引用本文: 韓濤, 蘇磊, 臧軻君, 楊雪, 王勝軍, 趙秀鶴, 曹麗麗, 遲兆富, 劉學伍. 肌陣攣性小腦協調不能的臨床及神經電生理特點. 癲癇雜志, 2016, 2(5): 401-405. doi: 10.7507/2096-0247.20160071 復制
版權信息: ?四川大學華西醫院華西期刊社《癲癇雜志》版權所有,未經授權不得轉載、改編
肌陣攣性小腦協調不能(Dyssynergia cerebellaris myoclonica,DCM)在1921年由Hunt首先報道,也稱Ramsay-Hunt綜合征(RHS)[1],是一組以肌陣攣、小腦性共濟失調及癲癇為特征的臨床綜合征。DCM具有家族遺傳性,為常染色體顯性遺傳或常染色體隱性遺傳,也可為散發,以散發居多,男女發病率相同。現本文報道兩個肌陣攣性小腦協調不能家系及一例散發患者,以探索其臨床及神經電生理特點。
資料與方法
1 家系一
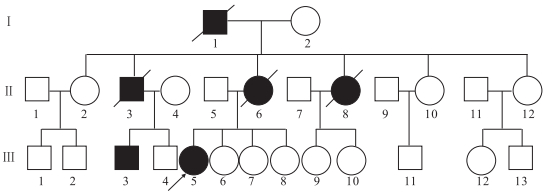
患者(先證者Ⅲ5)女,31歲。因發作性意識喪失4年,全身抖動伴行走不穩、言語蹙澀3年收入院。患者于2006年首次出現癲癇發作,家屬描述為全面性強直-陣攣發作,至入院前共有3次類似發作。入院前3年內患者漸出現發作性四肢不自主抖動,站立不穩,飲水嗆咳伴言語蹙澀。追問病史,患者家庭成員中共有6位患者有類似癥狀(家系譜見圖 1),其中6例患者中有5例有明顯的共濟失調及肌陣攣表現,1例共濟失調及肌陣攣較輕;僅有2例存在癲癇發作,且發作次數極少。先證者及同代患者神經系統查體均存在全身不自主短暫抖動,雙側指鼻試驗及跟膝脛試驗不能完成,雙側快復輪替動作差,直線行走不能,共濟失調步態,余未見明顯異常。腦電圖(EEG)檢查:全腦大量廣泛分布的棘慢波、多棘慢波及多棘波,雙側頂、中央區稍多尖慢波;顱腦磁共振(MRI)檢查:腦萎縮,以小腦及大腦皮層著。組織病理學檢查:肌肉、皮膚組織活檢未見異常。
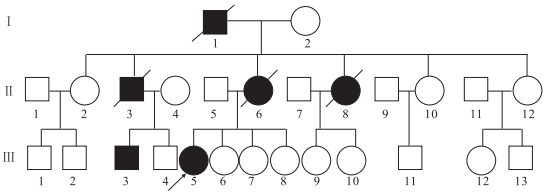 圖1
家系(一)肌陣攣性小腦協調不能圖譜
圖1
家系(一)肌陣攣性小腦協調不能圖譜
□健康男性Healthy male,○健康女性Healthy female,■男性患者Male patient,●女性患者Female patient,死亡者the dead,先證者Proband
Figure1. Pedigree chart of patients with dyssynergia cerebellaris myoclonica2 家系二
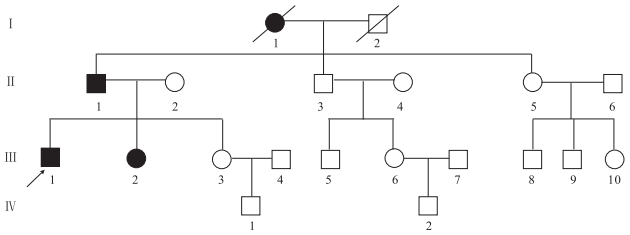
患者(先癥者Ⅲ1)男,21歲。因發作性肢體抽搐10年,身體不自主抖動、走路不穩4年入院。患者入院前10年無明顯誘因首次出現癲癇發作,家屬描述為全面性強直-陣攣發作,發作頻率約2~3次/年,未規律應用抗癲癇藥物(AEDs)。入院前4年內患者漸出現發作性四肢及腹部不自主抖動,行走不穩,飲水嗆咳伴言語蹙澀。追問病史,患者家庭成員中共有4位患者有類似癥狀(家系譜見圖 2),4例患者均存在明顯的共濟失調及肌陣孿發作。4例患者曾先后出現過癲癇發作,但發作次數較少。發作頻率在1~4次/年不等。在世的3例患者神經系統檢查均存在全身不自主短暫抖動,指鼻試驗及跟膝脛試驗不能完成,直線行走不能,共濟失調步態,余未見明顯異常。常規EEG檢查發作間期:全腦大量廣泛分布的棘慢波、多棘慢波及多棘波。1例行長時程視頻腦電(VEEG)檢查示發作間期及發作期全腦大量廣泛分布的棘慢波、多棘慢波及多棘波,有時以頂、中央區著;顱腦MRI檢查未見明顯異常。組織病理學檢查:肌肉、皮膚組織活檢未見異常。
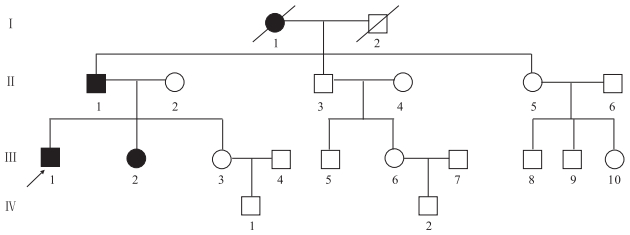 圖2
家系(二)常染色體顯性遺傳性肌陣攣性小腦協調不能圖譜
圖2
家系(二)常染色體顯性遺傳性肌陣攣性小腦協調不能圖譜
□健康男性Healthy male,○健康女性Healthy female,■男性患者Male patient,●女性患者Female patient,死亡者the dead,先證者Proband
Figure2. Pedigree chart of patients with autosomal dominant dyssynergia cerebellaris myoclonica3 散發患者
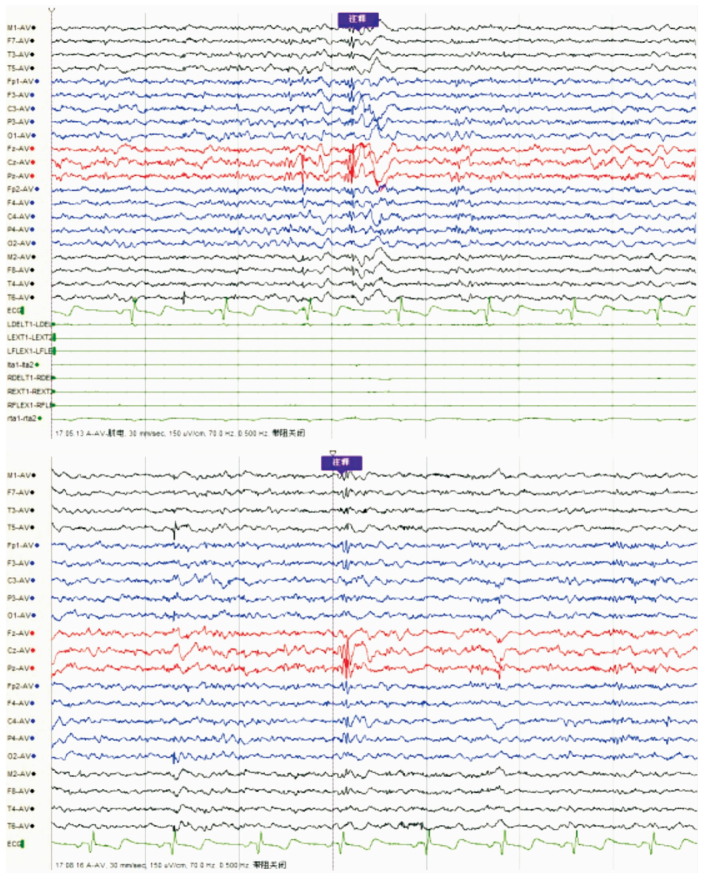
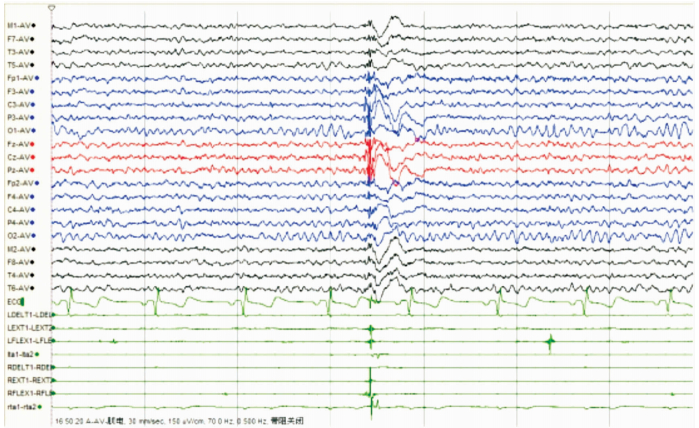
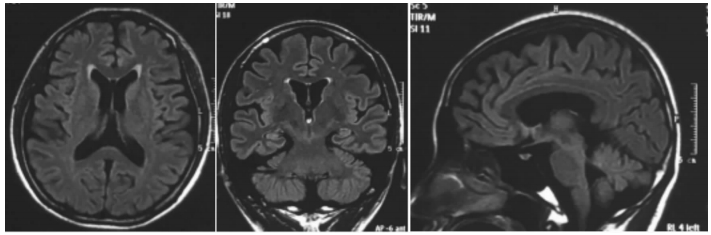
女,12歲。因行走不穩、言語欠清1年入院。患者入院前1年無明顯誘因出現行走不穩,以下肢為主,嚴重時突然跌倒,無意識喪失。癥狀進行性加重,漸言語障礙加重,偶伴飲水嗆咳。神經系統檢查:神志清,精神可。言語欠流利,眼球運動自如,無眼震,四肢肌力正常為5級,四肢肌張力正常。腱反射(++),病理征(-)。雙側指鼻試驗欠穩準,雙側跟膝脛試驗欠穩準,雙側快復輪替動作尚可完成,Romberg征(-)。深淺感覺正常。直線行走不能,共濟失調步態。VEEG檢查:發作間期及發作期EEG主要表現為全面性棘慢及多棘慢波,以頂、中央區及額區著。發作間期同時存在大量同步及異步出現的頂、中央區及額區棘慢波及多棘慢波(圖 3、4)。顱腦MRI檢查:大腦及小腦萎縮(圖 5)。
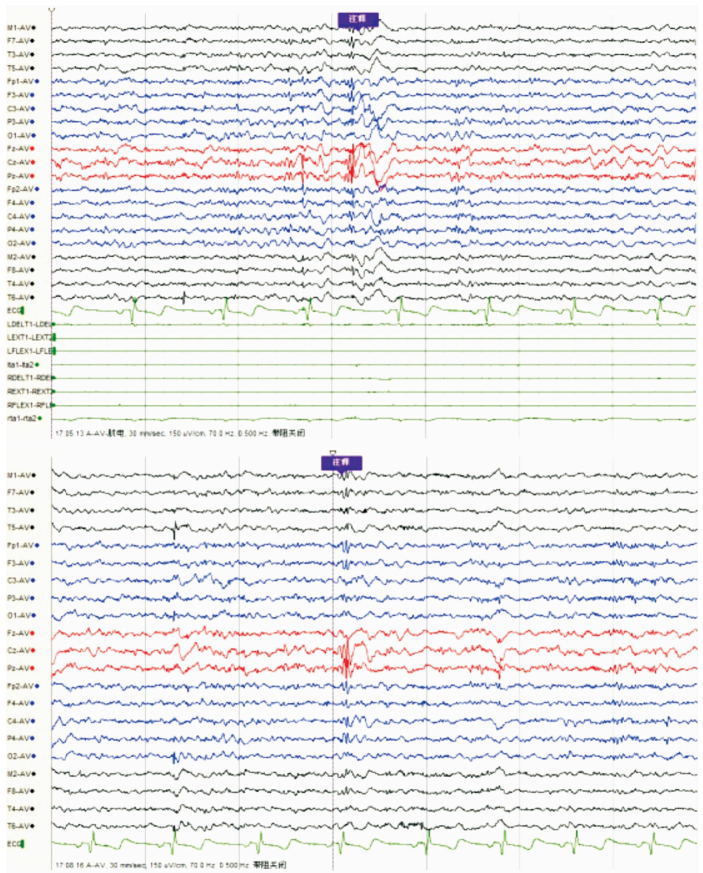 圖3
散發者發作間期EEG雙側頂、中央及額區著的全腦廣泛分布的棘慢波、多棘慢波及多棘波發放
Figure3.
EEG during interictal period spike slow wave, multiple spike slow wave, and multiple spike wave distributed on the whole brain, espacially in double parietal, central and frontal area
圖3
散發者發作間期EEG雙側頂、中央及額區著的全腦廣泛分布的棘慢波、多棘慢波及多棘波發放
Figure3.
EEG during interictal period spike slow wave, multiple spike slow wave, and multiple spike wave distributed on the whole brain, espacially in double parietal, central and frontal area
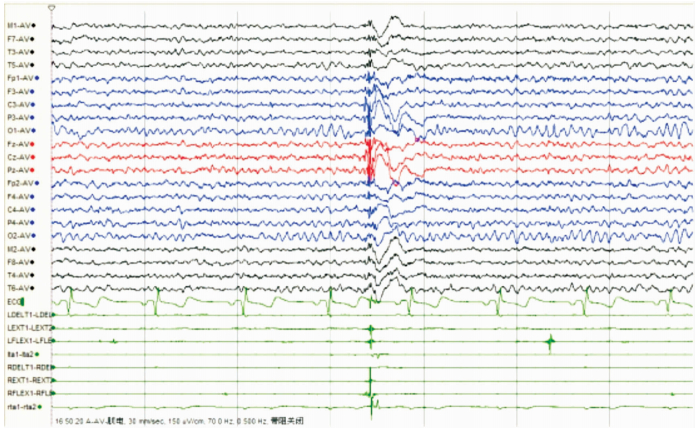 圖4
散發者發作期EEG雙側頂、中央及額區著的全腦廣泛分布的棘慢波、多棘慢波發放,同步肌電監測符合肌陣孿發作表現
Figure4.
EEG during ictal period spike slow wave, multiple spike slow wave distributed on the whole brain, espacially in double parietal, central and frontal area. and synchronous electrophysiological monitoring conformed myoclonus epilepsy.
圖4
散發者發作期EEG雙側頂、中央及額區著的全腦廣泛分布的棘慢波、多棘慢波發放,同步肌電監測符合肌陣孿發作表現
Figure4.
EEG during ictal period spike slow wave, multiple spike slow wave distributed on the whole brain, espacially in double parietal, central and frontal area. and synchronous electrophysiological monitoring conformed myoclonus epilepsy.
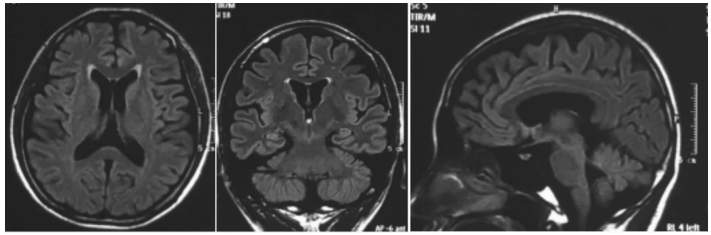 圖5
散發者??顱腦MRI T2 flair序列軸位、冠狀位及矢狀位顯示患者大腦及小腦皮層萎縮及明顯腦溝增寬
Figure5.
Brain MRI The axial, coronal and sagittal view in T2 flair sequence of brain MRI showed that cerebral and cerebellar cortex are atrophic and the brain sulci is widened obviously.
圖5
散發者??顱腦MRI T2 flair序列軸位、冠狀位及矢狀位顯示患者大腦及小腦皮層萎縮及明顯腦溝增寬
Figure5.
Brain MRI The axial, coronal and sagittal view in T2 flair sequence of brain MRI showed that cerebral and cerebellar cortex are atrophic and the brain sulci is widened obviously.
討論
1921年Ramsay-Hunt首先報道6例肌陣攣-小腦協調障礙-原發性齒狀核系統萎縮的患者。近百年來隨著對該病的不斷研究,學者們對其診斷漸有了統一認識,即肌陣攣、癲癇、進行性小腦共濟失調。該病臨床癥狀與進行性肌陣攣性癲癇(Progressive myoclonic epilepsy, PME)類似。有學者認為可將兩組疾病統稱為肌陣孿癲癇。Marseille等[2, 3]將該綜合征分成兩大類,即PME和進行性肌陣攣性共濟失調(Progressive myoclonic ataxia, PMA)。
此病肌陣攣發作多為動作性的,表現為突發短暫的、肢體為主的閃電樣肌肉收縮。癲癇可表現為肌陣攣癲癇,也可表現為全身強直陣攣性發作,但發作相對較少是該病的突出特征[4]。小腦性共濟失調是本病最突出的臨床癥狀,也是診斷的主要依據,表現為進行性的走路不穩、構音不清、意向性震顫等。除了上述癥狀外,本病與PME相比,多數無智能減退或僅有輕度的認知障礙[5]。腦MRI可以正常,也可以有大腦及小腦萎縮表現。
我們所報道的兩個家族性起病的患者皆以全身強直-陣攣性癲癇發作為首發癥狀,后漸出現小腦共濟失調表現,包括構音障礙、直線行走不穩、共濟失調步態等;此外,患者多數合并出現肌陣攣發作。散發患者以直線行走不能及構音障礙等小腦共濟失調癥狀就診,通過入院后VEEG檢查確診合并肌陣攣及肌陣攣癲癇;患者肌陣攣與小腦性共失調并存,強直-陣攣性癲癇發作次數極少,符合肌陣攣性小腦協調不能診斷。
關于肌陣攣性小腦協調不能患者神經電生理特點國內外研究較少。相關報道表明該病患者的EEG多表現為廣泛性棘波、多棘波、棘慢波及多棘慢波。我們所收治的患者發作間期及發作期EEG主要表現為全面性棘慢及多棘慢波發放,有時以頂、中央區及額區著,同時發作間期存在大量同步及異步出現的頂、中央區及額區棘慢波及多棘慢波;這在之前的文獻中鮮有報道。患者均無部分性癲癇發作,也沒有頭痛或癲癇發作前的感覺先兆;患者顱腦MRI檢查未發現頂額葉與其他皮層相比有明顯差異。我們推測出現局灶性癲癇樣放電的原因可能與不同部位的神經元損傷狀況不同有關,而這種差異通過目前的影像學檢查手段可能無法分辨。患者的發作期與發作間期的EEG表現并無明顯差異,本文中一例患者及家屬始終未發現患者的肌陣攣發作,通過VEEG及同步肌電監測確診。另有第二個家系中的3位患者常規EEG檢查僅發現發作間期全腦大量廣泛分布的棘慢波、多棘慢波及多棘波。后家系中一例患者行VEEG檢查示發作間期及發作期全腦大量廣泛分布的棘慢波、多棘慢波及多棘波,有時以頂、中央區著。這提示我們臨床中以VEEG聯合肌電監測對于該病的診斷有重要意義。我們將患者的VEEG監測結果與確診的肌陣攣癲癇伴蓬毛樣破碎紅纖維(Myoclonic epilepsy and ragged-red fiber disease)患者比較,癲癇樣放電的頻率及肌陣攣發作的次數均明顯偏少,這與肌陣攣性小腦協調不能患者癲癇發作少是一致的;同時也能部分解釋肌陣攣性小腦協調不能患者與PME患者相比,認知功能的損傷較輕。
針電極肌電圖及神經傳導監測的結果與肌陣攣性小腦協調不能患者的肌肉病變常常是一致的,但并不排除周圍神經病的存在。van Egmond等[6]對確診GOSR2基因突變的肌陣攣性小腦協調不能患者的相關研究表明,對于合并有腱反射消失的患者,其肌電圖檢查均存在明顯周圍神經損害,這種損害可同時累及背根神經節及前腳運動神經元,其中感覺神經的損害可能出現的更早。本組患者肌電圖檢查并未發現明顯異常,這可能與我們的患者發病時間較短或體質特殊性有關;同時患者并未出現明顯肌無力表現,查體也未發現腱反射的明顯改變,這可能也能解釋肌電檢查無陽性發現。
與PME相比,肌陣攣性小腦協調不能的分子遺傳學研究較少,僅有少量的研究表明SGCE、GOSR2、SCARB2等基因與肌陣攣性小腦協調不能存在相關性[6-8]。因此,臨床及電生理學特點成為該病診斷的重要手段。隨著分子生物學和遺傳學的快速發展,尋找更多的新的致病基因,可能對進一步明確本病的病因和類型有更大的意義。
肌陣攣性小腦協調不能(Dyssynergia cerebellaris myoclonica,DCM)在1921年由Hunt首先報道,也稱Ramsay-Hunt綜合征(RHS)[1],是一組以肌陣攣、小腦性共濟失調及癲癇為特征的臨床綜合征。DCM具有家族遺傳性,為常染色體顯性遺傳或常染色體隱性遺傳,也可為散發,以散發居多,男女發病率相同。現本文報道兩個肌陣攣性小腦協調不能家系及一例散發患者,以探索其臨床及神經電生理特點。
資料與方法
1 家系一
患者(先證者Ⅲ5)女,31歲。因發作性意識喪失4年,全身抖動伴行走不穩、言語蹙澀3年收入院。患者于2006年首次出現癲癇發作,家屬描述為全面性強直-陣攣發作,至入院前共有3次類似發作。入院前3年內患者漸出現發作性四肢不自主抖動,站立不穩,飲水嗆咳伴言語蹙澀。追問病史,患者家庭成員中共有6位患者有類似癥狀(家系譜見圖 1),其中6例患者中有5例有明顯的共濟失調及肌陣攣表現,1例共濟失調及肌陣攣較輕;僅有2例存在癲癇發作,且發作次數極少。先證者及同代患者神經系統查體均存在全身不自主短暫抖動,雙側指鼻試驗及跟膝脛試驗不能完成,雙側快復輪替動作差,直線行走不能,共濟失調步態,余未見明顯異常。腦電圖(EEG)檢查:全腦大量廣泛分布的棘慢波、多棘慢波及多棘波,雙側頂、中央區稍多尖慢波;顱腦磁共振(MRI)檢查:腦萎縮,以小腦及大腦皮層著。組織病理學檢查:肌肉、皮膚組織活檢未見異常。
圖1
家系(一)肌陣攣性小腦協調不能圖譜
□健康男性Healthy male,○健康女性Healthy female,■男性患者Male patient,●女性患者Female patient,死亡者the dead,先證者Proband
Figure1. Pedigree chart of patients with dyssynergia cerebellaris myoclonica2 家系二
患者(先癥者Ⅲ1)男,21歲。因發作性肢體抽搐10年,身體不自主抖動、走路不穩4年入院。患者入院前10年無明顯誘因首次出現癲癇發作,家屬描述為全面性強直-陣攣發作,發作頻率約2~3次/年,未規律應用抗癲癇藥物(AEDs)。入院前4年內患者漸出現發作性四肢及腹部不自主抖動,行走不穩,飲水嗆咳伴言語蹙澀。追問病史,患者家庭成員中共有4位患者有類似癥狀(家系譜見圖 2),4例患者均存在明顯的共濟失調及肌陣孿發作。4例患者曾先后出現過癲癇發作,但發作次數較少。發作頻率在1~4次/年不等。在世的3例患者神經系統檢查均存在全身不自主短暫抖動,指鼻試驗及跟膝脛試驗不能完成,直線行走不能,共濟失調步態,余未見明顯異常。常規EEG檢查發作間期:全腦大量廣泛分布的棘慢波、多棘慢波及多棘波。1例行長時程視頻腦電(VEEG)檢查示發作間期及發作期全腦大量廣泛分布的棘慢波、多棘慢波及多棘波,有時以頂、中央區著;顱腦MRI檢查未見明顯異常。組織病理學檢查:肌肉、皮膚組織活檢未見異常。
圖2
家系(二)常染色體顯性遺傳性肌陣攣性小腦協調不能圖譜
□健康男性Healthy male,○健康女性Healthy female,■男性患者Male patient,●女性患者Female patient,死亡者the dead,先證者Proband
Figure2. Pedigree chart of patients with autosomal dominant dyssynergia cerebellaris myoclonica3 散發患者
女,12歲。因行走不穩、言語欠清1年入院。患者入院前1年無明顯誘因出現行走不穩,以下肢為主,嚴重時突然跌倒,無意識喪失。癥狀進行性加重,漸言語障礙加重,偶伴飲水嗆咳。神經系統檢查:神志清,精神可。言語欠流利,眼球運動自如,無眼震,四肢肌力正常為5級,四肢肌張力正常。腱反射(++),病理征(-)。雙側指鼻試驗欠穩準,雙側跟膝脛試驗欠穩準,雙側快復輪替動作尚可完成,Romberg征(-)。深淺感覺正常。直線行走不能,共濟失調步態。VEEG檢查:發作間期及發作期EEG主要表現為全面性棘慢及多棘慢波,以頂、中央區及額區著。發作間期同時存在大量同步及異步出現的頂、中央區及額區棘慢波及多棘慢波(圖 3、4)。顱腦MRI檢查:大腦及小腦萎縮(圖 5)。
圖3
散發者發作間期EEG雙側頂、中央及額區著的全腦廣泛分布的棘慢波、多棘慢波及多棘波發放
Figure3.
EEG during interictal period spike slow wave, multiple spike slow wave, and multiple spike wave distributed on the whole brain, espacially in double parietal, central and frontal area
圖4
散發者發作期EEG雙側頂、中央及額區著的全腦廣泛分布的棘慢波、多棘慢波發放,同步肌電監測符合肌陣孿發作表現
Figure4.
EEG during ictal period spike slow wave, multiple spike slow wave distributed on the whole brain, espacially in double parietal, central and frontal area. and synchronous electrophysiological monitoring conformed myoclonus epilepsy.
圖5
散發者??顱腦MRI T2 flair序列軸位、冠狀位及矢狀位顯示患者大腦及小腦皮層萎縮及明顯腦溝增寬
Figure5.
Brain MRI The axial, coronal and sagittal view in T2 flair sequence of brain MRI showed that cerebral and cerebellar cortex are atrophic and the brain sulci is widened obviously.
討論
1921年Ramsay-Hunt首先報道6例肌陣攣-小腦協調障礙-原發性齒狀核系統萎縮的患者。近百年來隨著對該病的不斷研究,學者們對其診斷漸有了統一認識,即肌陣攣、癲癇、進行性小腦共濟失調。該病臨床癥狀與進行性肌陣攣性癲癇(Progressive myoclonic epilepsy, PME)類似。有學者認為可將兩組疾病統稱為肌陣孿癲癇。Marseille等[2, 3]將該綜合征分成兩大類,即PME和進行性肌陣攣性共濟失調(Progressive myoclonic ataxia, PMA)。
此病肌陣攣發作多為動作性的,表現為突發短暫的、肢體為主的閃電樣肌肉收縮。癲癇可表現為肌陣攣癲癇,也可表現為全身強直陣攣性發作,但發作相對較少是該病的突出特征[4]。小腦性共濟失調是本病最突出的臨床癥狀,也是診斷的主要依據,表現為進行性的走路不穩、構音不清、意向性震顫等。除了上述癥狀外,本病與PME相比,多數無智能減退或僅有輕度的認知障礙[5]。腦MRI可以正常,也可以有大腦及小腦萎縮表現。
我們所報道的兩個家族性起病的患者皆以全身強直-陣攣性癲癇發作為首發癥狀,后漸出現小腦共濟失調表現,包括構音障礙、直線行走不穩、共濟失調步態等;此外,患者多數合并出現肌陣攣發作。散發患者以直線行走不能及構音障礙等小腦共濟失調癥狀就診,通過入院后VEEG檢查確診合并肌陣攣及肌陣攣癲癇;患者肌陣攣與小腦性共失調并存,強直-陣攣性癲癇發作次數極少,符合肌陣攣性小腦協調不能診斷。
關于肌陣攣性小腦協調不能患者神經電生理特點國內外研究較少。相關報道表明該病患者的EEG多表現為廣泛性棘波、多棘波、棘慢波及多棘慢波。我們所收治的患者發作間期及發作期EEG主要表現為全面性棘慢及多棘慢波發放,有時以頂、中央區及額區著,同時發作間期存在大量同步及異步出現的頂、中央區及額區棘慢波及多棘慢波;這在之前的文獻中鮮有報道。患者均無部分性癲癇發作,也沒有頭痛或癲癇發作前的感覺先兆;患者顱腦MRI檢查未發現頂額葉與其他皮層相比有明顯差異。我們推測出現局灶性癲癇樣放電的原因可能與不同部位的神經元損傷狀況不同有關,而這種差異通過目前的影像學檢查手段可能無法分辨。患者的發作期與發作間期的EEG表現并無明顯差異,本文中一例患者及家屬始終未發現患者的肌陣攣發作,通過VEEG及同步肌電監測確診。另有第二個家系中的3位患者常規EEG檢查僅發現發作間期全腦大量廣泛分布的棘慢波、多棘慢波及多棘波。后家系中一例患者行VEEG檢查示發作間期及發作期全腦大量廣泛分布的棘慢波、多棘慢波及多棘波,有時以頂、中央區著。這提示我們臨床中以VEEG聯合肌電監測對于該病的診斷有重要意義。我們將患者的VEEG監測結果與確診的肌陣攣癲癇伴蓬毛樣破碎紅纖維(Myoclonic epilepsy and ragged-red fiber disease)患者比較,癲癇樣放電的頻率及肌陣攣發作的次數均明顯偏少,這與肌陣攣性小腦協調不能患者癲癇發作少是一致的;同時也能部分解釋肌陣攣性小腦協調不能患者與PME患者相比,認知功能的損傷較輕。
針電極肌電圖及神經傳導監測的結果與肌陣攣性小腦協調不能患者的肌肉病變常常是一致的,但并不排除周圍神經病的存在。van Egmond等[6]對確診GOSR2基因突變的肌陣攣性小腦協調不能患者的相關研究表明,對于合并有腱反射消失的患者,其肌電圖檢查均存在明顯周圍神經損害,這種損害可同時累及背根神經節及前腳運動神經元,其中感覺神經的損害可能出現的更早。本組患者肌電圖檢查并未發現明顯異常,這可能與我們的患者發病時間較短或體質特殊性有關;同時患者并未出現明顯肌無力表現,查體也未發現腱反射的明顯改變,這可能也能解釋肌電檢查無陽性發現。
與PME相比,肌陣攣性小腦協調不能的分子遺傳學研究較少,僅有少量的研究表明SGCE、GOSR2、SCARB2等基因與肌陣攣性小腦協調不能存在相關性[6-8]。因此,臨床及電生理學特點成為該病診斷的重要手段。隨著分子生物學和遺傳學的快速發展,尋找更多的新的致病基因,可能對進一步明確本病的病因和類型有更大的意義。