引用本文: 柯章敏, 潘航程, 王麗, 呂鏜烽, 張秀偉, 宋勇. 抑瘤素M在肺部疾病及惡病質中的研究進展. 中國呼吸與危重監護雜志, 2024, 23(12): 900-904. doi: 10.7507/1671-6205.202401039 復制
版權信息: ?四川大學華西醫院華西期刊社《中國呼吸與危重監護雜志》版權所有,未經授權不得轉載、改編
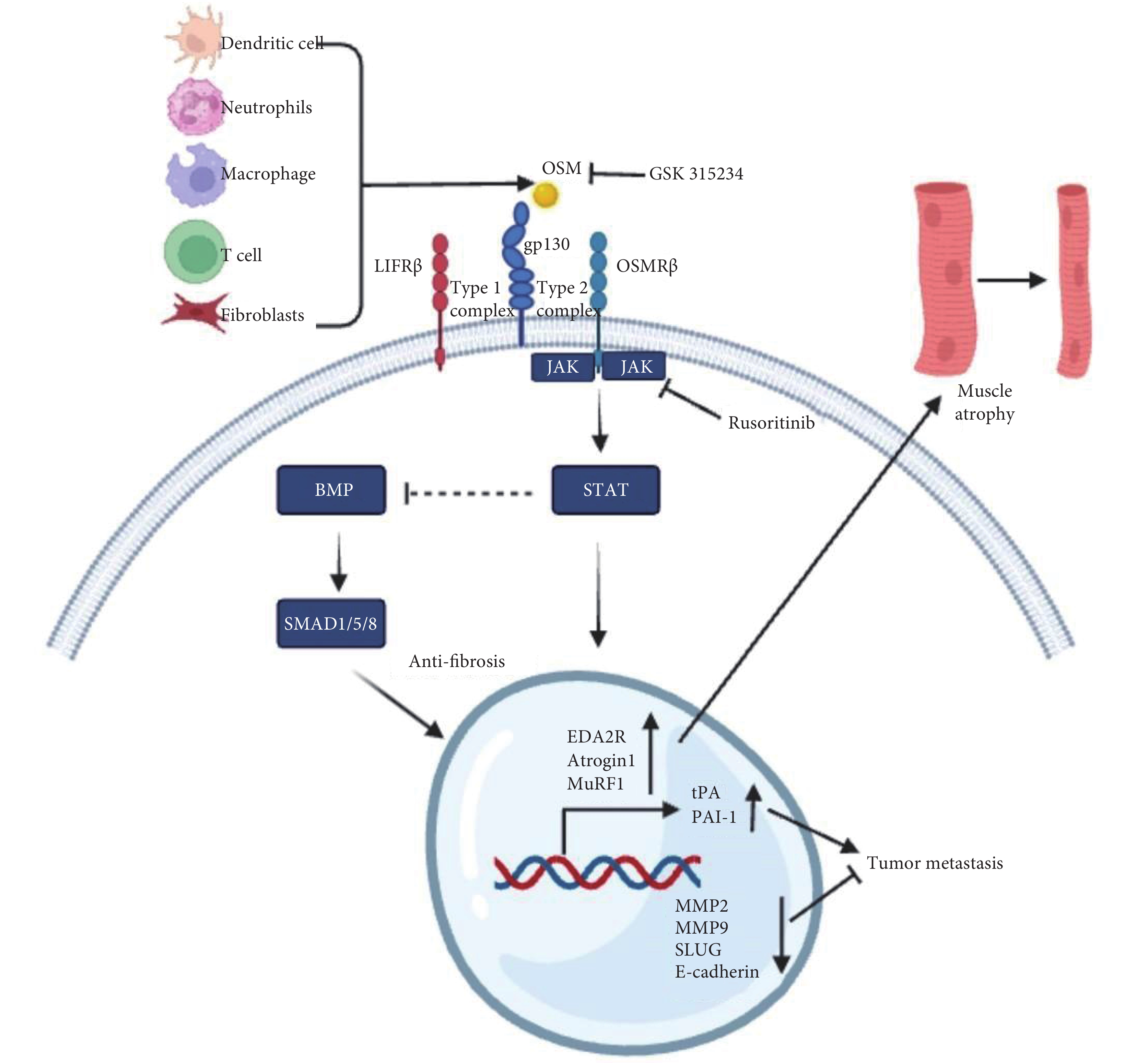
白細胞介素-6(Interleukin-6,IL-6)家族成員包括IL-6,抑瘤素M(oncostatin M,OSM),白血病抑制因子(Leukemia Inhibitory Factor,LIF),IL-11,IL-27,心肌營養素-1(cardiotrophin-1,CT-1)和心肌營養素樣細胞因子1(cardiotrophin-like cytokine factor 1,CLCF1)參與調節各種炎癥和腫瘤[1-3]。1986年,Zarling等[4]首次從人淋巴瘤U937細胞中分離一種生長調節蛋白,因其具有抑制黑色素瘤細胞的細胞生長而影響成纖維細胞因此得名OSM。成熟的人OSM分子大小約22 kDa,由196個氨基酸組成的可溶性蛋白[5]。OSM主要由活化的巨噬細胞、腫瘤相關成纖維細胞(cancer-associated fibroblasts,CAF)、T細胞、樹突狀細胞和中性粒細胞等多種細胞合成和分泌(圖1)[1,5]。LIF和OSM在結構上是IL-6家族最相似的成員,與其他IL-6家族細胞因子成員類似,OSM與gp130細胞外細胞因子結合同源區(cytokine-binding homology region,CHR)結構域結合,隨后招募LIFRβ形成I型受體復合物(LIFRβ/gp 130)或OSMR β形成II型受體復合物(OSMRβ/gp 130),且OSM與LIFR的結合親和力明顯低于OSMR(II型)復合物,激活Janus活化激酶/信號轉導子和轉錄激活子3(Janus-activated kinase/signal transducer and activator of transcription 3,JAK/STAT 3)、促分裂原活化蛋白激酶/細胞外調節激酶(mitogen-activated protein kinase/extracellular regulator kinase,MAPK/ERK)、c-Jun N末端激酶(c-Jun N-terminal Kinase,JNK)和磷脂酰肌醇-3-激酶/蛋白激酶B(phosphatidylinositol-3-kinase/protein kinase B,PI3K/AKT)等信號通路參與多種生理和病理功能,調控炎癥、腫瘤和肌肉萎縮等(圖1)[6-10]。對于部分細胞類型,OSM信號傳導亦可激活STAT1和STAT5通路[11-12]。目前OSM與OSMR形成的II型復合物通路在各種細胞系及動物實驗中進行了廣泛研究,LIFR(I型)復合物如何影響信號傳導或疾病進展尚不明確[1]。OSM已被證實在肺部炎癥和腫瘤中發揮重要作用,然而抗OSM治療目前尚在臨床前研究。本文綜述OSM與慢性肺部疾病的關聯, 介紹抗OSM通路對慢性肺部病變和肺部腫瘤的研究進展。
 圖1
OSM信號通路
圖1
OSM信號通路
1 OSM與慢性氣道疾病
細胞因子OSM在慢性氣道疾病過程中參與細胞外基質(Extracellular Matrix,ECM)蛋白沉積(如肺纖維化或嚴重哮喘)或分解代謝(如慢性阻塞性肺疾病)調控、在細胞增殖和細胞存活等方面發揮關鍵作用。肺成纖維細胞募集到氣道損傷部位被認為是導致ECM過度沉積的重要原因,且轉化生長因子-β(transforming growth factor-β,TGF-β)已被證實是纖維化發病機制的重要調節因子,TGF-β非依賴性途徑也可能參與其中[13]。
1.1 肺纖維化
過度的成纖維細胞活化、積聚和表型變化可能導致肺纖維化和組織功能喪失重要原因。Mozaffarian等[14]首次發現OSM在特發性肺纖維化和硬皮病患者的支氣管肺泡灌洗液中表達增高,募集炎癥細胞促進膠原沉積增加,該效應不依賴B和T淋巴細胞、嗜酸性粒細胞和肥大細胞且不通過經典IL-4/IL-13和TGF-β途徑,提示OSM可能作為獨立因素參與特發性肺纖維化的進程。Esnault[15]等通過獨創性途徑分析和qPCR進一步驗證表明OSM是成纖維細胞激活的潛在上游調節因子。過量的STAT-1/STAT 3信號傳導顯著加劇博萊霉素誘導的小鼠肺纖維化,而不通過經典的TGFβ-SMAD 3途徑[16]。因此,OSM通過何種機制誘導肺纖維化則成為研究的熱點。在BALB/c小鼠肺中,OSM誘導顯著的pSTAT 3活化,伴隨pSMAD 1/5抑制,且無TGFβ -SMAD 2活化[17]。由于pSMAD 1/5/8是BMP信號通路的下游,其在肺纖維化模型中表現出保護作用[18],因此pSTAT 3的過度活化可能通過調節BMP/pSMAD 1途徑間接介導纖維化。Ayaub等[19]通過博來霉素誘導的肺纖維化模型,并經過腺病毒轉染高表達IL-6和OSM,結果表明暴露于博來霉素和OSM或IL-6的小鼠肺中M2樣巨噬細胞的數量顯著增加,高表達IL-6Rα且缺乏OSMRβ,提出IL-6和OSM參與誘導巨噬細胞的選擇性M2活化,惡化博來霉素誘導的肺纖維化。進一步研究發現OSM增強成纖維細胞趨化性是由信號轉導子和轉錄激活子(STAT3)和p38絲裂原活化蛋白激酶介導的,而凝膠收縮和α-SMA表達的增強則由STAT3介導的[20]。與既往研究不同,Lan等[21]發現OSM預處理的間充質干細胞經旁分泌肝細胞生長因子,促進細胞增殖和遷移,博來霉素誘導的肺纖維化小鼠中移植OSM預處理的充質干細胞可顯著改善肺的呼吸功能,并下調了肺組織中炎癥因子轉化生長因子-β1和纖維化因子的表達。然而,OSM在特發性肺纖維化的作用仍需進一步闡述明確。
1.2 哮喘和慢性阻塞性肺疾病
OSM參與氣道重塑和肺功能下降相關,重癥哮喘由于氣道重塑致氣流不完全可逆,因此OSM可能調控重癥哮喘和慢性阻塞性肺疾病的病理改變。Simpsont等[22]發現相比于控制穩定不吸煙的哮喘患者,重癥哮喘伴不完全可逆氣流受限患者的痰液中巨噬細胞和中性粒細胞OSM表達水平明顯上升。近期經單細胞測序技術進一步證實氣道內OSM由肺炎克雷伯桿菌的脂多糖激活的巨噬細胞分泌[23]。Baines等[24]亦發現相比于健康志愿者,COPD患者的誘導痰中IL-8 和OSM表達明顯增高,提示OSM與重癥哮喘和慢性阻塞性肺疾病的慢性氣道炎癥和氣道重塑有關。Lin等[25]經生物信息學算法與單細胞或轉錄組數據進行交叉分析結果表明高水平的OSM,IL-18R1與重癥哮喘患者的肺功能和生存時間成負相關,因此OSM可作為重癥哮喘的監測和預后的標志物。在嚴重哮喘中,氣道上皮粘液產生細胞數量增加、肺間質纖維化及巨噬細胞高表達卵泡抑素樣1 促進OSM過度表達,誘導氣道上皮細胞中抵抗素樣分子α的增加,導致小鼠肺細胞外基質重塑[26],而抗OSM單抗可逆轉卵泡抑素樣1對氣道的重塑,提示OSM途徑可能是作用重癥哮喘和慢性阻塞性肺疾病氣道重塑新的靶點。
2 OSM與肺癌
肺癌是發病率和死亡率最高的惡性腫瘤,分子靶向和免疫的治療改善患者的生存時間,但是,多數患者在治療后仍死于腫瘤的耐藥、復發 [27],因此迫切需要尋找高效低毒新的治療靶點。OSM最初被發現在體外對黑色素瘤和其他癌癥細胞系具有直接的增殖抑制作用[4],Ouyang[28]等發現低濃度OSM在體外可抑制人肺癌細胞95-D細胞的增殖,減少基質金屬蛋白酶2(matrix metalloproteinase-2,MMP-2)和MMP-9的分泌降低細胞的粘附和侵襲能力。Spence等[29]發現OSM通過JAK3/STAT3通路促進組織型纖溶酶原激活劑(type plasminogen activator,tPA)和纖溶酶原激活物抑制物-1(plasminogen activator inhibitor-1,PAI-1)mRNA的表達促進腫瘤轉移。McCormick和Cichy團隊發現OSM較IL-6可更有效的促進肺癌細胞分化,OSM與TGF β 1因子共同調節透明質酸,促進肺癌轉移[30,31]。與之前的研究不同,Lauber等[32]發現OSM在體外對LLC細胞無增殖抑制作用,但在小鼠體內可促進腫瘤細胞顯著增長,通過轉染腺病毒致肺部OSM表達增高,可募集M2巨噬細胞改變腫瘤微環境,促進異位種植的腫瘤肺轉移增加,然而OSMRβ 敲除鼠則減少腫瘤肺轉移,提示OSM/OSMRβ通路調控腫瘤肺轉移。Shien等[33]亦發現OSM可在肺癌小鼠模型中經上皮間質轉化(epithelial–mesenchymal transition,EMT)促進肺轉移,肺癌細胞與CAF體外共孵育后,磷酸化STAT 3、OSMRβ和LIFRβ上調,同時E-鈣粘蛋白下調,CAF經OSMRβ/JAK 1/STAT 3通路促進腫瘤進展和耐藥,靶向選擇性JAK1抑制劑非戈替尼有效抑制STAT3激活,降低OSMR表達,抑制腫瘤增殖逆轉腫瘤耐藥。臨床前研究發現OSM及其通路在肺癌的發生和轉移中起到關鍵作用,那么OSM在真實世界肺癌患者中扮演什么樣的角色?Shien等[33]及其團隊分析TGCA和PROSPECT公共數據庫,高表達OSM,IL-6,LIF和OSMRβ的肺腺癌患者預后更差。另有研究表明放療和缺氧應激的肺癌細胞分泌大量微泡,誘導CAF分泌包括OSM,IL-8,IL-11,血管內皮生長因子等[34],OSM可能參與腫瘤的自我適應調控。部分研究發現OSM通過STAT1信號通路下調EMT起動子SLUG轉錄,抑制EMT及肺癌轉移[35]。綜上,臨床研究表明OSM可促進腫瘤增殖和轉移,而臨床前研究的結果并不完全一致,OSM通路網絡在肺癌中的具體機制需要進一步研究闡明。
3 OSM與惡病質
惡病質是一種以消耗為特征復雜的多因素綜合征,在肺癌和慢性肺部疾病患者中普遍存在,其主要特征表現為體重減輕、骨骼肌質量減少(伴或不伴有脂肪組織損失)以及全身炎癥[36-38]。全身性炎癥因子包括腫瘤壞死因子-α(TNF-α)、白細胞介素-1(IL-1)和IL-6 等水平升高可導致肌肉萎縮,針對其相應的中和抗體可抑制其萎縮效應[39,40]。在胰腺導管腺癌(高達80%患者發生惡病質)和肌營養不良患者的肌肉中OSMR和OSM靶基因的轉錄水平顯著增加,提示其在惡病質相關的肌肉萎縮中發揮重要作用[41]。Miki等[42]發現OSM處理C2C12肌管細胞24 h和48 h后,肌細胞生成素表達下降,atrogin-1和OMSR表達上升,肌管直徑比對照組分別減少18.7%和23.3%,STAT3抑制劑或敲除STAT3基因可逆轉其效應。與之類似,Domaniku等[41]通過敲除荷瘤小鼠特異性肌肉細胞OSMR,可抑制肌肉細胞萎縮恢復肌肉功能,近期其團隊進一步證實OSM可在體外通過JAK/STAT3通路上調原代肌管細胞表達Atrogin1致肌肉萎縮,明確了OSM信號通路在肌肉萎縮中的作用[43]。OSM誘導的肌肉萎縮依賴于JAK/STAT 3信號傳導,與Atrogin 1、Ampd 3、Serpina 3n、Cebpd、Sln和Mt 1/2多種萎縮相關基因的表達增加有關[42]。Bilgic等[43]近期工作發表在Nature雜志首次發現肺癌患者和Lewis小鼠惡病質模型的肌肉中組織外胚層發育不良A2受體(ectodysplasin A2 receptor,EDA2R)表達異常升高,與EDA2結合激活非經典NF-?B信號通路促進Atrogin1和MuRF1基因轉錄和蛋白表達,從而誘導肌肉萎縮,有趣的是,OSM不僅與OSMR結合發揮作用,可同時促進EDA-2R受體轉錄表達加劇肌肉細胞萎縮,敲除肌肉OSMR受體可逆轉EDA2R受體表達,抑制惡病質相關肌肉萎縮基因轉錄和緩解肌肉功能受損。相比與IL-6和LIF,OSM對萎縮基因的轉錄的調控作用更強,因此對肌管的萎縮效應更明顯。由于這三種細胞因子都與肌肉萎縮有關,因此理論上針對這些途徑的共同通路可能更有效。因此,近期一項I期針對IV期非小細胞肺癌合并惡病質的患者中使用JAK 1/2抑制劑魯索利替尼的安全性和有效性的臨床試驗(NCT 04906746)正在募集患者。一項II期隨機對照試驗,針對人源化單克隆抗OSM抗體(GSK
OSM在多種肺部疾病中發揮重要作用,部分抗OSM通路藥物臨床前研究取得優異的療效,然而目前尚無藥物獲批用于臨床疾病的治療。未來針對抗OSM或抗OSMRβ的藥物治療將成為肺部疾病研究中的熱門領域,我們期待著后續針對OSM信號通路更加深入的研究以及更多有效安全藥物的出現并應用于臨床。
利益沖突:本研究不涉及任何利益沖突。
白細胞介素-6(Interleukin-6,IL-6)家族成員包括IL-6,抑瘤素M(oncostatin M,OSM),白血病抑制因子(Leukemia Inhibitory Factor,LIF),IL-11,IL-27,心肌營養素-1(cardiotrophin-1,CT-1)和心肌營養素樣細胞因子1(cardiotrophin-like cytokine factor 1,CLCF1)參與調節各種炎癥和腫瘤[1-3]。1986年,Zarling等[4]首次從人淋巴瘤U937細胞中分離一種生長調節蛋白,因其具有抑制黑色素瘤細胞的細胞生長而影響成纖維細胞因此得名OSM。成熟的人OSM分子大小約22 kDa,由196個氨基酸組成的可溶性蛋白[5]。OSM主要由活化的巨噬細胞、腫瘤相關成纖維細胞(cancer-associated fibroblasts,CAF)、T細胞、樹突狀細胞和中性粒細胞等多種細胞合成和分泌(圖1)[1,5]。LIF和OSM在結構上是IL-6家族最相似的成員,與其他IL-6家族細胞因子成員類似,OSM與gp130細胞外細胞因子結合同源區(cytokine-binding homology region,CHR)結構域結合,隨后招募LIFRβ形成I型受體復合物(LIFRβ/gp 130)或OSMR β形成II型受體復合物(OSMRβ/gp 130),且OSM與LIFR的結合親和力明顯低于OSMR(II型)復合物,激活Janus活化激酶/信號轉導子和轉錄激活子3(Janus-activated kinase/signal transducer and activator of transcription 3,JAK/STAT 3)、促分裂原活化蛋白激酶/細胞外調節激酶(mitogen-activated protein kinase/extracellular regulator kinase,MAPK/ERK)、c-Jun N末端激酶(c-Jun N-terminal Kinase,JNK)和磷脂酰肌醇-3-激酶/蛋白激酶B(phosphatidylinositol-3-kinase/protein kinase B,PI3K/AKT)等信號通路參與多種生理和病理功能,調控炎癥、腫瘤和肌肉萎縮等(圖1)[6-10]。對于部分細胞類型,OSM信號傳導亦可激活STAT1和STAT5通路[11-12]。目前OSM與OSMR形成的II型復合物通路在各種細胞系及動物實驗中進行了廣泛研究,LIFR(I型)復合物如何影響信號傳導或疾病進展尚不明確[1]。OSM已被證實在肺部炎癥和腫瘤中發揮重要作用,然而抗OSM治療目前尚在臨床前研究。本文綜述OSM與慢性肺部疾病的關聯, 介紹抗OSM通路對慢性肺部病變和肺部腫瘤的研究進展。
圖1
OSM信號通路
1 OSM與慢性氣道疾病
細胞因子OSM在慢性氣道疾病過程中參與細胞外基質(Extracellular Matrix,ECM)蛋白沉積(如肺纖維化或嚴重哮喘)或分解代謝(如慢性阻塞性肺疾病)調控、在細胞增殖和細胞存活等方面發揮關鍵作用。肺成纖維細胞募集到氣道損傷部位被認為是導致ECM過度沉積的重要原因,且轉化生長因子-β(transforming growth factor-β,TGF-β)已被證實是纖維化發病機制的重要調節因子,TGF-β非依賴性途徑也可能參與其中[13]。
1.1 肺纖維化
過度的成纖維細胞活化、積聚和表型變化可能導致肺纖維化和組織功能喪失重要原因。Mozaffarian等[14]首次發現OSM在特發性肺纖維化和硬皮病患者的支氣管肺泡灌洗液中表達增高,募集炎癥細胞促進膠原沉積增加,該效應不依賴B和T淋巴細胞、嗜酸性粒細胞和肥大細胞且不通過經典IL-4/IL-13和TGF-β途徑,提示OSM可能作為獨立因素參與特發性肺纖維化的進程。Esnault[15]等通過獨創性途徑分析和qPCR進一步驗證表明OSM是成纖維細胞激活的潛在上游調節因子。過量的STAT-1/STAT 3信號傳導顯著加劇博萊霉素誘導的小鼠肺纖維化,而不通過經典的TGFβ-SMAD 3途徑[16]。因此,OSM通過何種機制誘導肺纖維化則成為研究的熱點。在BALB/c小鼠肺中,OSM誘導顯著的pSTAT 3活化,伴隨pSMAD 1/5抑制,且無TGFβ -SMAD 2活化[17]。由于pSMAD 1/5/8是BMP信號通路的下游,其在肺纖維化模型中表現出保護作用[18],因此pSTAT 3的過度活化可能通過調節BMP/pSMAD 1途徑間接介導纖維化。Ayaub等[19]通過博來霉素誘導的肺纖維化模型,并經過腺病毒轉染高表達IL-6和OSM,結果表明暴露于博來霉素和OSM或IL-6的小鼠肺中M2樣巨噬細胞的數量顯著增加,高表達IL-6Rα且缺乏OSMRβ,提出IL-6和OSM參與誘導巨噬細胞的選擇性M2活化,惡化博來霉素誘導的肺纖維化。進一步研究發現OSM增強成纖維細胞趨化性是由信號轉導子和轉錄激活子(STAT3)和p38絲裂原活化蛋白激酶介導的,而凝膠收縮和α-SMA表達的增強則由STAT3介導的[20]。與既往研究不同,Lan等[21]發現OSM預處理的間充質干細胞經旁分泌肝細胞生長因子,促進細胞增殖和遷移,博來霉素誘導的肺纖維化小鼠中移植OSM預處理的充質干細胞可顯著改善肺的呼吸功能,并下調了肺組織中炎癥因子轉化生長因子-β1和纖維化因子的表達。然而,OSM在特發性肺纖維化的作用仍需進一步闡述明確。
1.2 哮喘和慢性阻塞性肺疾病
OSM參與氣道重塑和肺功能下降相關,重癥哮喘由于氣道重塑致氣流不完全可逆,因此OSM可能調控重癥哮喘和慢性阻塞性肺疾病的病理改變。Simpsont等[22]發現相比于控制穩定不吸煙的哮喘患者,重癥哮喘伴不完全可逆氣流受限患者的痰液中巨噬細胞和中性粒細胞OSM表達水平明顯上升。近期經單細胞測序技術進一步證實氣道內OSM由肺炎克雷伯桿菌的脂多糖激活的巨噬細胞分泌[23]。Baines等[24]亦發現相比于健康志愿者,COPD患者的誘導痰中IL-8 和OSM表達明顯增高,提示OSM與重癥哮喘和慢性阻塞性肺疾病的慢性氣道炎癥和氣道重塑有關。Lin等[25]經生物信息學算法與單細胞或轉錄組數據進行交叉分析結果表明高水平的OSM,IL-18R1與重癥哮喘患者的肺功能和生存時間成負相關,因此OSM可作為重癥哮喘的監測和預后的標志物。在嚴重哮喘中,氣道上皮粘液產生細胞數量增加、肺間質纖維化及巨噬細胞高表達卵泡抑素樣1 促進OSM過度表達,誘導氣道上皮細胞中抵抗素樣分子α的增加,導致小鼠肺細胞外基質重塑[26],而抗OSM單抗可逆轉卵泡抑素樣1對氣道的重塑,提示OSM途徑可能是作用重癥哮喘和慢性阻塞性肺疾病氣道重塑新的靶點。
2 OSM與肺癌
肺癌是發病率和死亡率最高的惡性腫瘤,分子靶向和免疫的治療改善患者的生存時間,但是,多數患者在治療后仍死于腫瘤的耐藥、復發 [27],因此迫切需要尋找高效低毒新的治療靶點。OSM最初被發現在體外對黑色素瘤和其他癌癥細胞系具有直接的增殖抑制作用[4],Ouyang[28]等發現低濃度OSM在體外可抑制人肺癌細胞95-D細胞的增殖,減少基質金屬蛋白酶2(matrix metalloproteinase-2,MMP-2)和MMP-9的分泌降低細胞的粘附和侵襲能力。Spence等[29]發現OSM通過JAK3/STAT3通路促進組織型纖溶酶原激活劑(type plasminogen activator,tPA)和纖溶酶原激活物抑制物-1(plasminogen activator inhibitor-1,PAI-1)mRNA的表達促進腫瘤轉移。McCormick和Cichy團隊發現OSM較IL-6可更有效的促進肺癌細胞分化,OSM與TGF β 1因子共同調節透明質酸,促進肺癌轉移[30,31]。與之前的研究不同,Lauber等[32]發現OSM在體外對LLC細胞無增殖抑制作用,但在小鼠體內可促進腫瘤細胞顯著增長,通過轉染腺病毒致肺部OSM表達增高,可募集M2巨噬細胞改變腫瘤微環境,促進異位種植的腫瘤肺轉移增加,然而OSMRβ 敲除鼠則減少腫瘤肺轉移,提示OSM/OSMRβ通路調控腫瘤肺轉移。Shien等[33]亦發現OSM可在肺癌小鼠模型中經上皮間質轉化(epithelial–mesenchymal transition,EMT)促進肺轉移,肺癌細胞與CAF體外共孵育后,磷酸化STAT 3、OSMRβ和LIFRβ上調,同時E-鈣粘蛋白下調,CAF經OSMRβ/JAK 1/STAT 3通路促進腫瘤進展和耐藥,靶向選擇性JAK1抑制劑非戈替尼有效抑制STAT3激活,降低OSMR表達,抑制腫瘤增殖逆轉腫瘤耐藥。臨床前研究發現OSM及其通路在肺癌的發生和轉移中起到關鍵作用,那么OSM在真實世界肺癌患者中扮演什么樣的角色?Shien等[33]及其團隊分析TGCA和PROSPECT公共數據庫,高表達OSM,IL-6,LIF和OSMRβ的肺腺癌患者預后更差。另有研究表明放療和缺氧應激的肺癌細胞分泌大量微泡,誘導CAF分泌包括OSM,IL-8,IL-11,血管內皮生長因子等[34],OSM可能參與腫瘤的自我適應調控。部分研究發現OSM通過STAT1信號通路下調EMT起動子SLUG轉錄,抑制EMT及肺癌轉移[35]。綜上,臨床研究表明OSM可促進腫瘤增殖和轉移,而臨床前研究的結果并不完全一致,OSM通路網絡在肺癌中的具體機制需要進一步研究闡明。
3 OSM與惡病質
惡病質是一種以消耗為特征復雜的多因素綜合征,在肺癌和慢性肺部疾病患者中普遍存在,其主要特征表現為體重減輕、骨骼肌質量減少(伴或不伴有脂肪組織損失)以及全身炎癥[36-38]。全身性炎癥因子包括腫瘤壞死因子-α(TNF-α)、白細胞介素-1(IL-1)和IL-6 等水平升高可導致肌肉萎縮,針對其相應的中和抗體可抑制其萎縮效應[39,40]。在胰腺導管腺癌(高達80%患者發生惡病質)和肌營養不良患者的肌肉中OSMR和OSM靶基因的轉錄水平顯著增加,提示其在惡病質相關的肌肉萎縮中發揮重要作用[41]。Miki等[42]發現OSM處理C2C12肌管細胞24 h和48 h后,肌細胞生成素表達下降,atrogin-1和OMSR表達上升,肌管直徑比對照組分別減少18.7%和23.3%,STAT3抑制劑或敲除STAT3基因可逆轉其效應。與之類似,Domaniku等[41]通過敲除荷瘤小鼠特異性肌肉細胞OSMR,可抑制肌肉細胞萎縮恢復肌肉功能,近期其團隊進一步證實OSM可在體外通過JAK/STAT3通路上調原代肌管細胞表達Atrogin1致肌肉萎縮,明確了OSM信號通路在肌肉萎縮中的作用[43]。OSM誘導的肌肉萎縮依賴于JAK/STAT 3信號傳導,與Atrogin 1、Ampd 3、Serpina 3n、Cebpd、Sln和Mt 1/2多種萎縮相關基因的表達增加有關[42]。Bilgic等[43]近期工作發表在Nature雜志首次發現肺癌患者和Lewis小鼠惡病質模型的肌肉中組織外胚層發育不良A2受體(ectodysplasin A2 receptor,EDA2R)表達異常升高,與EDA2結合激活非經典NF-?B信號通路促進Atrogin1和MuRF1基因轉錄和蛋白表達,從而誘導肌肉萎縮,有趣的是,OSM不僅與OSMR結合發揮作用,可同時促進EDA-2R受體轉錄表達加劇肌肉細胞萎縮,敲除肌肉OSMR受體可逆轉EDA2R受體表達,抑制惡病質相關肌肉萎縮基因轉錄和緩解肌肉功能受損。相比與IL-6和LIF,OSM對萎縮基因的轉錄的調控作用更強,因此對肌管的萎縮效應更明顯。由于這三種細胞因子都與肌肉萎縮有關,因此理論上針對這些途徑的共同通路可能更有效。因此,近期一項I期針對IV期非小細胞肺癌合并惡病質的患者中使用JAK 1/2抑制劑魯索利替尼的安全性和有效性的臨床試驗(NCT 04906746)正在募集患者。一項II期隨機對照試驗,針對人源化單克隆抗OSM抗體(GSK
OSM在多種肺部疾病中發揮重要作用,部分抗OSM通路藥物臨床前研究取得優異的療效,然而目前尚無藥物獲批用于臨床疾病的治療。未來針對抗OSM或抗OSMRβ的藥物治療將成為肺部疾病研究中的熱門領域,我們期待著后續針對OSM信號通路更加深入的研究以及更多有效安全藥物的出現并應用于臨床。
利益沖突:本研究不涉及任何利益沖突。