞衰竭并增強細胞因子反應[49]。慢阻肺的一個主要復雜因素是氣道感染在急性加重以及疾病進展中的重要作用。研究發現感染使PD-1在慢阻肺和對照組T細胞上的表達上調。與對照組相比,慢阻肺巨噬細胞上病毒誘導的配體PD-L1表達降低,感染導致慢阻肺的IFN-γ釋放量相應增加。同時,流感顯著上調了對照組CD8+ T細胞上的細胞毒性脫顆粒(CD107a)標志物,但對慢阻肺患者的細胞毒性脫顆粒物(CD107a)沒有上調。說明慢阻肺急性加重患者CD8+ T細胞功能障礙[48]。另外一項研究發現與對照組相比,慢阻肺患者抗原刺激的IFN-γ產生減少,PD-1表達增加[50]。動物實驗證實,香煙煙霧誘導的和細菌誘導的炎癥與表達PD-1和PD-L1的肺CD4+、CD8+ T細胞數量增加有關。阻斷PD-1可以減少肺損傷和中性粒細胞炎癥。并且慢阻肺急性加重的血清中PD-L1濃度增加。PD-L1濃度與血清和肺泡灌洗液中PD-1濃度呈正相關[51]。還有研究表明PD-1抑制劑治療可通過降低PD-1表達和增加PD-L1表達來減輕慢阻肺的肺部炎癥[52]。
3.3 PD-1/PD-L1介導慢阻肺中T細胞衰竭
T細胞衰竭是T細胞功能障礙的一種獨特的分化狀態。在衰竭過程中,功能的喪失以分層的方式發生。通常從細胞增殖能力、細胞因子的產生和體外殺傷能力的喪失開始,然后是脫顆粒等細胞毒性功能的喪失,最后是T細胞數量的減少[53]。T細胞衰竭模式已在人類的許多慢性感染和癌癥期間出現[54-56]。免疫調節是T細胞衰竭的核心,而PD-1及其配體的軸似乎是參與T細胞衰竭的主要抑制性受體途徑。然而衰竭的T細胞并不是完全終結的狀態,通過阻斷PD-1通路可以逆轉T細胞衰竭[57]。
研究表明與健康對照組相比,慢阻肺患者的初始T細胞數量顯著減少,而表達衰竭標記物(PD-1)的初始和記憶T細胞比例增加[58]。另外一項研究發現與健康志愿者相比,CD8+初始T細胞的頻率在慢阻肺中顯著降低。慢阻肺和慢阻肺急性加重患者活化的CD4+ T細胞的比例降低,效應記憶CD8+ T細胞(effector memory CD8+ T cells,CD8+ TEM)的頻率顯著增加,并且PD1+的CD8+ TEM的頻率顯著高于健康組。與健康組相比,慢阻肺急性加重中PD1+的CD8+ TEM中CD38的表達顯著增加。CD38是獲得性抵抗PD-1/PD-L1阻斷的主要機制,導致CD8+ T細胞抗病毒活性的保留[59]。此外,研究證明PD-1在呼吸道病毒再感染過程中損害次級效應肺CD8+ T細胞。他們發現肺部CD8+ T細胞損傷類似于在慢性病毒感染期間觀察到的衰竭表型,其中CD8+ T細胞上調PD-1,并且當被病毒抗原再次刺激時沒有反應。CD8+ T細胞衰竭發生在感染后幾周,產生IFN-γ和TNF-α的能力首先喪失,隨后是脫顆粒和細胞毒性能力受損[60-61]。而阻斷PD-1可以逆轉T細胞衰竭并恢復細胞因子反應[49,57],從而減少慢阻肺的肺損傷和肺部炎癥[51-52]。
綜上,慢阻肺中存在T細胞衰竭,PD-1誘導慢阻肺中T細胞衰竭,阻斷PD-1可以逆轉T細胞衰竭。
3.4 慢阻肺、PD-1/PD-L1和免疫代謝
免疫反應高度依賴于代謝過程中的主要變化。不同的免疫細胞利用不同的代謝程序來實現其效應功能。正常生理情況下,大多數靜息免疫細胞消耗能量以三磷酸腺苷(adenosine triphosphate,ATP)的形式,主要通過糖的有氧氧化產生。記憶T細胞、Treg細胞和未成熟骨髓來源的DCs也可以利用脂肪酸氧化。抑制糖酵解可以促進記憶T細胞的形成,而抑制脂肪酸氧化和氧化磷酸化可減少記憶T細胞的形成。免疫細胞被激活后將從靜止狀態轉化為效應模式,進入細胞的葡萄糖轉運的增加驅動糖酵解活性的提高,導致糖酵解中間分子作為生物合成過程的前體分子的過度利用,例如磷酸戊糖途徑,脂質合成以及乳酸生成的增加[62]。
3.4.1 慢阻肺中的免疫代謝變化
慢阻肺患者與健康對照組相比,外周血單核細胞(peripheral blood mononuclear cells,PBMCs)在利用葡萄糖時細胞外酸化率(extracellular acidification rate,ECAR)和棕櫚酸或丙酮酸的線粒體耗氧率(oxygen consumption rate,OCR)顯著降低。在葡萄糖或棕櫚酸存在下,細胞體外實驗顯示暴露于香煙煙霧后產生類似的代謝變化和細胞吞噬能力的降低。并且觀察到健康吸煙者中PBMCs的棕櫚酸OCR與FEV1和FVC呈正相關。健康吸煙者向脂肪酸代謝的轉變促進了炎性細胞因子反應[63]。而另一項研究表明非吸煙者對照組、吸煙組和慢阻肺組的肺泡巨噬細胞(alveolar macrophages,AMs)基礎氧化應激反應和ECAR相似,但也觀察到慢阻肺患者有較低的線粒體呼吸和有缺陷的代償性糖酵解,并且與較低的FEV1%pred相關[64]。還有研究發現慢阻肺患者的中性粒細胞糖原循環、糖異生和糖酵解減少,導致功能受損[65]。動物實驗表明,香煙煙霧暴露改變了肺葡萄糖代謝,導致糖酵解速率降低和戊糖磷酸途徑增加,這分別由甘油醛-3-磷酸脫氫酶和葡萄糖-6-磷酸脫氫酶的表達和活性改變所致[66]。香煙煙霧暴露使Ⅱ型肺泡細胞代謝葡萄糖時細胞外酸化率降低,而棕櫚酸代謝增加,并且肉毒堿-棕櫚酰轉移酶1A(carnitine palmitoyl-transferase 1A,CPT1A)在Ⅱ型肺泡細胞中的表達增加[67]。慢阻肺患者AMs的轉錄組和脂質組譜顯示了GOLD等級依賴的變化,如膽固醇代謝和干擾素-α和γ反應。據推測,膽固醇的細胞內積聚是細胞炎癥反應的觸發因素。而香煙煙霧可以通過降低ATP結合盒轉運蛋白A1的轉運活性導致膽固醇在細胞內積聚[68]。
3.4.2 PD-1/PD-L1在免疫代謝中的作用
兩個主要的靶通路包括Ras-絲裂原活化蛋白激酶和磷脂酰肌醇3激酶/蛋白激酶B,它們是T細胞代謝的關鍵調節因子,主要整合來自TCR、共刺激因子和細胞因子受體信號來促進糖酵解表型。而PD-1與PD-L1結合可以抑制這些途徑,從而抑制T細胞的糖酵解以及氨基酸轉運和代謝,最終導致T細胞發揮效應功能所需的能量和物質合成減少。但同時也減少效應T細胞由糖酵解引起的快速死亡和細胞終末分化,使T細胞壽命得以延長。這與前面我們提到的慢阻肺患者效應記憶T細胞的比例增加但功能受損相符合。同時,PD-1/PD-L1信號通路可以通過增加CPT1A的表達促進內源性脂肪酸氧化,并通過增加脂肪酶的表達誘導脂肪分解,而使代謝平衡向脂肪酸代謝模式傾斜[69-70]。這與前文提到在健康吸煙者和香煙煙霧暴露的肺泡細胞中觀察到的向脂肪酸代謝的轉變一致,而在慢阻肺中仍缺乏相關研究。
炎癥性T細胞反應依賴于有氧糖酵解代謝。葡萄糖代謝的調節對免疫細胞的激活和功能具有關鍵作用,葡萄糖的缺乏直接阻礙了細胞因子的產生。研究發現與對照組相比,慢阻肺急性加重表現出代謝活性減弱,包括糖酵解、膽固醇代謝、脂肪生成等,慢阻肺急性加重中的CD4+或CD8+ T細胞攝取葡萄糖的能力降低。此外,慢阻肺急性加重中的T細胞表現出線粒體質量的損失和糖酵解代謝的依賴性,使它們產生更少的ATP和在激活中執行關鍵細胞功能的能力受損。并且,與健康對照組相比,PD-1在慢阻肺急性加重中過度表達。為了證實糖酵解調節慢阻肺急性加重的適應性免疫系統,他們用mTOR激活劑和抗PD-1抑制劑處理慢阻肺急性加重的T細胞,發現T細胞產生IFN-γ、葡萄糖轉運蛋白1表達和葡萄糖攝取增加[71]。
綜上,目前有限的研究表明慢阻肺患者存在免疫代謝重編程,其中葡萄糖代謝方面均提示糖酵解減少,而脂肪酸代謝方面研究較少而不能得出結論。糖酵解的減少將導致慢阻肺患者免疫細胞的能量和物質合成不足,尤其是慢阻肺急性加重時T細胞顯示出對糖酵解代謝的依賴性。而PD-1/PD-L1信號通路在抑制T細胞的糖酵解方面發揮重要作用。因此,我們假設PD-1在慢阻肺的上調導致免疫代謝紊亂,不能為T細胞發揮效應功能提供足夠的能量和物質基礎,從而在慢阻肺的發病機制中發揮作用。
4 抗PD-1/PD-L1在慢阻肺中的治療前景
PD-1/PD-L1抑制劑治療主要集中在腫瘤領域,目前各種PD-1和(或)PD-L1單抗藥物已經獲批應用于PD-L1高表達(腫瘤細胞≥1%~50%或免疫細胞≥1%~10%)的患者[72]。穩定型慢阻肺患者肺中CD8+ T細胞中PD-1表達的百分比高達16.2%,CD4+ T細胞中PD-1表達的百分比高達12.5%[48,50],急性加重時PD-1+ T細胞的頻率要高于穩定型慢阻肺患者[49]。并且合并慢阻肺的肺癌患者PD-L1陽性表達要高于非慢阻肺肺癌患者[73]。肺癌合并慢阻肺患者可能從免疫治療中獲益更多,具有更好的預后和肺功能改善[74]。此外,PD-1/PD-L1抑制劑可增強T細胞反應,提供針對多種感染的免疫防御[75]。在2019年新型冠狀病毒大流行期間,抗PD-1/PD-L1治療顯示其收益和風險并存。一方面通過抗PD-1/PD-L1干擾抑制信號的轉導,可提高COVID -19患者體內T細胞的數量和功能,進而提高T細胞對病毒的清除率。另一方面衰竭T細胞的再激活導致炎癥細胞因子釋放加劇,引起免疫相關不良事件[76]。
慢阻肺中存在的免疫紊亂和T細胞功能障礙為靶向PD-1/PD-L1治療提供了理論依據,但同時仍然有許多問題需要思考。首先是治療時機的選擇,PD-1或PD-L1表達的百分比高達多少時需要治療,是否需要對穩定期慢阻肺患者進行治療,還是僅僅在急性加重時給予治療。其次是安全性如何,免疫相關不良事件的危害不容忽視。
5 總結與展望
慢阻肺的發病機制迄今尚未完全闡明,大量的研究結果表明慢阻肺中存在免疫紊亂,包括免疫細胞數量變化、比例失衡、功能障礙以及代謝改變。PD-1是一個關鍵的免疫檢查點抑制受體,越來越多的研究發現慢阻肺患者中PD-1的表達上調。PD-1/PD-L1軸可以誘導T細胞衰竭,使T細胞的增殖、活化和效應功能降低。不管是在穩定型慢阻肺的慢性炎癥環境還是在慢阻肺急性加重中,幾乎所有研究均發現了PD-1+ T細胞的數量增加并且功能受損。阻斷PD-1后T細胞的功能得到恢復,肺部炎癥和損傷減輕。此外,PD-1/PD-L1信號通路還可以介導慢阻肺中的免疫代謝紊亂,然而相關研究較少,這將是未來的研究方向之一。總的來說,PD-1/PD-L1在慢阻肺慢性炎癥環境和急性加重中均發揮一定的作用。但是PD-1/PD-L1信號通路對慢阻肺炎癥、免疫和代謝的調控具有復雜性,其對慢阻肺的確切作用與機制仍有待進一步研究。
利益沖突:本文不涉及任何利益沖突