引用本文: 董尚勝, 黃小麗, 陳艷娟. 以癲癇為主要表現的16p11.2末端微缺失綜合征病例遺傳學分析. 癲癇雜志, 2024, 10(3): 274-277. doi: 10.7507/2096-0247.202401009 復制
版權信息: ?四川大學華西醫院華西期刊社《癲癇雜志》版權所有,未經授權不得轉載、改編
人類染色體16p11.2區域位于16號染色體的近端短臂上,16p11.2微缺失和微重復與孤獨癥、智力障礙、發育障礙、癲癇、精神分裂癥和肥胖等密切相關[1]。16p11.2微缺失的發生率為0.028%~0.043%,16p11.2微重復的發生率為0.035%~0.053%[2]。對國內16p11.2微缺失綜合征進行文獻回顧,共16篇關于16p11.2微缺失的病例報道[3-18],其中6篇針對產前診斷[3,5,7,10,11,18],10篇關于產后的病例分析[4,6,8,9,12-17],4篇針對癲癇表型分析[6,8,9,16],1篇關于疑似16p11.2微缺失所致糖尿病[14],1篇關于發作性運動障礙[12],未發現16p11.2末端微缺失綜合征病例報道。現報道通過基因檢查確診的一例以癲癇為主要表型的16p11.2末端微缺失綜合征,以期為相關疾病臨床診治提供一定參考。
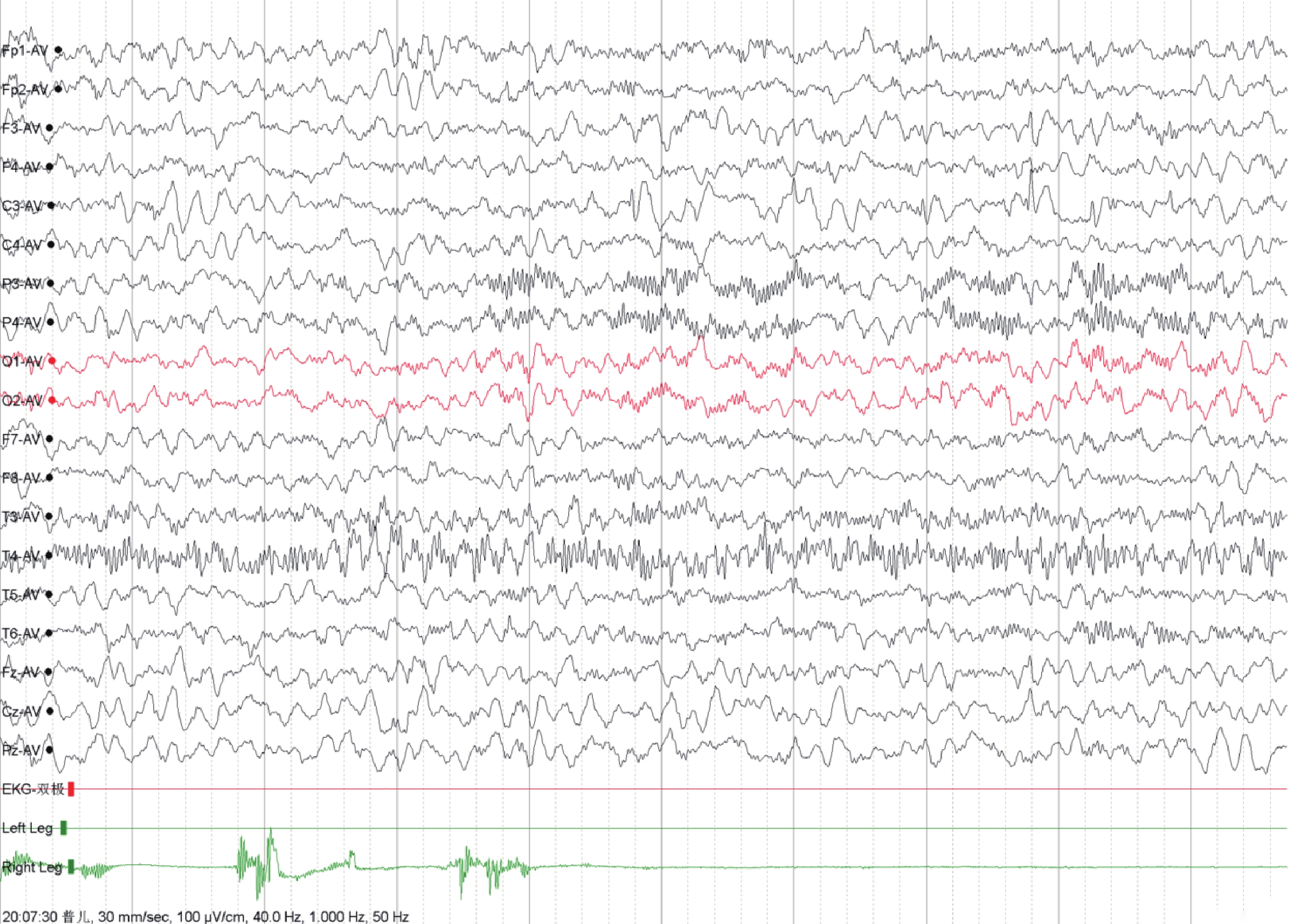
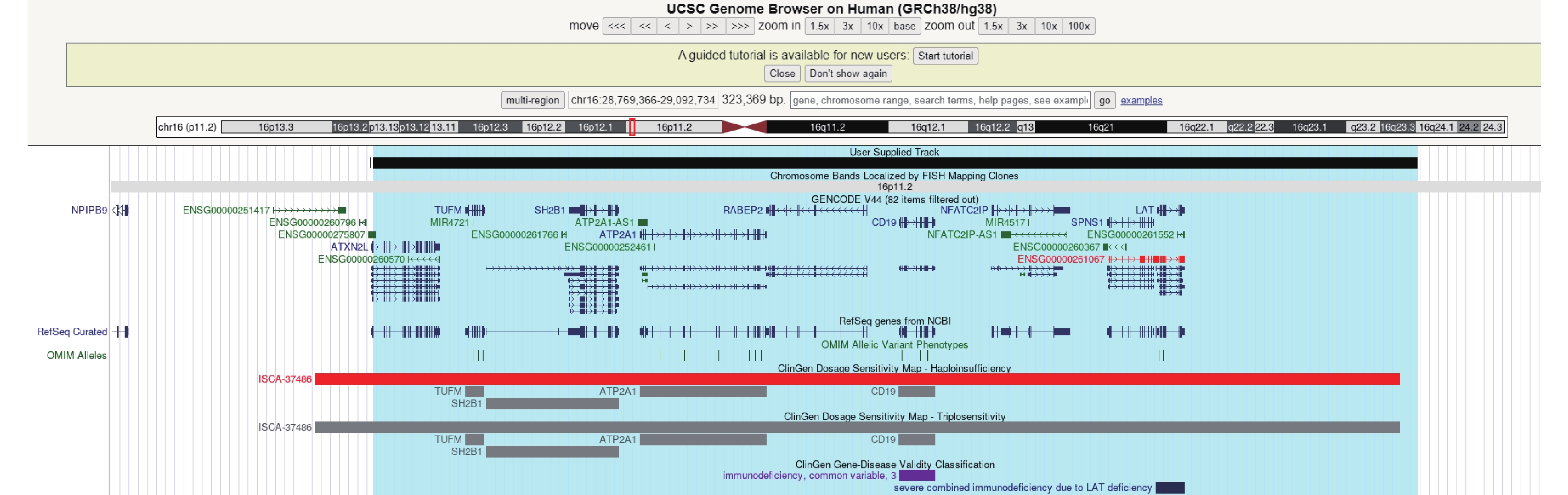
病例資料 患兒 男,2歲8月齡。因“反復發作性抽搐1年7個月多次有熱或無熱抽搐”就診江門市婦幼保健院癲癇專科。1歲1月齡出現發熱,最高體溫39.5℃,予布洛芬混懸液口服10 min后仍發熱明顯并出現抽搐發作,表現為四肢強直屈曲1 min左右,伴雙目上視、口唇發紺、流涎,后出現肢體節律性抽搐數十秒后停止,發作后患兒精神疲倦,閉目,呼之可應,數分鐘后如發作前狀態,2歲前共有5次發熱抽搐史,表現基本同前;2歲3月齡時,出現無熱抽搐發作3次,表現為右下肢抖動,眨眼,神志清醒,可以對答,后演變為意識不清,肢體強直及抖動,持續約2~3 min后自行緩解,無發熱,無嘔吐、腹瀉,無咳嗽、流涕。家長拒絕抗癲癇治療,定期門診復診,發育與同齡兒相似。第1胎第1產,胎齡37周,剖宮產,無窒息史,出生體重2.4 kg,身長49 cm。神經精神發育:3月齡抬頭,3月齡會笑,6月齡會坐,6月齡出牙,10月齡會站,12月齡會走、會說話。否認家族抽搐及神經系統疾病史,否認反復肺炎或感染病史。查體:脈搏:112次/min,呼吸:26次/min,血壓:92/54 mmHg,身高:94.5 cm,體重:13 kg,頭圍:45 cm。無特殊面容,肢體活動正常,皮膚未見牛奶咖啡斑,顱神經查體未見異常。心肺腹查體未見明顯異常。輔助檢查:血氣分析、生化、血常規、血清鎂離子、肝功四項、腎功四項、心功能四項、糞便常規、尿常規、心電圖、頭顱核磁共振成像:未見異常(圖1)。12 h腦電圖:左側中央區多量棘慢波發放,可波及前、中顳區(圖2)。0~6歲兒童心理行為測試:總發育商88.7,粗大運動發育商93.4,精細運動發育商88.7,適應能力發育商84,語言能力發育商79.4,社交能力發育商98.1。家系全外顯子檢查:chr16:28823260-29038839(gh38),16p11.2區域0.22Mb新生致病性缺失,與16p11.2末端微缺失綜合征相關,家長拒絕進一步進行染色體微陣列(CMA)驗證。患兒CNV片段詳見圖3。
 圖1
顱腦磁共振成像
圖1
顱腦磁共振成像
T1加權像示結構未見異常,腦髓鞘化正常
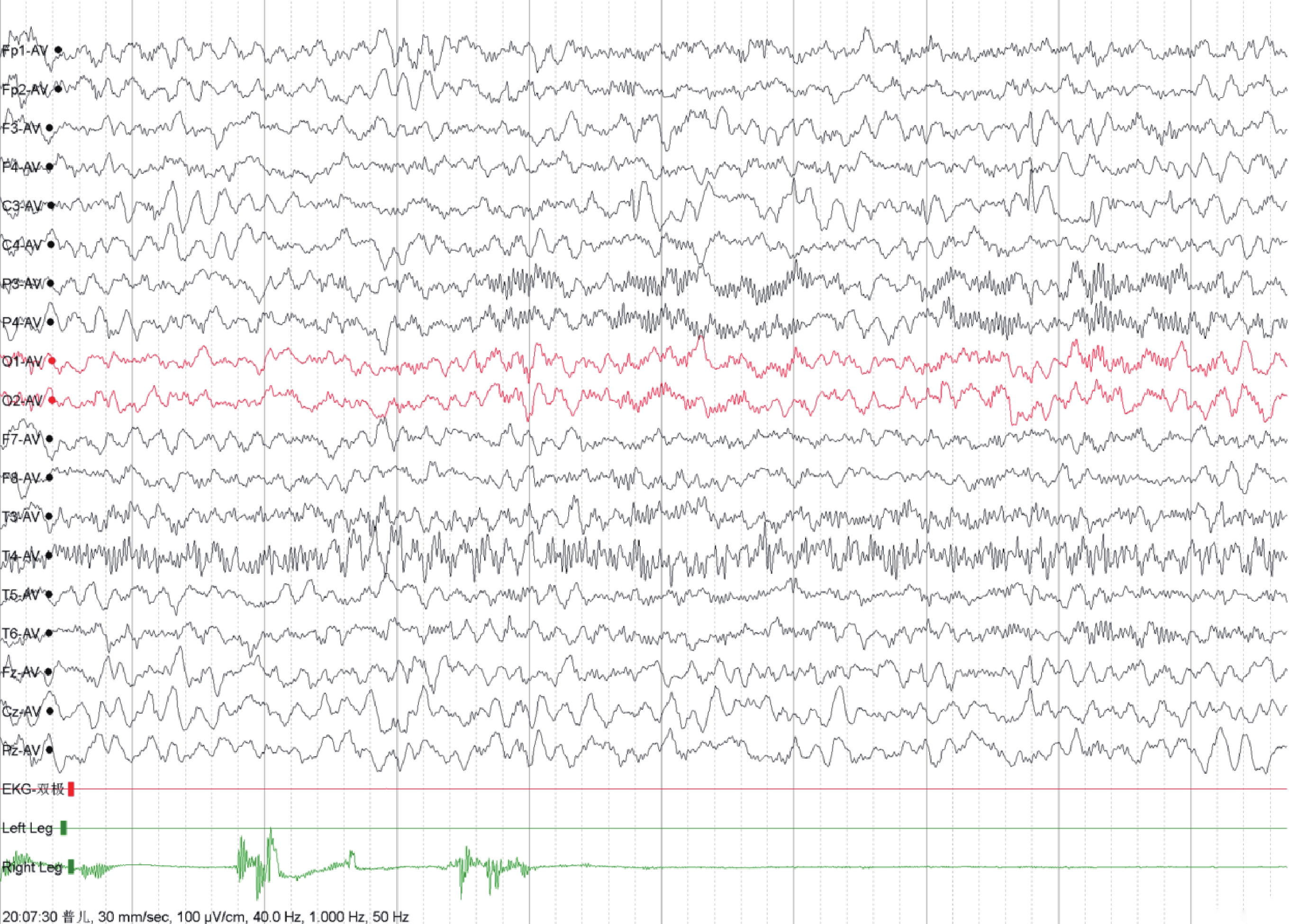 圖2
視頻腦電圖
圖2
視頻腦電圖
左側中央區多量棘慢波發放,可波及前顳及中顳區
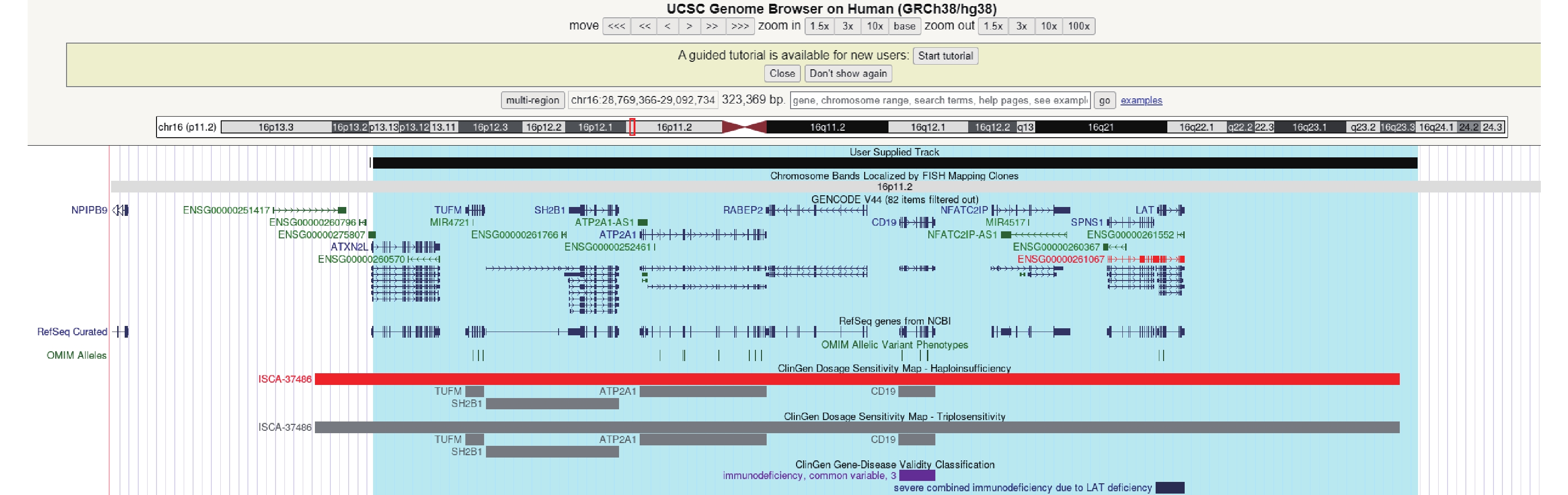 圖3
患者CNV片段(chr16:28823260-29038839)
圖3
患者CNV片段(chr16:28823260-29038839)
在UCSC映射情況,涉及9個蛋白編碼基因,4基因與OMIM疾病表型相關,覆蓋了16p11.2 (distal,BP2-BP3)94.66%的區域
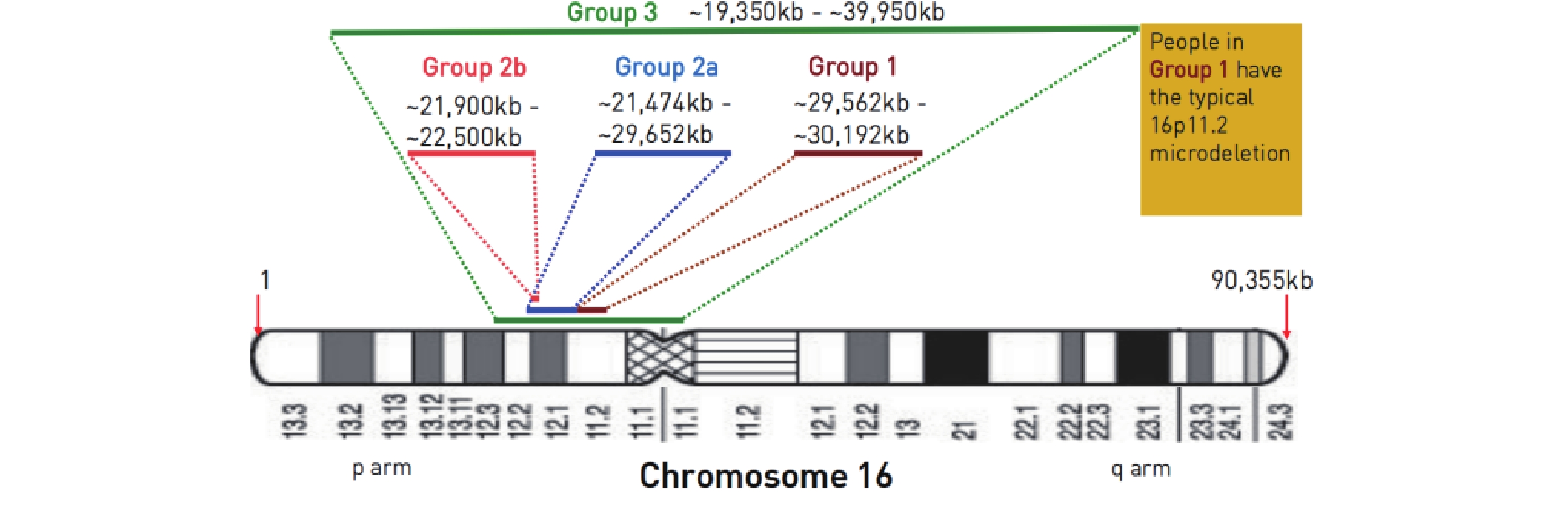
討論 根據orphan數據庫的總結,16p11.2區域的CNV變異包含5個相關微缺失/微重復綜合征,16p11.2-12.2微缺失綜合征(ORPHA ID 261211)、16p11.2-12.2微重復綜合征(ORPHA ID 261204)、16p11.2末端微缺失綜合征(ORPHA ID 261222)、16p11.2近端微缺失綜合征(ORPHA ID 261197)、16p11.2近端微重復綜合征(ORPHA ID 370079)。患兒CNV片段中區域和基因情況詳見表1。16p11.2微缺失綜合征包括典型16p11.2微缺失綜合征(BP4-BP5區域)及非典型16p11.2微缺失綜合征(BP2-BP3區域)兩大類(圖4)[19]。典型組約為600 kb大小缺失,包含29個基因,非典型組可分為2a型和2b型,與典型組不重疊且更靠近16p11.2末端,這個區域也稱為16p11.2末端區域。第3組為大片段缺失,包含前面兩組所有遺傳物質。
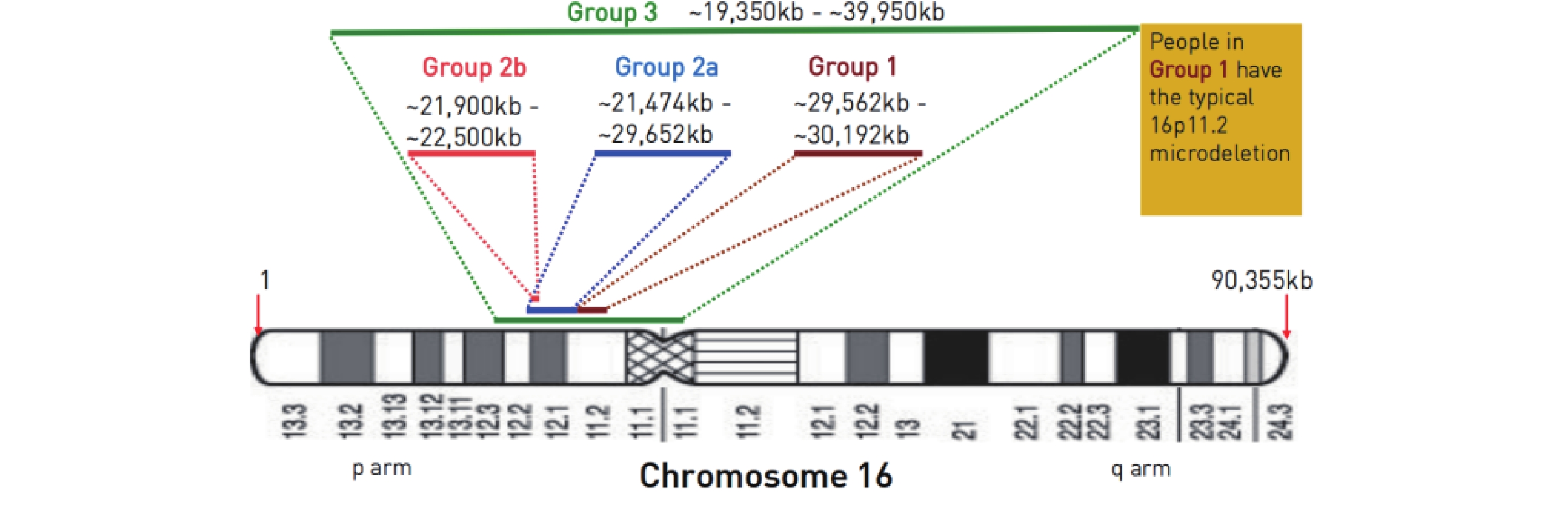 圖4
16p11.2微缺失綜合分型示意圖[19]
圖4
16p11.2微缺失綜合分型示意圖[19]
16p11.2末端微缺失綜合征(distal 16p11.2 microdeletion syndrome,MIM 613444)是一種罕見的染色體異常綜合征,與染色體16p11.2上220 kb區域的缺失相關,該區間包含9個基因:ATXN2L(MIM:607931)、TUFM(MIM:602389)、SH2B1(MIM:608937)、ATP2A1(MIM: 108730)、RABEP2(MIM:611869)、CD19(MIM:107265)、NFATC2IP(MIM:614525)、SPNS1(MIM:612583)和LAT(MIM:602354)。其中,SH2B1編碼參與瘦素和胰島素信號傳導,在全基因組關聯研究中發現SH2B1基因缺失與肥胖、發育遲緩及代謝相關疾病有關[20]。Genesio等[21]在一例關于先天性膈疝(CDH)和遠端16p11.2微缺失的產前病例提示,ATP2A1可能是一種參與膈肌發育的基因。LAT過表達引起腦增殖細胞減少和小頭畸形,LAT基因敲除小鼠出現腦體積變化,LAT除了在T細胞發育起作用外,還是16p11.2(BP2-BP3)片段相關神經發育表型的主要貢獻因子[22]。CD19基因突變與先天性免疫缺陷相關。Tabet總結[23]了11例重疊16p11.2末端微缺失病例,這些病例的表型高度可變,包括發育遲緩(n=5)、學習困難(n=1)、行為問題[如ASD(n=2)和ADHD(n=1)]、異常面部特征(n=6)、肥胖(n=4)、糖尿病(n=2)、癲癇發作(n=1)、先天性腎臟和尿路畸形(n=2)和先天性巨結腸病(n=2)。本患兒神經行為發育正常,是否隨著年齡增長會出現學習困難或異常行為問題,仍需進一步隨訪。
文獻報道16p11.2染色體攜帶者中21.8%~26.8%有癲癇表,癲癇的發生與PRRT2基因的單倍劑量不足有關,并且16p11.2微缺失造成的癲癇較單純PRRT2基因突變的癲癇表型嚴重[8]。國內學者總結了11例16p11.2微缺失相關癲癇情況,11例患兒均以抽搐為首發癥狀,均為嬰幼兒期起病,發作類型均為局灶性發作,11例都涉及PRRT2基因缺失,常用的抗癲癇發作藥物為奧卡西平或丙戊酸鈉,癲癇癥狀控制良好,未見難性癲癇病例[8]。關于16p11.2遠端微缺失是否會引起癲癇發作,目前相關病例較少[23]。本患兒主要表現為左側Rolandic區放電,臨床癥狀是右側肢體乏力后,出現頭暈感及全面性發作,符合左側中央區起源的局灶性癲癇發作,演變為全面性強直陣攣發作。發作形式與典型的16p11.2微缺失綜合征相似[8,9]。本患兒CNV片段不包括PRTT2基因,也未見明顯運動障礙表現,心理行為測試表明患兒無發育障礙表現,片段中引起癲癇的基因暫不清楚,16p11.2末端區域是否存在癲癇相關基因或結構仍需更多病例證明[23]。
總之,16p11.2末端微缺失綜合征是一個非常罕見的疾病,基因型與表型的關系仍需進一步總結。
利益沖突聲明 所有作者無利益沖突。
人類染色體16p11.2區域位于16號染色體的近端短臂上,16p11.2微缺失和微重復與孤獨癥、智力障礙、發育障礙、癲癇、精神分裂癥和肥胖等密切相關[1]。16p11.2微缺失的發生率為0.028%~0.043%,16p11.2微重復的發生率為0.035%~0.053%[2]。對國內16p11.2微缺失綜合征進行文獻回顧,共16篇關于16p11.2微缺失的病例報道[3-18],其中6篇針對產前診斷[3,5,7,10,11,18],10篇關于產后的病例分析[4,6,8,9,12-17],4篇針對癲癇表型分析[6,8,9,16],1篇關于疑似16p11.2微缺失所致糖尿病[14],1篇關于發作性運動障礙[12],未發現16p11.2末端微缺失綜合征病例報道。現報道通過基因檢查確診的一例以癲癇為主要表型的16p11.2末端微缺失綜合征,以期為相關疾病臨床診治提供一定參考。
病例資料 患兒 男,2歲8月齡。因“反復發作性抽搐1年7個月多次有熱或無熱抽搐”就診江門市婦幼保健院癲癇專科。1歲1月齡出現發熱,最高體溫39.5℃,予布洛芬混懸液口服10 min后仍發熱明顯并出現抽搐發作,表現為四肢強直屈曲1 min左右,伴雙目上視、口唇發紺、流涎,后出現肢體節律性抽搐數十秒后停止,發作后患兒精神疲倦,閉目,呼之可應,數分鐘后如發作前狀態,2歲前共有5次發熱抽搐史,表現基本同前;2歲3月齡時,出現無熱抽搐發作3次,表現為右下肢抖動,眨眼,神志清醒,可以對答,后演變為意識不清,肢體強直及抖動,持續約2~3 min后自行緩解,無發熱,無嘔吐、腹瀉,無咳嗽、流涕。家長拒絕抗癲癇治療,定期門診復診,發育與同齡兒相似。第1胎第1產,胎齡37周,剖宮產,無窒息史,出生體重2.4 kg,身長49 cm。神經精神發育:3月齡抬頭,3月齡會笑,6月齡會坐,6月齡出牙,10月齡會站,12月齡會走、會說話。否認家族抽搐及神經系統疾病史,否認反復肺炎或感染病史。查體:脈搏:112次/min,呼吸:26次/min,血壓:92/54 mmHg,身高:94.5 cm,體重:13 kg,頭圍:45 cm。無特殊面容,肢體活動正常,皮膚未見牛奶咖啡斑,顱神經查體未見異常。心肺腹查體未見明顯異常。輔助檢查:血氣分析、生化、血常規、血清鎂離子、肝功四項、腎功四項、心功能四項、糞便常規、尿常規、心電圖、頭顱核磁共振成像:未見異常(圖1)。12 h腦電圖:左側中央區多量棘慢波發放,可波及前、中顳區(圖2)。0~6歲兒童心理行為測試:總發育商88.7,粗大運動發育商93.4,精細運動發育商88.7,適應能力發育商84,語言能力發育商79.4,社交能力發育商98.1。家系全外顯子檢查:chr16:28823260-29038839(gh38),16p11.2區域0.22Mb新生致病性缺失,與16p11.2末端微缺失綜合征相關,家長拒絕進一步進行染色體微陣列(CMA)驗證。患兒CNV片段詳見圖3。
圖1
顱腦磁共振成像
T1加權像示結構未見異常,腦髓鞘化正常
圖2
視頻腦電圖
左側中央區多量棘慢波發放,可波及前顳及中顳區
圖3
患者CNV片段(chr16:28823260-29038839)
在UCSC映射情況,涉及9個蛋白編碼基因,4基因與OMIM疾病表型相關,覆蓋了16p11.2 (distal,BP2-BP3)94.66%的區域
討論 根據orphan數據庫的總結,16p11.2區域的CNV變異包含5個相關微缺失/微重復綜合征,16p11.2-12.2微缺失綜合征(ORPHA ID 261211)、16p11.2-12.2微重復綜合征(ORPHA ID 261204)、16p11.2末端微缺失綜合征(ORPHA ID 261222)、16p11.2近端微缺失綜合征(ORPHA ID 261197)、16p11.2近端微重復綜合征(ORPHA ID 370079)。患兒CNV片段中區域和基因情況詳見表1。16p11.2微缺失綜合征包括典型16p11.2微缺失綜合征(BP4-BP5區域)及非典型16p11.2微缺失綜合征(BP2-BP3區域)兩大類(圖4)[19]。典型組約為600 kb大小缺失,包含29個基因,非典型組可分為2a型和2b型,與典型組不重疊且更靠近16p11.2末端,這個區域也稱為16p11.2末端區域。第3組為大片段缺失,包含前面兩組所有遺傳物質。
圖4
16p11.2微缺失綜合分型示意圖[19]
16p11.2末端微缺失綜合征(distal 16p11.2 microdeletion syndrome,MIM 613444)是一種罕見的染色體異常綜合征,與染色體16p11.2上220 kb區域的缺失相關,該區間包含9個基因:ATXN2L(MIM:607931)、TUFM(MIM:602389)、SH2B1(MIM:608937)、ATP2A1(MIM: 108730)、RABEP2(MIM:611869)、CD19(MIM:107265)、NFATC2IP(MIM:614525)、SPNS1(MIM:612583)和LAT(MIM:602354)。其中,SH2B1編碼參與瘦素和胰島素信號傳導,在全基因組關聯研究中發現SH2B1基因缺失與肥胖、發育遲緩及代謝相關疾病有關[20]。Genesio等[21]在一例關于先天性膈疝(CDH)和遠端16p11.2微缺失的產前病例提示,ATP2A1可能是一種參與膈肌發育的基因。LAT過表達引起腦增殖細胞減少和小頭畸形,LAT基因敲除小鼠出現腦體積變化,LAT除了在T細胞發育起作用外,還是16p11.2(BP2-BP3)片段相關神經發育表型的主要貢獻因子[22]。CD19基因突變與先天性免疫缺陷相關。Tabet總結[23]了11例重疊16p11.2末端微缺失病例,這些病例的表型高度可變,包括發育遲緩(n=5)、學習困難(n=1)、行為問題[如ASD(n=2)和ADHD(n=1)]、異常面部特征(n=6)、肥胖(n=4)、糖尿病(n=2)、癲癇發作(n=1)、先天性腎臟和尿路畸形(n=2)和先天性巨結腸病(n=2)。本患兒神經行為發育正常,是否隨著年齡增長會出現學習困難或異常行為問題,仍需進一步隨訪。
文獻報道16p11.2染色體攜帶者中21.8%~26.8%有癲癇表,癲癇的發生與PRRT2基因的單倍劑量不足有關,并且16p11.2微缺失造成的癲癇較單純PRRT2基因突變的癲癇表型嚴重[8]。國內學者總結了11例16p11.2微缺失相關癲癇情況,11例患兒均以抽搐為首發癥狀,均為嬰幼兒期起病,發作類型均為局灶性發作,11例都涉及PRRT2基因缺失,常用的抗癲癇發作藥物為奧卡西平或丙戊酸鈉,癲癇癥狀控制良好,未見難性癲癇病例[8]。關于16p11.2遠端微缺失是否會引起癲癇發作,目前相關病例較少[23]。本患兒主要表現為左側Rolandic區放電,臨床癥狀是右側肢體乏力后,出現頭暈感及全面性發作,符合左側中央區起源的局灶性癲癇發作,演變為全面性強直陣攣發作。發作形式與典型的16p11.2微缺失綜合征相似[8,9]。本患兒CNV片段不包括PRTT2基因,也未見明顯運動障礙表現,心理行為測試表明患兒無發育障礙表現,片段中引起癲癇的基因暫不清楚,16p11.2末端區域是否存在癲癇相關基因或結構仍需更多病例證明[23]。
總之,16p11.2末端微缺失綜合征是一個非常罕見的疾病,基因型與表型的關系仍需進一步總結。
利益沖突聲明 所有作者無利益沖突。