引用本文: 吳曉鈺, 劉瑞寒, 孔鈺, 孔慶霞. KCND3 新發突變引起的癲癇發育性腦病一例并文獻復習. 癲癇雜志, 2024, 10(4): 368-371. doi: 10.7507/2096-0247.202405010 復制
版權信息: ?四川大學華西醫院華西期刊社《癲癇雜志》版權所有,未經授權不得轉載、改編
癲癇性腦病是由于癲癇頻繁發作導致發育遲緩或減退[1],癲癇在小兒神經系統發生率較高,對于接受藥物治療的兒童患者,癲癇不受控發作是重大問題,難治性癲癇是一種病因十分復雜的疾病,遺傳是難治性癲癇最常見的病因之一,其中編碼離子通道或神經遞質受體的基因突變最常見,鉀離子通道是構成離子通道家族中數量最多、最復雜的離子通道家族,鉀離子通道在神經元的電活動中起著至關重要的作用,并直接參與癲癇發作的機制[2],KCND3 編碼電壓門控鉀離子通道亞家族 D 成員 3(Voltage-gated Potassium Channel,KV4.3)是一種六跨膜蛋白,KV4.3 編碼離子通道亞基共組裝成功能性四聚體通道,形成孔結構域(跨膜螺旋 S5-S6),被形成電壓傳感器結構域的 S1-S4 段包圍[3]。KCND3 缺陷可引起心臟和神經系統綜合征[4]。其中心臟表型主要表現為早期復極綜合征和陣發性房顫,大腦表型主要表現為癲癇和智力缺陷,而本案例患者僅以癲癇為主要表型。
病例資料 患兒 男,4歲。因“抽搐”就診于濟寧醫學院附屬醫院癲癇門診。患兒足月產,出生體重3.65 kg,哭聲強,否認窒息、青紫,Apgar評分不詳。3月齡會抬頭,7月齡會坐,1歲2月齡會走,現上幼兒園小班,目前說話欠清晰。父母,姐姐身體健康無同類疾病史,患兒于1歲4月齡出現發熱時抽搐,表現為意識喪失、喚之不應、雙眼斜視、雙手握拳、面色無發紺、偶有口吐白沫、無大小便失禁,伴四肢抖動,持續約1 min緩解,發作后體溫38~39℃,共8~9次,患兒2歲出現無熱抽搐,表現為意識喪失、喚之不應、四肢抖動、無口吐白沫、無大小便失禁,持續約3~10 min緩解,緩解后入睡,每月約抽搐1次,無發熱、無咳嗽、無嘔吐及腹瀉,完善腦電圖等檢查診斷為"癲癇",先后加用丙戊酸鈉、左乙拉西坦、拉考沙胺、拉莫三嗪、唑尼沙胺、氯硝西灃、吡侖帕奈等抗癲癇發作藥物治療(具體加用順序及加藥時間家屬描述不清),因控制不佳停用"左乙拉西坦、拉考沙胺"。自2023-09-06近半年口服丙戊酸鈉,拉莫三嗪、唑尼沙胺、氯硝西灃、吡侖帕奈治療,目前無抽搐全面性發作,間斷出現雙眼上翻,持續1~2 s緩解,數天發作1次。2023-08就診于外院,完善腦電圖后,調整用藥,丙戊酸鈉加量,余抗癲癇發作藥物劑量未變,患兒仍有間斷雙眼上翻。2023-09-06患兒目前服用拉莫三嗪 50 mg 每日兩次、唑尼沙胺 100 mg 每日兩次、丙戊酸鈉緩釋片(0.25 g片劑+2 mL丙戊酸鈉藥水)、氯硝西泮0.5 mg 每晚一次、吡侖帕奈8 mg 每晚一次,抗癲癇治療。患兒自發病以來,神志清、精神欠佳、飲食可、睡眠欠佳、大小便無異常、體重無明顯變化
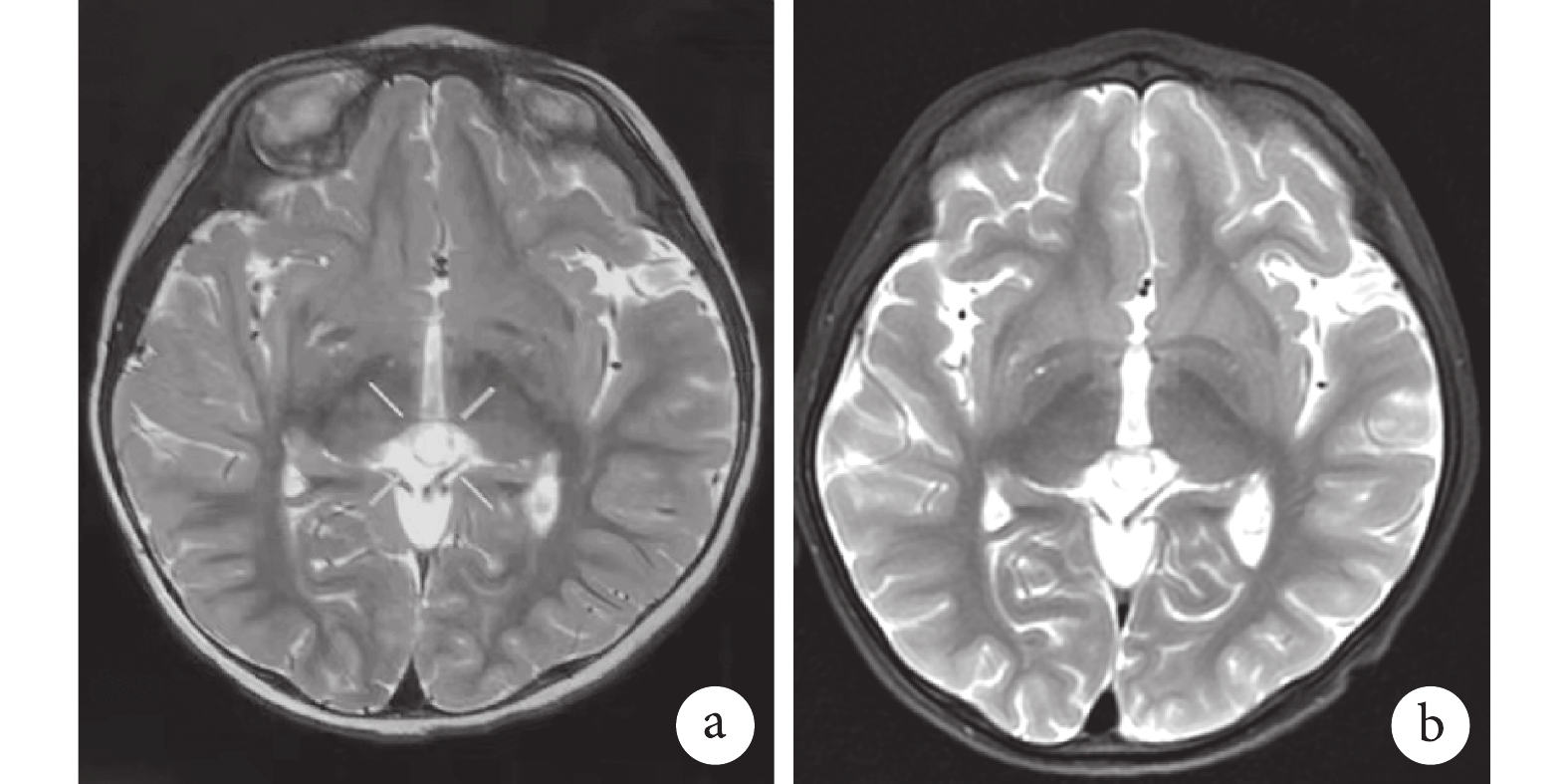
輔助檢查 心臟彩超心電圖無異常。影像學檢查:顱腦核磁共振檢查(Magnetic resonance imaging,MRI):雙側大腦半球結構對稱,腦灰白質對比正常,腦實質未見明顯異常;松果體呈囊性影,直徑約8.6 mm。幕下小腦半球及腦干形態、大小及信號未見明顯異常,雙橋小腦角未見明顯異常。各腦室、腦池、腦裂及腦溝對稱,大小、形態正常,中線結構居中。考慮為囊性松果體(圖1)。實驗室檢查:血常規,肝功、腎功、鉀、鈉、氯、鈣無明顯異常。腦電圖檢查:2021-03-06我院腦電圖示多量左側中央區放電,稍多量右側中央區,少許右側頂區放電。2022-05-23我院腦電圖示異常學齡前兒童視頻腦電圖,多灶性放電。2023-04-25我院腦電圖示異常學齡前兒童視頻腦電圖 多灶性放電(左右中顳、額區為著)。2023-08-22外院視頻腦電圖示兒童期異常腦電圖,雙側導聯可見廣泛性棘波、棘慢波發放,雙側額極、額、枕、中顳、后顳、額中線著。2023-09-05我院腦電圖示不正常腦電圖,高度異常(全導慢棘慢波持續發放)。2023-09-06我院腦電圖可見大量全導2 Hz左右中高﹣極高波幅慢尖慢波連續性發放,前頭部明顯;可見少許左側枕﹣中顳區、右側枕區、右側中央區高波幅尖慢波散發或簇發,向臨近導聯擴散。2023-10-20我院腦電圖示醒睡各期可見大量 雙側枕區、顳區、中央區、頂區、額區 同步或不同步中﹣高波幅尖慢波散發、簇發或連續發放。重度異常學齡前兒童視頻腦電圖,大量多灶性放電,以雙側枕區、中額區為著。2023-11-09我院腦電圖可見連續全導2.7~3.5 Hz左右中高﹣極高波幅慢尖慢波發放,雙側枕區明顯,異常學齡前兒童普通腦電圖,無發作期表現,發作間期異常(圖2)。
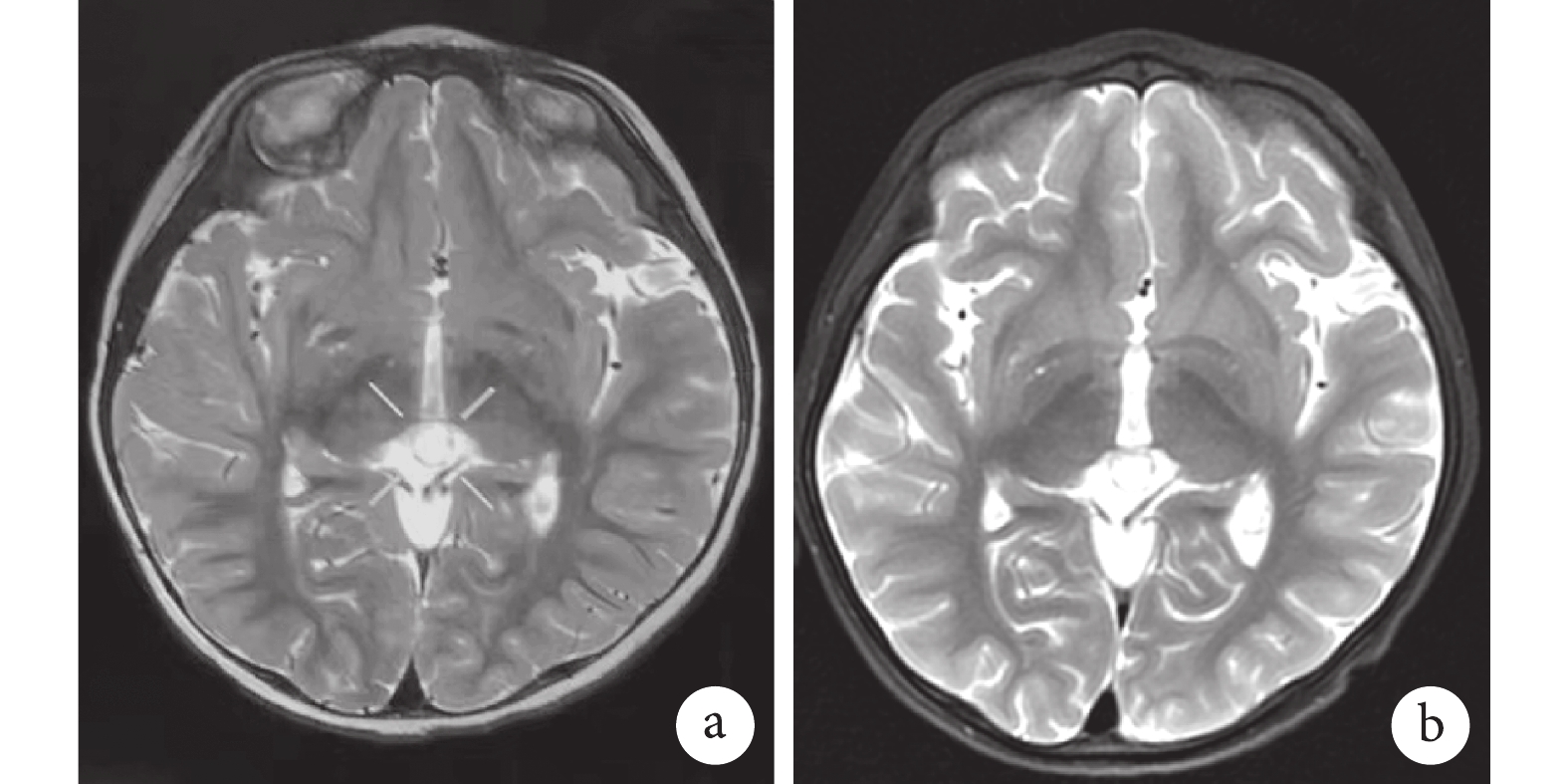 圖1
患兒腦部MRI
圖1
患兒腦部MRI
a. 2021-3-1;b. 2023-7-22
 圖2
患兒腦電圖
圖2
患兒腦電圖
a. 背景活動;b. 發作間期異常腦電圖;c. 發作間期異常腦電圖;d. 發作間期異常腦電圖
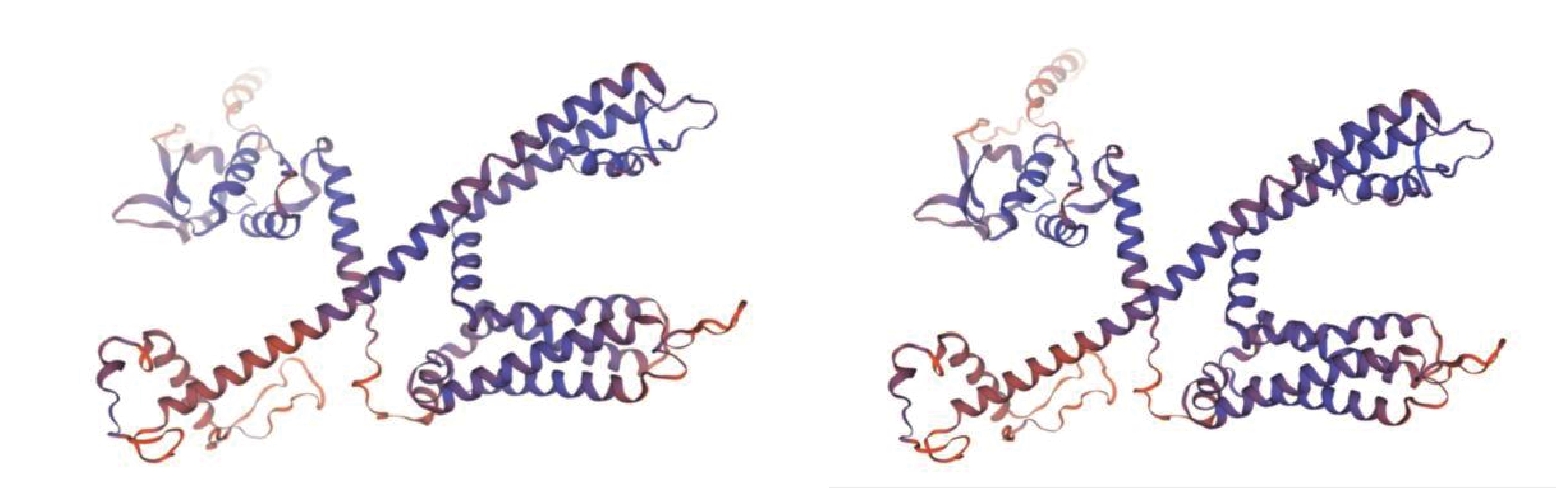
全外顯子基因檢測:KCND3 (c.1174G>A ,p.Val392Ile)致病性變異(新發變異),父母均無異常。致病性證據:c.1174G>A(p.Val392lle);根據 ACMG 指南,該變異初步判定為致病性變異(Pathogenic)PP3_Moderate+PS4+PM2_SupportingPSPP3Modere1REVEL 預測結果為有害,SIFT、PolyPhen_2、MutationTaster 、GERP+預測結果分別為良性、有害、有害、有害;PS4文獻數據庫已有該位點(Sudden unexplained death syndrome)的病例報道,變異標簽為 DM(致病突變),ClinVar 數據庫對該位點的致病性分析為 Pathogenic/Likely pathogenic,Spinocerebellar ataxia type 19/22|Brugada syndrome 9|not provided : PM2 Supporting 常人群數據庫中的頻率為﹣:PS2 經家系驗證分析,受檢人之父該位點無變異,受檢人之母該位點無變異,此變異為自發突變。KNCD3蛋白三級結構預測見圖3。
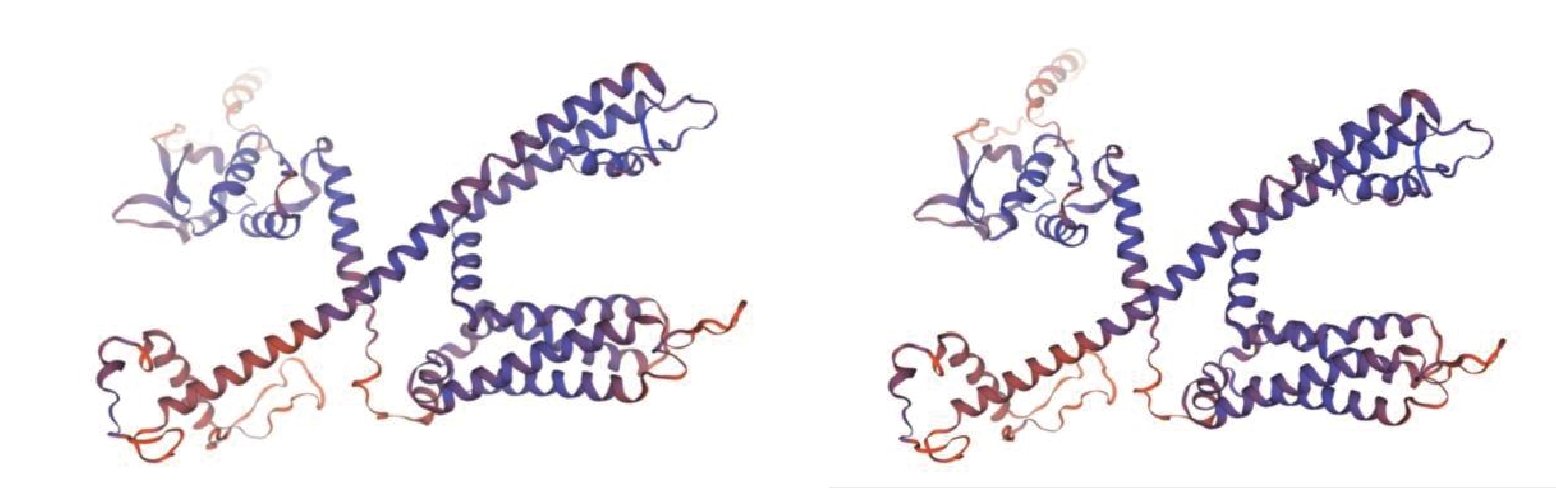 圖3
KNCD3蛋白三級結構預測
圖3
KNCD3蛋白三級結構預測
討論 Kv4通道是鉀離子通道家族中的一員:分為不同的亞型,包括Kv4.1、Kv4.2和Kv4.3,Kv4是Kvs家族中表達于腦、心臟和平滑肌的一個亞群[5],Kv4.3由KCND3編碼(3),Kv4.3在中樞神經系統中高度表達,尤其在小腦浦肯野細胞、深核、顆粒細胞和中間神經元中表達。KCND3 c.1174G>A 致病性變異(新發變異),KCND3基因突變導致的相關疾病有Brugada綜合征9型,臨床表型 :心ST段抬高,暈厥;脊髓小腦性共濟失調(Spinocerebellarataxia,SCA)19型 ;阿爾茲海默病(Alzheimer's disease,AD ),臨床表型:構音障礙;腱反射減弱,小腦萎縮,肌陣攣,腱反射亢進;吞咽困難,共濟失調等。 KCND3 Val392Ile突變已被報道4例,然而,關于這種突變攜帶者的臨床特征的信息是有限的, Wang等[6]報道患者5歲,發病年齡為1歲6月齡,癲癇發作時表現為四肢僵硬,屈曲,發紺,意識喪失,持續2~3 min,發作1~2次/月,且多在發熱后發作,先后使用奧卡西平和丙戊酸鈉,未見改善,發作時出現另一種癲癇表現,眼球轉動,伴單側或雙側肢體陣攣,在3歲3月齡時加用左乙拉西坦,癲癇發作頻率降低至1次/月,3歲11月齡時出現視力下降,無法精準拿起小物體,癲癇發作前發育指標正常,但發病后認知記憶能力下降,4歲時核磁共振正常,無癲癇或其他神經系統疾病。該案例與本病例均為KCND3從頭自發突變(c.1174G > A, p.Val392Ile),與本案例不同點在于本例患兒發作時無發紺,無視力下降,且發病年齡較早。
KCND3突變還可導致心腦通道病[7],Giudicessi等[8]首次報道KCND3 p. val392ile陽性的心臟驟停病例是一例20歲男性,有暈厥發作史,暈厥發作后心肺驟停后死亡。在死亡前幾天沒有任何明顯的疾病跡象,尿液藥物和毒素檢測呈陰性。死者生前未行心電圖檢查,在青少年時期有過兩次可疑的暈厥發作,家族中無心臟驟停或心律失常相關心臟事件的記錄,據報道死者父母和姐姐的12導聯心電圖檢查均正常[8]。Zhang等[7]報道的一例11歲女性患兒,2 歲時診斷出患有熱性驚厥,從 4 歲開始有規律性熱性驚厥;癲癇發作時意識喪失。通常會持續幾秒鐘,然后突然消失,6歲時診斷智力障礙(韋氏量表評分 < 75)。心電圖可檢測到有效不應期(Effective refractive period,ERP)下導聯 (II、III、AVF) 和側導聯 (V5、V6) 的 J 點升高。24小時動態心電圖監測儀顯示陣發性心房顫動、早發性心房復合體(Premature atrial contraction,PAC)、成對PAC和陣發性房性心動過速(Atrial tachycardia,AT),超聲心動圖結果正常。9歲時,診斷為早期復極綜合癥(Early repolarization syndrome,ERS)。Tadashi Nakajima等[9]報道的KCND3 Val392Ile突變相關的新型心腦通道病,表現出心臟(早期復極化綜合征和陣發性心房顫動)和腦表型(癲癇和智力障礙)。該患者因陣發性心房顫動(Paroxysmal Atrial Fibrillation,PAF)引起心悸,于夜間和清晨發作,持續兩個月,3~9歲發生過幾次局灶性意識損害發作,伴有智力殘疾(Intellectual Disabilities,ID)(WISC-III,FIQ:13歲時64),但無任何局灶性體征或SCA表型。13歲時記錄的腦電圖顯示雙側枕骨區有局灶性峰值。被診斷為病因不明的局灶性癲癇,MRI未見腦結構異常。先證者妹妹,體格檢查和血液檢查沒有發現異常。超聲心動圖顯示無結構性心臟病的跡象,在2~7歲時有過幾次局性意識受損癲癇發作,伴有ID (WISC-III,12歲時FIQ:41),但無任何局灶性體征或SCA表型。14歲時記錄的腦電圖顯示雙側枕區交替出現局灶性峰值,先證者的母親中發現了KCND3 V3921突變,在7歲和40歲時發生過兩次暈厥,先證者的父親未發現KCND3突變。先證者的外祖父和外祖母不攜帶KCND3 V3921突變。因此,這是在先證者的母親身上發生的新生突變。其被診斷為病因不明的局灶性癲癇。MRI未見腦結構異常。
KCND3 Val392Ile突變引起癲癇的機制尚不明確,但與KCND3相似的KCND2突變已被報道與癲癇發作有關,KCND2突變導致KV4.2的升高或降低導致癲癇的發作[2],KCND3突變引起的Kv4.3升高是否會導致癲癇目前尚不清楚。在KCND3 Val392Ile突變的情況下,Kv4.3的增加可能與癲癇有關(9)。同時KCND3是SCA 19/22的致病基因,這是一種常染色體顯性小腦共濟失調,定位于染色體1p21-q23[10-11],SCA 是一組遺傳性神經退行性疾病,其特征是進行性小腦性共濟失調和所有年齡段的可變錐體、錐體外系、大腦或脊髓癥狀,包括帕金森綜合征和癲癇癥狀患者[12-14]。此外,Singh等[13]綜述報道的15例SCA患者中有7例在嬰兒期或兒童期發病,且以癲癇為首發癥狀。
綜上,本文報道了一例KCND3自發性變異(c.1174G > A,p.Val392Ile),以癲癇為主要表現,發病年齡小,無心臟表型,父母無表型,藥物難以控制其發作,其臨床表現的特殊性豐富了該基因突變的臨床表型。
利益沖突聲明 所有作者無利益沖突。
癲癇性腦病是由于癲癇頻繁發作導致發育遲緩或減退[1],癲癇在小兒神經系統發生率較高,對于接受藥物治療的兒童患者,癲癇不受控發作是重大問題,難治性癲癇是一種病因十分復雜的疾病,遺傳是難治性癲癇最常見的病因之一,其中編碼離子通道或神經遞質受體的基因突變最常見,鉀離子通道是構成離子通道家族中數量最多、最復雜的離子通道家族,鉀離子通道在神經元的電活動中起著至關重要的作用,并直接參與癲癇發作的機制[2],KCND3 編碼電壓門控鉀離子通道亞家族 D 成員 3(Voltage-gated Potassium Channel,KV4.3)是一種六跨膜蛋白,KV4.3 編碼離子通道亞基共組裝成功能性四聚體通道,形成孔結構域(跨膜螺旋 S5-S6),被形成電壓傳感器結構域的 S1-S4 段包圍[3]。KCND3 缺陷可引起心臟和神經系統綜合征[4]。其中心臟表型主要表現為早期復極綜合征和陣發性房顫,大腦表型主要表現為癲癇和智力缺陷,而本案例患者僅以癲癇為主要表型。
病例資料 患兒 男,4歲。因“抽搐”就診于濟寧醫學院附屬醫院癲癇門診。患兒足月產,出生體重3.65 kg,哭聲強,否認窒息、青紫,Apgar評分不詳。3月齡會抬頭,7月齡會坐,1歲2月齡會走,現上幼兒園小班,目前說話欠清晰。父母,姐姐身體健康無同類疾病史,患兒于1歲4月齡出現發熱時抽搐,表現為意識喪失、喚之不應、雙眼斜視、雙手握拳、面色無發紺、偶有口吐白沫、無大小便失禁,伴四肢抖動,持續約1 min緩解,發作后體溫38~39℃,共8~9次,患兒2歲出現無熱抽搐,表現為意識喪失、喚之不應、四肢抖動、無口吐白沫、無大小便失禁,持續約3~10 min緩解,緩解后入睡,每月約抽搐1次,無發熱、無咳嗽、無嘔吐及腹瀉,完善腦電圖等檢查診斷為"癲癇",先后加用丙戊酸鈉、左乙拉西坦、拉考沙胺、拉莫三嗪、唑尼沙胺、氯硝西灃、吡侖帕奈等抗癲癇發作藥物治療(具體加用順序及加藥時間家屬描述不清),因控制不佳停用"左乙拉西坦、拉考沙胺"。自2023-09-06近半年口服丙戊酸鈉,拉莫三嗪、唑尼沙胺、氯硝西灃、吡侖帕奈治療,目前無抽搐全面性發作,間斷出現雙眼上翻,持續1~2 s緩解,數天發作1次。2023-08就診于外院,完善腦電圖后,調整用藥,丙戊酸鈉加量,余抗癲癇發作藥物劑量未變,患兒仍有間斷雙眼上翻。2023-09-06患兒目前服用拉莫三嗪 50 mg 每日兩次、唑尼沙胺 100 mg 每日兩次、丙戊酸鈉緩釋片(0.25 g片劑+2 mL丙戊酸鈉藥水)、氯硝西泮0.5 mg 每晚一次、吡侖帕奈8 mg 每晚一次,抗癲癇治療。患兒自發病以來,神志清、精神欠佳、飲食可、睡眠欠佳、大小便無異常、體重無明顯變化
輔助檢查 心臟彩超心電圖無異常。影像學檢查:顱腦核磁共振檢查(Magnetic resonance imaging,MRI):雙側大腦半球結構對稱,腦灰白質對比正常,腦實質未見明顯異常;松果體呈囊性影,直徑約8.6 mm。幕下小腦半球及腦干形態、大小及信號未見明顯異常,雙橋小腦角未見明顯異常。各腦室、腦池、腦裂及腦溝對稱,大小、形態正常,中線結構居中。考慮為囊性松果體(圖1)。實驗室檢查:血常規,肝功、腎功、鉀、鈉、氯、鈣無明顯異常。腦電圖檢查:2021-03-06我院腦電圖示多量左側中央區放電,稍多量右側中央區,少許右側頂區放電。2022-05-23我院腦電圖示異常學齡前兒童視頻腦電圖,多灶性放電。2023-04-25我院腦電圖示異常學齡前兒童視頻腦電圖 多灶性放電(左右中顳、額區為著)。2023-08-22外院視頻腦電圖示兒童期異常腦電圖,雙側導聯可見廣泛性棘波、棘慢波發放,雙側額極、額、枕、中顳、后顳、額中線著。2023-09-05我院腦電圖示不正常腦電圖,高度異常(全導慢棘慢波持續發放)。2023-09-06我院腦電圖可見大量全導2 Hz左右中高﹣極高波幅慢尖慢波連續性發放,前頭部明顯;可見少許左側枕﹣中顳區、右側枕區、右側中央區高波幅尖慢波散發或簇發,向臨近導聯擴散。2023-10-20我院腦電圖示醒睡各期可見大量 雙側枕區、顳區、中央區、頂區、額區 同步或不同步中﹣高波幅尖慢波散發、簇發或連續發放。重度異常學齡前兒童視頻腦電圖,大量多灶性放電,以雙側枕區、中額區為著。2023-11-09我院腦電圖可見連續全導2.7~3.5 Hz左右中高﹣極高波幅慢尖慢波發放,雙側枕區明顯,異常學齡前兒童普通腦電圖,無發作期表現,發作間期異常(圖2)。
圖1
患兒腦部MRI
a. 2021-3-1;b. 2023-7-22
圖2
患兒腦電圖
a. 背景活動;b. 發作間期異常腦電圖;c. 發作間期異常腦電圖;d. 發作間期異常腦電圖
全外顯子基因檢測:KCND3 (c.1174G>A ,p.Val392Ile)致病性變異(新發變異),父母均無異常。致病性證據:c.1174G>A(p.Val392lle);根據 ACMG 指南,該變異初步判定為致病性變異(Pathogenic)PP3_Moderate+PS4+PM2_SupportingPSPP3Modere1REVEL 預測結果為有害,SIFT、PolyPhen_2、MutationTaster 、GERP+預測結果分別為良性、有害、有害、有害;PS4文獻數據庫已有該位點(Sudden unexplained death syndrome)的病例報道,變異標簽為 DM(致病突變),ClinVar 數據庫對該位點的致病性分析為 Pathogenic/Likely pathogenic,Spinocerebellar ataxia type 19/22|Brugada syndrome 9|not provided : PM2 Supporting 常人群數據庫中的頻率為﹣:PS2 經家系驗證分析,受檢人之父該位點無變異,受檢人之母該位點無變異,此變異為自發突變。KNCD3蛋白三級結構預測見圖3。
圖3
KNCD3蛋白三級結構預測
討論 Kv4通道是鉀離子通道家族中的一員:分為不同的亞型,包括Kv4.1、Kv4.2和Kv4.3,Kv4是Kvs家族中表達于腦、心臟和平滑肌的一個亞群[5],Kv4.3由KCND3編碼(3),Kv4.3在中樞神經系統中高度表達,尤其在小腦浦肯野細胞、深核、顆粒細胞和中間神經元中表達。KCND3 c.1174G>A 致病性變異(新發變異),KCND3基因突變導致的相關疾病有Brugada綜合征9型,臨床表型 :心ST段抬高,暈厥;脊髓小腦性共濟失調(Spinocerebellarataxia,SCA)19型 ;阿爾茲海默病(Alzheimer's disease,AD ),臨床表型:構音障礙;腱反射減弱,小腦萎縮,肌陣攣,腱反射亢進;吞咽困難,共濟失調等。 KCND3 Val392Ile突變已被報道4例,然而,關于這種突變攜帶者的臨床特征的信息是有限的, Wang等[6]報道患者5歲,發病年齡為1歲6月齡,癲癇發作時表現為四肢僵硬,屈曲,發紺,意識喪失,持續2~3 min,發作1~2次/月,且多在發熱后發作,先后使用奧卡西平和丙戊酸鈉,未見改善,發作時出現另一種癲癇表現,眼球轉動,伴單側或雙側肢體陣攣,在3歲3月齡時加用左乙拉西坦,癲癇發作頻率降低至1次/月,3歲11月齡時出現視力下降,無法精準拿起小物體,癲癇發作前發育指標正常,但發病后認知記憶能力下降,4歲時核磁共振正常,無癲癇或其他神經系統疾病。該案例與本病例均為KCND3從頭自發突變(c.1174G > A, p.Val392Ile),與本案例不同點在于本例患兒發作時無發紺,無視力下降,且發病年齡較早。
KCND3突變還可導致心腦通道病[7],Giudicessi等[8]首次報道KCND3 p. val392ile陽性的心臟驟停病例是一例20歲男性,有暈厥發作史,暈厥發作后心肺驟停后死亡。在死亡前幾天沒有任何明顯的疾病跡象,尿液藥物和毒素檢測呈陰性。死者生前未行心電圖檢查,在青少年時期有過兩次可疑的暈厥發作,家族中無心臟驟停或心律失常相關心臟事件的記錄,據報道死者父母和姐姐的12導聯心電圖檢查均正常[8]。Zhang等[7]報道的一例11歲女性患兒,2 歲時診斷出患有熱性驚厥,從 4 歲開始有規律性熱性驚厥;癲癇發作時意識喪失。通常會持續幾秒鐘,然后突然消失,6歲時診斷智力障礙(韋氏量表評分 < 75)。心電圖可檢測到有效不應期(Effective refractive period,ERP)下導聯 (II、III、AVF) 和側導聯 (V5、V6) 的 J 點升高。24小時動態心電圖監測儀顯示陣發性心房顫動、早發性心房復合體(Premature atrial contraction,PAC)、成對PAC和陣發性房性心動過速(Atrial tachycardia,AT),超聲心動圖結果正常。9歲時,診斷為早期復極綜合癥(Early repolarization syndrome,ERS)。Tadashi Nakajima等[9]報道的KCND3 Val392Ile突變相關的新型心腦通道病,表現出心臟(早期復極化綜合征和陣發性心房顫動)和腦表型(癲癇和智力障礙)。該患者因陣發性心房顫動(Paroxysmal Atrial Fibrillation,PAF)引起心悸,于夜間和清晨發作,持續兩個月,3~9歲發生過幾次局灶性意識損害發作,伴有智力殘疾(Intellectual Disabilities,ID)(WISC-III,FIQ:13歲時64),但無任何局灶性體征或SCA表型。13歲時記錄的腦電圖顯示雙側枕骨區有局灶性峰值。被診斷為病因不明的局灶性癲癇,MRI未見腦結構異常。先證者妹妹,體格檢查和血液檢查沒有發現異常。超聲心動圖顯示無結構性心臟病的跡象,在2~7歲時有過幾次局性意識受損癲癇發作,伴有ID (WISC-III,12歲時FIQ:41),但無任何局灶性體征或SCA表型。14歲時記錄的腦電圖顯示雙側枕區交替出現局灶性峰值,先證者的母親中發現了KCND3 V3921突變,在7歲和40歲時發生過兩次暈厥,先證者的父親未發現KCND3突變。先證者的外祖父和外祖母不攜帶KCND3 V3921突變。因此,這是在先證者的母親身上發生的新生突變。其被診斷為病因不明的局灶性癲癇。MRI未見腦結構異常。
KCND3 Val392Ile突變引起癲癇的機制尚不明確,但與KCND3相似的KCND2突變已被報道與癲癇發作有關,KCND2突變導致KV4.2的升高或降低導致癲癇的發作[2],KCND3突變引起的Kv4.3升高是否會導致癲癇目前尚不清楚。在KCND3 Val392Ile突變的情況下,Kv4.3的增加可能與癲癇有關(9)。同時KCND3是SCA 19/22的致病基因,這是一種常染色體顯性小腦共濟失調,定位于染色體1p21-q23[10-11],SCA 是一組遺傳性神經退行性疾病,其特征是進行性小腦性共濟失調和所有年齡段的可變錐體、錐體外系、大腦或脊髓癥狀,包括帕金森綜合征和癲癇癥狀患者[12-14]。此外,Singh等[13]綜述報道的15例SCA患者中有7例在嬰兒期或兒童期發病,且以癲癇為首發癥狀。
綜上,本文報道了一例KCND3自發性變異(c.1174G > A,p.Val392Ile),以癲癇為主要表現,發病年齡小,無心臟表型,父母無表型,藥物難以控制其發作,其臨床表現的特殊性豐富了該基因突變的臨床表型。
利益沖突聲明 所有作者無利益沖突。