


引用本文: 趙彤, 陳芳. SPTAN1基因突變所致伴中央顳區棘波的自限性癲癇患兒一例并文獻復習. 癲癇雜志, 2024, 10(3): 278-280. doi: 10.7507/2096-0247.202402004 復制
版權信息: ?四川大學華西醫院華西期刊社《癲癇雜志》版權所有,未經授權不得轉載、改編
SPTAN1基因位于染色體9q34.11,編碼非紅細胞αII血影蛋白(non-erythrocyteαII spectrin),該蛋白與β血影蛋白形成異源四聚體[1],對維持細胞膜骨架起重要作用,并對神經元突觸形成、遞質釋放起到調節作用[2],其致病性變異可致翻譯出的蛋白結構、功能發生變化,進一步影響大腦結構形成、發育進程,從而引發一系列神經系統疾病。伴中央顳區棘波的自限性癲癇(Self-limited epilepsy with centrotemporal spikes,SeLECTS)通常發病年齡特定、癲癇發作在青春期前自行緩解,大多數癥狀呈良性、抗癲癇治療效果良好,極少數患者在缺乏適當治療的情況下會出現認知障礙等不良后果。本研究回顧1例SPTAN1基因突變致SeLECTS患兒的臨床特征,結合相關文獻進行討論。
病例資料 患兒 女,8歲4月齡。因“1月內抽搐發作2次”于2023年6月就診于河北省兒童醫院。患兒8歲3月齡起病,表現為入睡后不久后出現意識喪失、面部肌肉抽搐、口齒不清、流涎、全身強直陣攣發作,持續約1分鐘后自行緩解,緩解后無肢體活動障礙等不適,共發作2次,癥狀相似,無發熱,無咳嗽、咳痰,無鼻塞、流涕,無頭痛、頭暈,無惡心、嘔吐,無腹痛、腹瀉,無煩躁、嗜睡、精神行為異常,無大小便失禁,未予以治療。患兒既往體健。患兒為母親第1胎第1產,孕足月順產,出生體重3 270 g,否認窒息搶救史。患兒父母均體健,否認近期婚配。患兒生長發育同正常同齡兒。否認發育障礙、癲癇及其他疾病家族史。
體格檢查:神清語利,全身皮膚未見皮疹、異常色素沉著。淺表淋巴結未觸及腫大,雙側瞳孔直徑3 mm正大等圓,對光反射靈敏。口腔黏膜光滑,咽無充血,雙側扁桃體無腫大。頸部無抵抗;雙肺呼吸音清,未聞及干濕性啰音;心律規整,心音有力,未聞及雜音;腹軟,肝脾肋下未觸及,腸鳴音存在;脊柱無側彎、壓痛,四肢活動可,四肢肌力、肌張力正常,雙側肱二、三頭肌腱反射存在,雙側膝腱反射存在,雙側巴氏征、克氏征、布氏征陰性。輔助檢查:血常規、肝功能、腎功能、血氨、乳酸、血及尿遺傳代謝串聯質譜篩查、頭顱核磁未見異常。韋氏智力量表測試示:正常。視頻腦電圖示:異常兒童腦電圖,醒睡各期左側顳區、右側Rolandic區棘波、棘慢波不同步發放(圖1)。
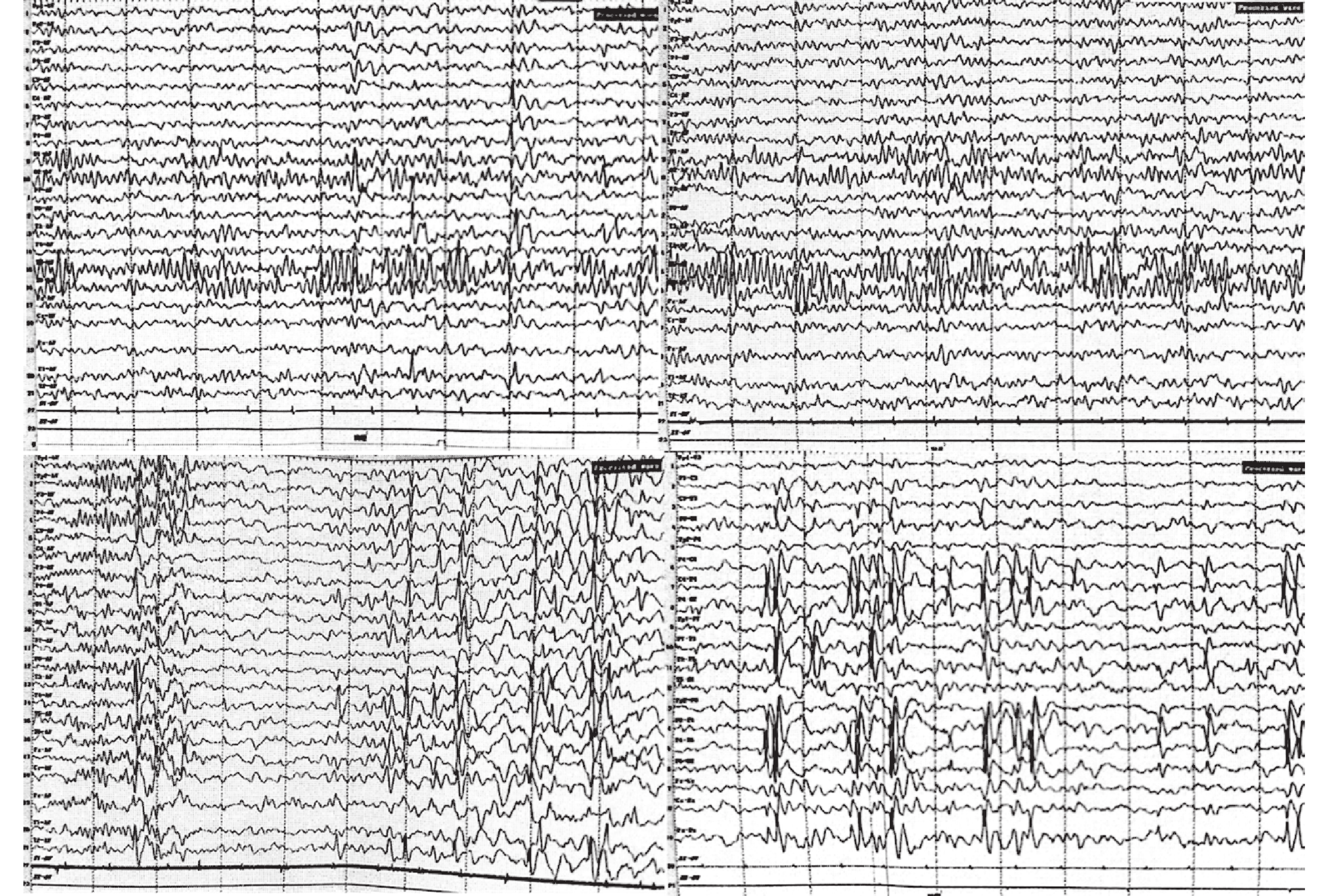 圖1
患兒視頻腦電圖
圖1
患兒視頻腦電圖
醒睡各期左側顳區、右側Rolandic區棘波、棘慢波不同步發放
患兒在學齡期出現癲癇發作,頭顱核磁、血尿遺傳代謝篩查未見異常,考慮存在遺傳病因。經患兒父母簽署知情同意書后,分別抽取患兒、父母外周血2mL提取DNA行家系全外基因組檢測,并對分析結果行Sanger驗證。全基因組檢測提示患兒SPTAN1基因存在1個雜合變異(NM_001130438.2): c.6926T>C(p.Met2309Thr)。此變異導致編碼蛋白的第2 309位氨基酸由甲硫氨酸突變成蘇氨酸。經Sanger驗證,父母均未見變異,為新發變異(PS2)(圖2)。該變異在人類基因頻率數據庫(Genome Aggregation Database,GnomAD)基因頻率未被報道(PM2)。根據美國醫學遺傳學與基因組學學會(American College of Medical Genetics and Genomics,ACMG)指南判定,根據美國醫學遺傳學與基因組學學會指南,該變異為可能致病變異。
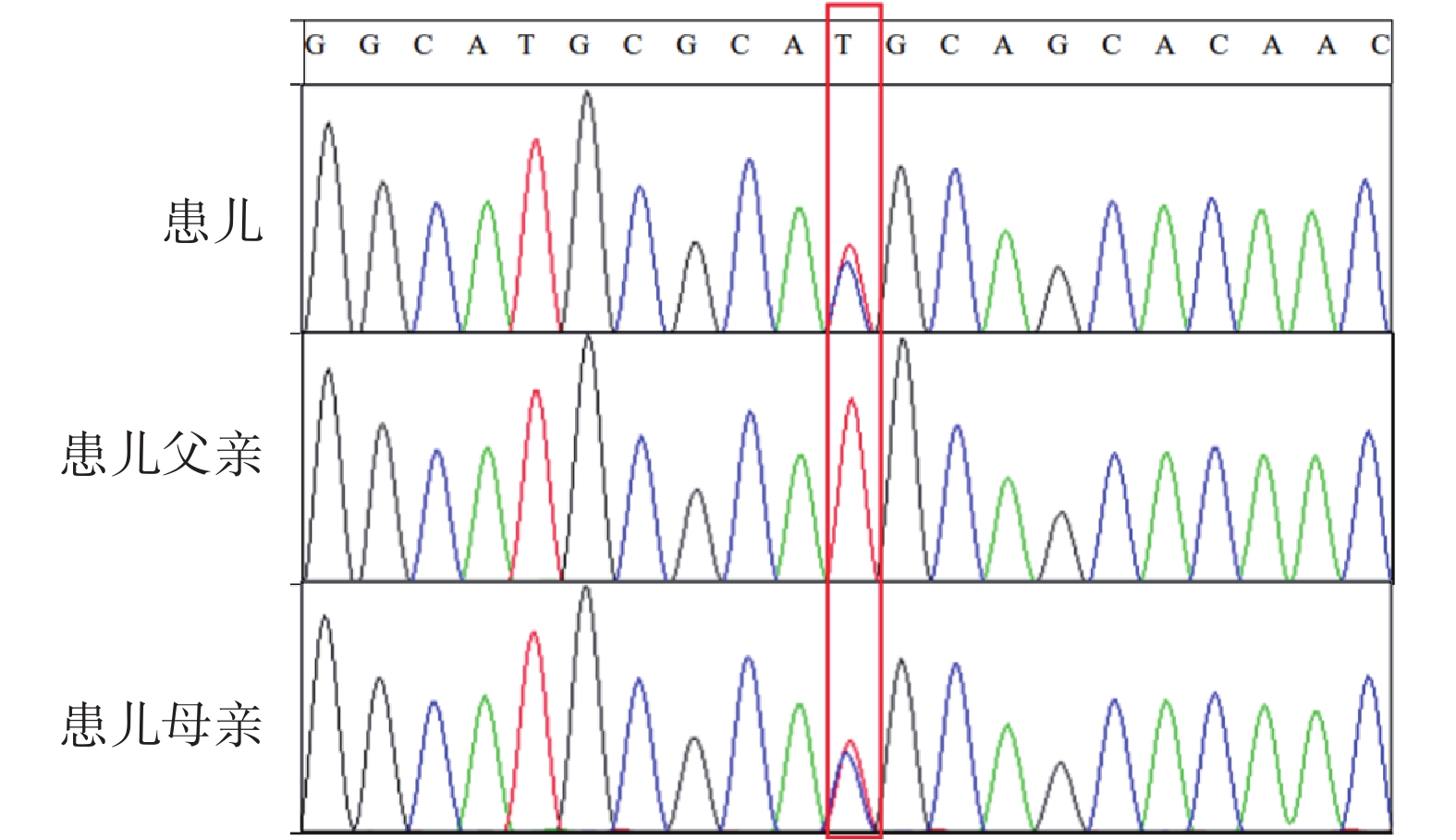 圖2
SPTAN1基因檢測
圖2
SPTAN1基因檢測
c.6926T>C位點雜合錯義變異,未在受檢者父母外周血中檢出,紅框提示變異位點
根據患兒臨床癥狀、視頻腦電圖及基因檢查結果,診斷SeLECTS,給予口服左乙拉西坦片0.25 g(10 mg/kg),每日2次。1個月后復查視頻腦電圖示:正常兒童腦電圖。隨訪7個月,患兒無抽搐發作。
討論 伴中央顳區棘波的自限性癲癇又被稱為Rolandic癲癇,是一種兒童常見的良性、局灶性癲癇綜合征,為常染色體顯性遺傳病[3]。Jabbari等[4]在一項大型SeLECTS患者中進行遺傳基因突變篩查研究,發現了SPTAN1基因,但目前無SPTAN1基因變異致BECTs的病例報道,大多數報道的相關表型包括伴進行性腦萎縮的早發性癲癇性腦病、伴小腦畸形的嚴重智力低下,以及伴或不伴癲癇的相對較輕表型[5]。
SPTAN1基因位于編碼一種廣泛分布于細胞膜上的絲狀骨架蛋白,由αII和β血影蛋白構成,具有高度保守的氨基酸結構,對神經元樹突、軸突的發育、神經元突觸的形成中起到關鍵作用 [5]。SeLECTS是兒童時期最常見的癲癇綜合征之一,約占兒童癲癇的 15%~24%,發病年齡以5~10歲最常見,其特征是癲癇發作隨著年齡的增長而逐漸消失[6],腦電圖示大腦中央和顳區存在局部放電[7],存在一定程度的認知障礙、行為和社交障礙等合并癥[8,9]。其病因尚不清楚,Samaitien?等[10]提出其病因與睡眠相關,還與遺傳有關。
目前已報道的SPTAN1基因變異相關疾病表型大致分為4種,分別為發育性癲癇性腦病(Developmental epileptic encephalopathy,DEE)、遠端遺傳性運動神經病、痙攣性截癱、發育遲緩伴或不伴癲癇[11]。以上表型中DEE表型例數最多,目前已有24例被報道[12-17],這類表型多數為新生重復突變,87.5%的病例診斷為West綜合征,合并有小頭畸形、肌張力下降、藥物難治性癲癇,頭顱核磁有特征性變化,大多數表現為髓鞘化減退及大腦或小腦萎縮。已報道28例SPTAN1基因變異致痙攣性截癱相關表型病例均有共濟失調[16,18],伴或不伴癲癇發作,均不存在無智力障礙、發育遲緩,頭顱核磁、腦電圖結果大多數無異常。SPTAN1基因變異致發育遲緩伴或不伴癲癇表型共有21例被報道[15,16],這類表型給予抗癲癇發作藥物治療可以得到良好的效果,腦電圖以局灶性棘波、多灶性棘波或全面性棘波為主,可伴有輕度智力障礙、學習障礙等合并癥,頭顱核磁基本正常。本例患兒為SPTAN1基因變異所致SeLECTS表型,目前國內外尚無報道,與既往已報道的表型相比,臨床癥狀溫和,無智力、認知落后等合并癥,口服藥物控制癲癇發作效果良好。
已發現SPTAN1基因重復突變與嚴重DEE表型具有相關性,而錯義、移碼突變與共濟失調、痙攣性截癱及溫和癲癇表型存在相關性[18]。另外,已有研究提出,突變位點位于αII及β血影蛋白的異源二聚體區域時,會影響最終翻譯的蛋白質空間分布而致聚集現象,并發現與最嚴重的癲癇表型相關[15]。位于αII和β血影蛋白的重復結構域時,一般不致早發型癲癇性腦病,而與共濟失調表型有關[19]。Morsy等[16]通過對SPTAN1基因變異所致突變體的突變位點進行蛋白質建模實驗,發現錯義突變位點p.(Arg2124Cys)和p.(Ser2448Phe)影響αII和β血影蛋白的異聚化過程,未對蛋白質的結構、空間效應產生影響,2個突變體表現為輕微癲癇。本例患兒為錯義突變、突變位點c.6926T>C(p.Met2309Thr),與上述位點接近,考慮良性癲癇表型的形成與上述機制有關聯。
本研究報道了國內首例SPTAN1基因變異致SeLECTS,擴大了SPTAN1基因表型譜,并為這類罕見病的診治及遺傳咨詢提供了理論基礎。
利益沖突聲明 所有作者無利益沖突。
SPTAN1基因位于染色體9q34.11,編碼非紅細胞αII血影蛋白(non-erythrocyteαII spectrin),該蛋白與β血影蛋白形成異源四聚體[1],對維持細胞膜骨架起重要作用,并對神經元突觸形成、遞質釋放起到調節作用[2],其致病性變異可致翻譯出的蛋白結構、功能發生變化,進一步影響大腦結構形成、發育進程,從而引發一系列神經系統疾病。伴中央顳區棘波的自限性癲癇(Self-limited epilepsy with centrotemporal spikes,SeLECTS)通常發病年齡特定、癲癇發作在青春期前自行緩解,大多數癥狀呈良性、抗癲癇治療效果良好,極少數患者在缺乏適當治療的情況下會出現認知障礙等不良后果。本研究回顧1例SPTAN1基因突變致SeLECTS患兒的臨床特征,結合相關文獻進行討論。
病例資料 患兒 女,8歲4月齡。因“1月內抽搐發作2次”于2023年6月就診于河北省兒童醫院。患兒8歲3月齡起病,表現為入睡后不久后出現意識喪失、面部肌肉抽搐、口齒不清、流涎、全身強直陣攣發作,持續約1分鐘后自行緩解,緩解后無肢體活動障礙等不適,共發作2次,癥狀相似,無發熱,無咳嗽、咳痰,無鼻塞、流涕,無頭痛、頭暈,無惡心、嘔吐,無腹痛、腹瀉,無煩躁、嗜睡、精神行為異常,無大小便失禁,未予以治療。患兒既往體健。患兒為母親第1胎第1產,孕足月順產,出生體重3 270 g,否認窒息搶救史。患兒父母均體健,否認近期婚配。患兒生長發育同正常同齡兒。否認發育障礙、癲癇及其他疾病家族史。
體格檢查:神清語利,全身皮膚未見皮疹、異常色素沉著。淺表淋巴結未觸及腫大,雙側瞳孔直徑3 mm正大等圓,對光反射靈敏。口腔黏膜光滑,咽無充血,雙側扁桃體無腫大。頸部無抵抗;雙肺呼吸音清,未聞及干濕性啰音;心律規整,心音有力,未聞及雜音;腹軟,肝脾肋下未觸及,腸鳴音存在;脊柱無側彎、壓痛,四肢活動可,四肢肌力、肌張力正常,雙側肱二、三頭肌腱反射存在,雙側膝腱反射存在,雙側巴氏征、克氏征、布氏征陰性。輔助檢查:血常規、肝功能、腎功能、血氨、乳酸、血及尿遺傳代謝串聯質譜篩查、頭顱核磁未見異常。韋氏智力量表測試示:正常。視頻腦電圖示:異常兒童腦電圖,醒睡各期左側顳區、右側Rolandic區棘波、棘慢波不同步發放(圖1)。
圖1
患兒視頻腦電圖
醒睡各期左側顳區、右側Rolandic區棘波、棘慢波不同步發放
患兒在學齡期出現癲癇發作,頭顱核磁、血尿遺傳代謝篩查未見異常,考慮存在遺傳病因。經患兒父母簽署知情同意書后,分別抽取患兒、父母外周血2mL提取DNA行家系全外基因組檢測,并對分析結果行Sanger驗證。全基因組檢測提示患兒SPTAN1基因存在1個雜合變異(NM_001130438.2): c.6926T>C(p.Met2309Thr)。此變異導致編碼蛋白的第2 309位氨基酸由甲硫氨酸突變成蘇氨酸。經Sanger驗證,父母均未見變異,為新發變異(PS2)(圖2)。該變異在人類基因頻率數據庫(Genome Aggregation Database,GnomAD)基因頻率未被報道(PM2)。根據美國醫學遺傳學與基因組學學會(American College of Medical Genetics and Genomics,ACMG)指南判定,根據美國醫學遺傳學與基因組學學會指南,該變異為可能致病變異。
圖2
SPTAN1基因檢測
c.6926T>C位點雜合錯義變異,未在受檢者父母外周血中檢出,紅框提示變異位點
根據患兒臨床癥狀、視頻腦電圖及基因檢查結果,診斷SeLECTS,給予口服左乙拉西坦片0.25 g(10 mg/kg),每日2次。1個月后復查視頻腦電圖示:正常兒童腦電圖。隨訪7個月,患兒無抽搐發作。
討論 伴中央顳區棘波的自限性癲癇又被稱為Rolandic癲癇,是一種兒童常見的良性、局灶性癲癇綜合征,為常染色體顯性遺傳病[3]。Jabbari等[4]在一項大型SeLECTS患者中進行遺傳基因突變篩查研究,發現了SPTAN1基因,但目前無SPTAN1基因變異致BECTs的病例報道,大多數報道的相關表型包括伴進行性腦萎縮的早發性癲癇性腦病、伴小腦畸形的嚴重智力低下,以及伴或不伴癲癇的相對較輕表型[5]。
SPTAN1基因位于編碼一種廣泛分布于細胞膜上的絲狀骨架蛋白,由αII和β血影蛋白構成,具有高度保守的氨基酸結構,對神經元樹突、軸突的發育、神經元突觸的形成中起到關鍵作用 [5]。SeLECTS是兒童時期最常見的癲癇綜合征之一,約占兒童癲癇的 15%~24%,發病年齡以5~10歲最常見,其特征是癲癇發作隨著年齡的增長而逐漸消失[6],腦電圖示大腦中央和顳區存在局部放電[7],存在一定程度的認知障礙、行為和社交障礙等合并癥[8,9]。其病因尚不清楚,Samaitien?等[10]提出其病因與睡眠相關,還與遺傳有關。
目前已報道的SPTAN1基因變異相關疾病表型大致分為4種,分別為發育性癲癇性腦病(Developmental epileptic encephalopathy,DEE)、遠端遺傳性運動神經病、痙攣性截癱、發育遲緩伴或不伴癲癇[11]。以上表型中DEE表型例數最多,目前已有24例被報道[12-17],這類表型多數為新生重復突變,87.5%的病例診斷為West綜合征,合并有小頭畸形、肌張力下降、藥物難治性癲癇,頭顱核磁有特征性變化,大多數表現為髓鞘化減退及大腦或小腦萎縮。已報道28例SPTAN1基因變異致痙攣性截癱相關表型病例均有共濟失調[16,18],伴或不伴癲癇發作,均不存在無智力障礙、發育遲緩,頭顱核磁、腦電圖結果大多數無異常。SPTAN1基因變異致發育遲緩伴或不伴癲癇表型共有21例被報道[15,16],這類表型給予抗癲癇發作藥物治療可以得到良好的效果,腦電圖以局灶性棘波、多灶性棘波或全面性棘波為主,可伴有輕度智力障礙、學習障礙等合并癥,頭顱核磁基本正常。本例患兒為SPTAN1基因變異所致SeLECTS表型,目前國內外尚無報道,與既往已報道的表型相比,臨床癥狀溫和,無智力、認知落后等合并癥,口服藥物控制癲癇發作效果良好。
已發現SPTAN1基因重復突變與嚴重DEE表型具有相關性,而錯義、移碼突變與共濟失調、痙攣性截癱及溫和癲癇表型存在相關性[18]。另外,已有研究提出,突變位點位于αII及β血影蛋白的異源二聚體區域時,會影響最終翻譯的蛋白質空間分布而致聚集現象,并發現與最嚴重的癲癇表型相關[15]。位于αII和β血影蛋白的重復結構域時,一般不致早發型癲癇性腦病,而與共濟失調表型有關[19]。Morsy等[16]通過對SPTAN1基因變異所致突變體的突變位點進行蛋白質建模實驗,發現錯義突變位點p.(Arg2124Cys)和p.(Ser2448Phe)影響αII和β血影蛋白的異聚化過程,未對蛋白質的結構、空間效應產生影響,2個突變體表現為輕微癲癇。本例患兒為錯義突變、突變位點c.6926T>C(p.Met2309Thr),與上述位點接近,考慮良性癲癇表型的形成與上述機制有關聯。
本研究報道了國內首例SPTAN1基因變異致SeLECTS,擴大了SPTAN1基因表型譜,并為這類罕見病的診治及遺傳咨詢提供了理論基礎。
利益沖突聲明 所有作者無利益沖突。

