引用本文: 黃小麗, 董尚勝, 潘光毅. 母源KCNQ3基因突變致自限性家族性新生兒癲癇一例. 癲癇雜志, 2024, 10(4): 364-367. doi: 10.7507/2096-0247.202404003 復制
版權信息: ?四川大學華西醫院華西期刊社《癲癇雜志》版權所有,未經授權不得轉載、改編
自限性家族性新生兒癲癇(Self-limited familial neonatal epilepsy,SLFNE),也稱良性家族性新生兒驚厥(Benign familial neonatalconvusions,BFNC),是一種罕見的、常染色體顯性遺傳的原發性癲癇綜合征,外顯率為80%~90%,國外報道發病率為1/10萬,其臨床特點為患兒在出生后2~3 d出現全身性或部分性驚厥發作,一般數周或數月可自行消失,預后良好[1,2]。BFNE是由編碼電壓門控K+通道亞基的KCNQ2/3基因突變所致,與KCNQ2基因相比,迄今為止描述KCNQ3基因變異的病例相對較少[3,4]。本研究對江門市婦幼保健院一例新生兒驚厥患兒進行家系全外顯子檢測,發現KCNQ3基因突變,來源于母親。患兒母親成年后發現癲癇發作,智力正常,有輕度雙相障礙,對拉莫三嗪抗癲癇治療高度敏感,考慮患兒為母源KCNQ3基因突變致自限性家族性新生兒癲癇,給予苯巴比妥治療后驚厥完全控制,生長發育正常,現報道如下。
臨床資料 患兒 男,49天,因“反復抽搐1個月”于江門市婦幼保健院門診就診。患兒母親為孕1產1,胎齡37+1周出生,出生時Apgar評分8分-9分-10分,出生1 min時心率約90次/min,肌力減弱,予正壓通氣后心率、肌力可恢復。出生體重2.57 ㎏,身長46 cm,頭圍:33 cm。出生后測臍血pH值7.031,曾因患兒臍血pH值低在新生兒科住院3天。住院期間患兒哭聲大,反應好,無氣促紫紺,吃奶正常,二便正常。出生后第4天患兒出現發作性紫紺2次,伴有肢體抖動,持續約2~3 min緩解,再次入住新生兒科。入院后患兒仍有反復抽搐,表現為睡眠中醒來,四肢劃動,繼而出現深大呼吸,雙目凝視,紫紺,雙上肢抖動,或下肢抽動,最后出現四肢不對稱強直,約3 min左右緩解,緩解后入睡。發作間期患兒反應好,吃奶好,無氣促,四肢肌張力、肌力正常。入院后完善相關檢查,給予低流量吸氧、苯巴比妥抗驚厥等對癥治療,抽搐逐漸減少,無氣促紫紺,吃奶好,予帶藥出院。出院后患兒一直口服苯巴比妥無發作(7.5 mg/次, 每日兩次)。父母否認近親婚配,患兒母親既往有癲癇病史,約25歲曾出現抽搐發作,表現為意識不清,肢體強直,發作頻次約1~2次/月,腦電圖(Electroencephalography,EEG)及核磁共振(Magnetic resonance imaging,MRI)無異常,予拉莫三嗪后無發作,妊娠前停藥,2023年7月患兒母親妊娠2個月再次抽搐發作,發作前無異常,發作時突然意識不清,肢體強直樣表現,持續1~2 min緩解,重新服用拉莫三嗪治療,規律服藥后發作控制。體格檢查:頭圍36 cm,神清,精神反應好,無特殊面容,心肺腹查體無特殊,生長發育正常,四肢肌張力、肌力正常。
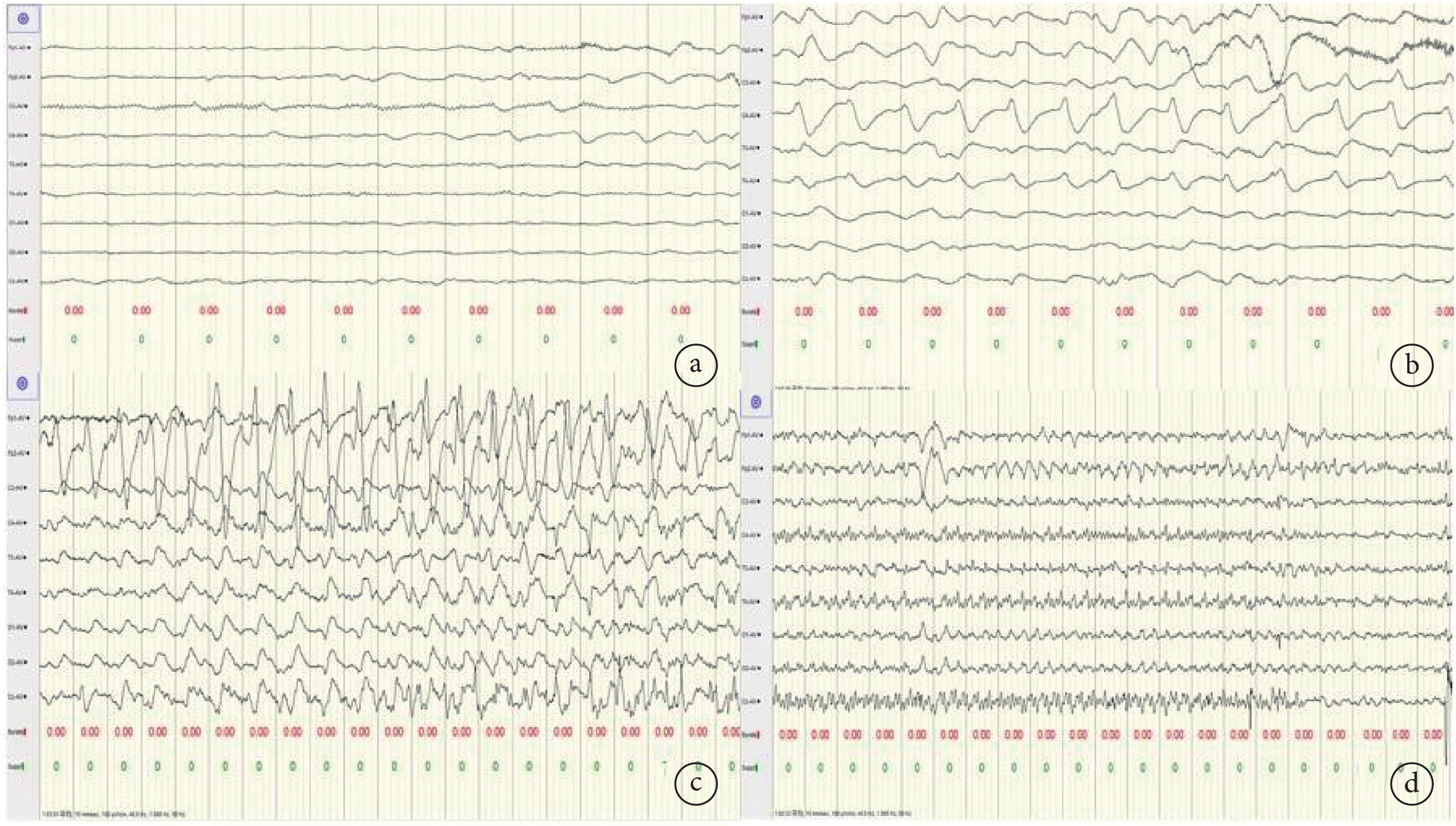
輔助檢查:入院后查血氣分析+生化九項:pH值7.47,二氧化碳分壓26.90 mmHg,氧分壓108.30 mmHg,實際碳酸氫鹽19.00 mmol/L,鈉離子142.20 mmol/L,鉀離子3.90 mmol/L,鈣離子1.30 mmol/L,葡萄糖3.10 mmol/L,乳酸1.80 mmol/L。血氨 54 umol/L。肝功能八項:總膽紅素241.20 umol/L,直接膽紅素14.00 umol/L,間接膽紅素227.20 umol/L。血常規、C-反應蛋白、降鈣素原(Procalcitonin,PCT)、心腎功能、腦脊液常規、腦脊液生化、腦脊液培養、二便常規、痰培養均無異常。顱腦MRI平掃:腦成熟髓鞘形成水平相當于足月新生兒;顱腦MRI平掃未見明顯異常。胸片無異常。心臟彩超:卵圓孔未閉。2024-2-5 aEEG(圖1)示:重度異常新生兒腦電圖,① 生理波少,發育延遲;② 左側中央區少量尖(慢)波;③ 8次局灶性發作,其中6次為左側中央區起始,2次為右側中央區起始),發作期圖形見圖2。2024-2-8 aEEG示:重度異常腦電圖,① 生理波少,發育延遲;② 右側中央區少量尖(慢)波;③ 兩次驚厥發作。2024-2-13復查aEEG示:大致正常腦電圖。家系全外顯子檢測:KCNQ3,NM_004519.4:c.1018G>C:p.G340R,雜合突變,父親為野生型,母親為雜合突變,詳見圖3。
 圖1
患兒2024-2-5 aEEG
圖1
患兒2024-2-5 aEEG
振幅:AS期(6~23uV),QS期(5~40uV),睡眠-覺醒周期(SWC):可見10個周期,8次臨床發作(黑色剪頭)
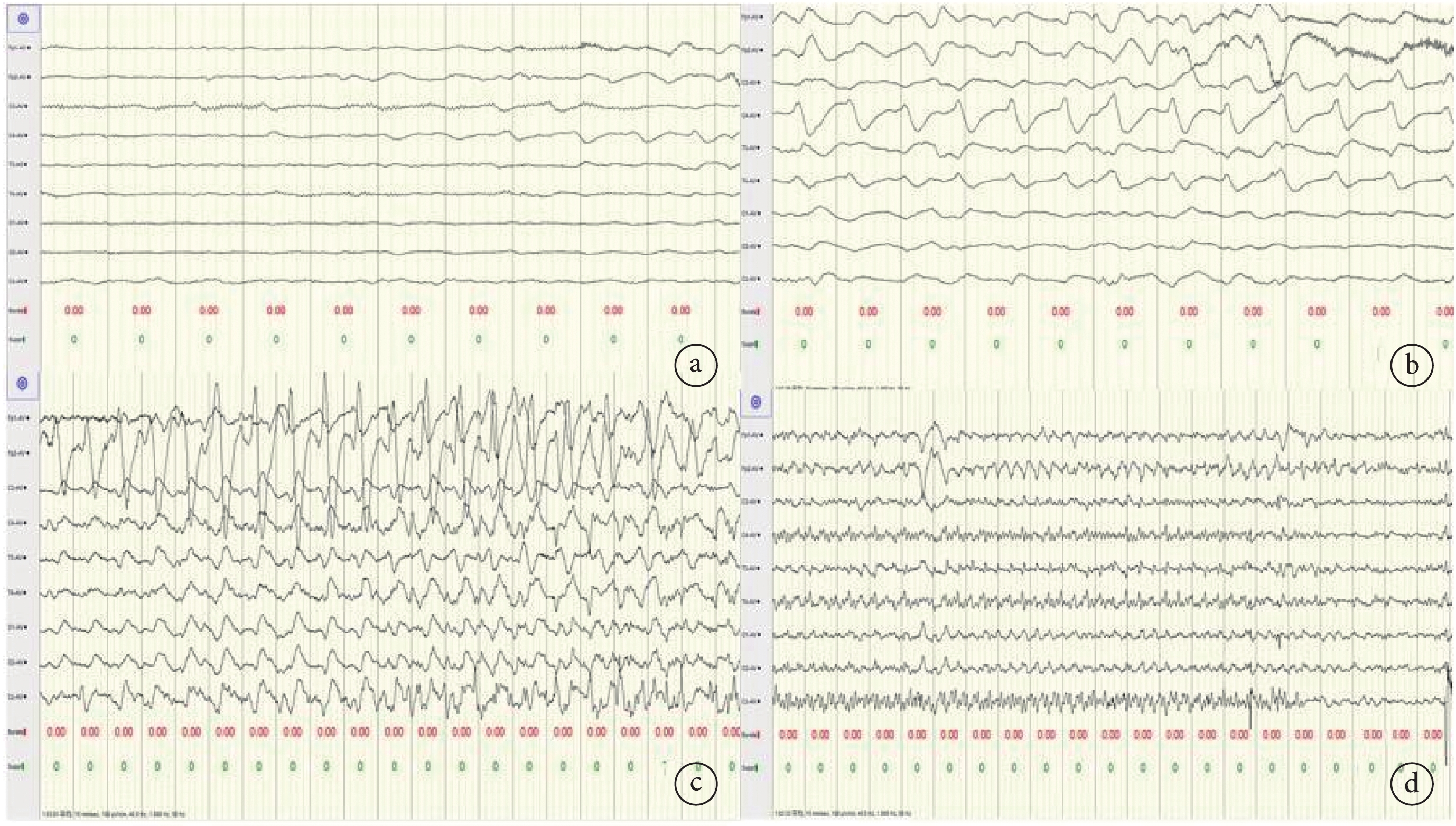 圖2
發作期腦電圖演變過程
圖2
發作期腦電圖演變過程
a為發作開始中央區起源(6次左側中央區起源,2次右側中央區起源) b廣泛性慢波 頻率逐漸增快,演變為c、d
 圖3
KCNQ3蛋白第340位甘氨酸保守性評估
圖3
KCNQ3蛋白第340位甘氨酸保守性評估
討論 KCNQ3相關疾病包括SLFNE、自限性家族性嬰兒癲癇(Self-limited familial epilepsy,SLFIE)及KCNQ3相關的神經發育障礙(Neurodevelopmental disorder,NDD)[5]。在KCNQ3-SLFNE中,新生兒于出生后2~8 d出現癲癇發作,大部分于1歲內癲癇發作自然消失。癲癇發作時間短暫但呈叢集樣發作,持續1~2 min不等,發作類型包括強直發作、局灶性陣攣發作,伴或不伴自主神經改變,運動發作可能局限于身體的一個部位,或遷移到其他部位,或泛化[6]。在KCNQ3-SLFIE中,癲癇發作在1月齡~1歲內出現,1~2歲后消失。癲癇發作一般短暫,持續2 min,每天集群形式發作,發作類型大部分為局灶性發作,少部分為全面性發作,表現為彌漫性肌張力強直,伴四肢抽搐、頭部偏斜,或運動停止伴意識喪失和發紺[7]。在KCNQ3-NDD中,患者表現為智力障礙伴自閉癥等全面發育障礙表現,可伴/不伴癲癇發作及皮質視覺障礙[4,8,9]。SCN2A及PRRT2基因與良性家族性新生兒嬰兒驚厥相關,與部分KCNQ3-BFNE表現相重疊。3月齡前起病的SCN2A相關癲癇多為功能獲得型(Gain-of-function,GOF)為主,KCNQ2、KCNQ3相關的BFNE多為輕度功能喪失型(Loss-of-function,LOF),兩者對鈉通道阻滯劑(Sodium channel blockers,SCB)治療都高度敏感,如卡馬西平、奧卡西平和拉莫三嗪等[10,11]。新生兒驚厥是新生兒神經系統疾病中最常見疾病之一,目前尚無明確的用藥指南。國外一項大樣本研究結果顯示,苯巴比妥為新生兒驚厥一線用藥[12]。國內學者也有報道,苯巴比妥治療KCNQ2基因突變致新生兒驚厥有一定療效[13]。本例患兒新生兒期起病,在未明確具體病因之前,首選苯巴比妥治療。明確為KCNQ3-BFNE后,若苯巴比妥治療效果不理想,或僅部分控制,可換用鈉通道阻滯劑。
根據美國醫學遺傳學與基因組學學會指南,KCNQ3基因c.1018G>C(p.G340R)為可能致病性變異。頻率證據PM2方面,該變異在千人基因組(1 000G)、人類外顯子數據庫(ExAC)和人群基因組突變頻率數據庫(gnomAD)中等多個人群頻率庫中頻率為0。
KCNQ3基因p.G340V在良性新生驚厥中有報道[14],符合PM5證據。該變異在多種哺乳動物之間均為高度保守(圖3),有害性預測軟件REVEL為有害0.987分,SIFT為有害0分,CADD為有害32分,符合PP3條件。患兒臨床表型高度符合自限性家族性新生兒癲癇,母親成年癲癇并對鈉離子阻滯劑高度敏感,符合KCNQ3相關癲癇表現,符合PP4條件,但關于成人KCNQ3相關癲癇暫未見文獻報告,或關于良性新生兒驚厥的患兒成年后是否仍有癲癇發作方面還未有相關文獻報道。有研究提示KCNQ2的S6節段內高度保守的殘基Gly-301(相關KCNQ3的Gly-340位點)對KCNQ通道門控至關重要[15],也支持本位點變異為致病性變異證據。
KCNQ3基因編碼鉀離子電壓門控通道超家族亞基Kv7.3,位于神經細胞軸突后段,在大腦皮層、海馬、尾狀核、杏仁核和丘腦中廣泛表達,主要與KCNQ2基因編碼的同源Kv7.2組裝成M通道,M通道在保持細胞膜靜息電位的穩定性中發揮重要作用,并調節神經元興奮性[16,17]。KCNQ3基因功能喪失性變異通常會改變鉀離子通道的門控作用、運輸及亞細胞定位功能,鉀通道功能喪失性變異可致神經興奮性增高。KCNQ3變異體(c.719T>G;p.M240R)倉鼠細胞中的細胞膜片鉗記錄顯示,M240R的Kv7.3通道無功能改變,而包含M240R突變亞基的Kv7.3與Kv7.2結合的異聚通道顯示激活門控中約10 mV的去極化位移,與體外LOF效應一致[18]。KCNQ鉀通道與電壓門控鈉通道共同定位于神經元細胞膜,二者相互作用并調節[19],因此鉀通道功能障礙者可能對SCB有反應[20]。奧卡西平通過靶向鈉通道阻斷鈉離子在動作電位過程中的移動從而防止癲癇活動的發生[21],拉考沙胺通過增強鈉通道的慢失活起到抗癲癇作用[22]。因此,對于GOF鈉通道變異和LOF鉀通道變異的患者,SCB是對發病機制的一種精準治療[23]。
GOF KCNQ3亞基改變導致神經發育性障礙相關表型及LOF改變造成新生兒期的良性驚厥的機制不明確,大部分研究基于KCNQ2和KCNQ3形成異聚體(KCNQ2/3)模型進行探討。部分研究示,在癲癇性腦病和自閉癥譜系障礙患者中發現了多個KCNQ3的GOF變異。Kv7.3亞基包含6個跨膜片段(S1-S6)和胞質N端和C端,在S1-S4電壓傳感域中,S4跨膜段包含一系列帶正電荷的精氨酸殘基,這些殘基使該通道能夠隨著膜電位的變化而進行開合[18]。KCNQ3基因R227和R230是S4段最外的2個精氨酸,使其產生的鉀離子電流減少,減慢細胞的復極化或產生復極化不完全,導致細胞的興奮性增加或動作電位的自發放。Tristan等[9]對11例神經發育障礙患者進行研究,發現KCNQ3基因多個個體出現新生雜合子變異R230(R230C、R230H、R230S)和R227(R227Q),所有患者在出生2年內都表現出全面發育遲緩,大多數(8/11,73%)表現為語言障礙,所有患者都有自閉癥特征,5例(5/11,45%)被診斷為自閉癥譜系障礙,11例中有8例(73%)經常出現睡眠激活的多灶性癲癇樣放電,大多數患者(9/11,82%)未出現癲癇發作,在新生兒期也無患者出現癲癇發作,突變體KCNQ3通道的電壓鉗記錄顯示這些變異為GOF效應。多個研究示KCNQ3的P574位點異常與自閉癥一定相關[24-26],其機制與Kv7.3/Kv7.2異聚體模型有明顯的區別。Miceli等[26]在3例無驚厥史的自閉癥譜系障礙兒童個體中發現c.1720C>T(p.P574S)核苷酸變化,p.P574S的Kv7.3變異與Kv7.5共同表達時,顯著降低了非洲爪蛙卵母細胞中鉀電流的幅度,但與Kv7.2或Kv7.4無電流改變,異聚體Kv7.3/Kv7.5通道的功能障礙與某些形式的自閉癥譜系障礙、癲癇和可能的其他精神疾病的發病機制有關。
大部分KCNQ2或KCNQ3相關BFNE兒童的癲癇發作可通過常規抗癲癇發作藥物(尤其鈉通道阻滯劑)控制[27]。相反,對于KCNQ2或KCNQ3造成的嚴重發育性癲癇腦病,還未有很好的抗癲癇方案[28],依佐加濱(ezogabine)及瑞替加賓(retigabine)是一種選擇性Kv7通道激活劑,已被證明對功能喪失性Kv7.2變異患者的癲癇發作控制及發育起積極作用[29,30],但由于其不利的風險/效益比限制了其臨床應用。在非藥物療法中,生酮飲食被證明對KCNQ2變異引起的神經發育障礙兒童特別有效,但作用機制尚未完全了解,也暫無數據表明對KCNQ3相關疾病有影響[31]。
利益沖突聲明 所有作者無利益沖突。
自限性家族性新生兒癲癇(Self-limited familial neonatal epilepsy,SLFNE),也稱良性家族性新生兒驚厥(Benign familial neonatalconvusions,BFNC),是一種罕見的、常染色體顯性遺傳的原發性癲癇綜合征,外顯率為80%~90%,國外報道發病率為1/10萬,其臨床特點為患兒在出生后2~3 d出現全身性或部分性驚厥發作,一般數周或數月可自行消失,預后良好[1,2]。BFNE是由編碼電壓門控K+通道亞基的KCNQ2/3基因突變所致,與KCNQ2基因相比,迄今為止描述KCNQ3基因變異的病例相對較少[3,4]。本研究對江門市婦幼保健院一例新生兒驚厥患兒進行家系全外顯子檢測,發現KCNQ3基因突變,來源于母親。患兒母親成年后發現癲癇發作,智力正常,有輕度雙相障礙,對拉莫三嗪抗癲癇治療高度敏感,考慮患兒為母源KCNQ3基因突變致自限性家族性新生兒癲癇,給予苯巴比妥治療后驚厥完全控制,生長發育正常,現報道如下。
臨床資料 患兒 男,49天,因“反復抽搐1個月”于江門市婦幼保健院門診就診。患兒母親為孕1產1,胎齡37+1周出生,出生時Apgar評分8分-9分-10分,出生1 min時心率約90次/min,肌力減弱,予正壓通氣后心率、肌力可恢復。出生體重2.57 ㎏,身長46 cm,頭圍:33 cm。出生后測臍血pH值7.031,曾因患兒臍血pH值低在新生兒科住院3天。住院期間患兒哭聲大,反應好,無氣促紫紺,吃奶正常,二便正常。出生后第4天患兒出現發作性紫紺2次,伴有肢體抖動,持續約2~3 min緩解,再次入住新生兒科。入院后患兒仍有反復抽搐,表現為睡眠中醒來,四肢劃動,繼而出現深大呼吸,雙目凝視,紫紺,雙上肢抖動,或下肢抽動,最后出現四肢不對稱強直,約3 min左右緩解,緩解后入睡。發作間期患兒反應好,吃奶好,無氣促,四肢肌張力、肌力正常。入院后完善相關檢查,給予低流量吸氧、苯巴比妥抗驚厥等對癥治療,抽搐逐漸減少,無氣促紫紺,吃奶好,予帶藥出院。出院后患兒一直口服苯巴比妥無發作(7.5 mg/次, 每日兩次)。父母否認近親婚配,患兒母親既往有癲癇病史,約25歲曾出現抽搐發作,表現為意識不清,肢體強直,發作頻次約1~2次/月,腦電圖(Electroencephalography,EEG)及核磁共振(Magnetic resonance imaging,MRI)無異常,予拉莫三嗪后無發作,妊娠前停藥,2023年7月患兒母親妊娠2個月再次抽搐發作,發作前無異常,發作時突然意識不清,肢體強直樣表現,持續1~2 min緩解,重新服用拉莫三嗪治療,規律服藥后發作控制。體格檢查:頭圍36 cm,神清,精神反應好,無特殊面容,心肺腹查體無特殊,生長發育正常,四肢肌張力、肌力正常。
輔助檢查:入院后查血氣分析+生化九項:pH值7.47,二氧化碳分壓26.90 mmHg,氧分壓108.30 mmHg,實際碳酸氫鹽19.00 mmol/L,鈉離子142.20 mmol/L,鉀離子3.90 mmol/L,鈣離子1.30 mmol/L,葡萄糖3.10 mmol/L,乳酸1.80 mmol/L。血氨 54 umol/L。肝功能八項:總膽紅素241.20 umol/L,直接膽紅素14.00 umol/L,間接膽紅素227.20 umol/L。血常規、C-反應蛋白、降鈣素原(Procalcitonin,PCT)、心腎功能、腦脊液常規、腦脊液生化、腦脊液培養、二便常規、痰培養均無異常。顱腦MRI平掃:腦成熟髓鞘形成水平相當于足月新生兒;顱腦MRI平掃未見明顯異常。胸片無異常。心臟彩超:卵圓孔未閉。2024-2-5 aEEG(圖1)示:重度異常新生兒腦電圖,① 生理波少,發育延遲;② 左側中央區少量尖(慢)波;③ 8次局灶性發作,其中6次為左側中央區起始,2次為右側中央區起始),發作期圖形見圖2。2024-2-8 aEEG示:重度異常腦電圖,① 生理波少,發育延遲;② 右側中央區少量尖(慢)波;③ 兩次驚厥發作。2024-2-13復查aEEG示:大致正常腦電圖。家系全外顯子檢測:KCNQ3,NM_004519.4:c.1018G>C:p.G340R,雜合突變,父親為野生型,母親為雜合突變,詳見圖3。
圖1
患兒2024-2-5 aEEG
振幅:AS期(6~23uV),QS期(5~40uV),睡眠-覺醒周期(SWC):可見10個周期,8次臨床發作(黑色剪頭)
圖2
發作期腦電圖演變過程
a為發作開始中央區起源(6次左側中央區起源,2次右側中央區起源) b廣泛性慢波 頻率逐漸增快,演變為c、d
圖3
KCNQ3蛋白第340位甘氨酸保守性評估
討論 KCNQ3相關疾病包括SLFNE、自限性家族性嬰兒癲癇(Self-limited familial epilepsy,SLFIE)及KCNQ3相關的神經發育障礙(Neurodevelopmental disorder,NDD)[5]。在KCNQ3-SLFNE中,新生兒于出生后2~8 d出現癲癇發作,大部分于1歲內癲癇發作自然消失。癲癇發作時間短暫但呈叢集樣發作,持續1~2 min不等,發作類型包括強直發作、局灶性陣攣發作,伴或不伴自主神經改變,運動發作可能局限于身體的一個部位,或遷移到其他部位,或泛化[6]。在KCNQ3-SLFIE中,癲癇發作在1月齡~1歲內出現,1~2歲后消失。癲癇發作一般短暫,持續2 min,每天集群形式發作,發作類型大部分為局灶性發作,少部分為全面性發作,表現為彌漫性肌張力強直,伴四肢抽搐、頭部偏斜,或運動停止伴意識喪失和發紺[7]。在KCNQ3-NDD中,患者表現為智力障礙伴自閉癥等全面發育障礙表現,可伴/不伴癲癇發作及皮質視覺障礙[4,8,9]。SCN2A及PRRT2基因與良性家族性新生兒嬰兒驚厥相關,與部分KCNQ3-BFNE表現相重疊。3月齡前起病的SCN2A相關癲癇多為功能獲得型(Gain-of-function,GOF)為主,KCNQ2、KCNQ3相關的BFNE多為輕度功能喪失型(Loss-of-function,LOF),兩者對鈉通道阻滯劑(Sodium channel blockers,SCB)治療都高度敏感,如卡馬西平、奧卡西平和拉莫三嗪等[10,11]。新生兒驚厥是新生兒神經系統疾病中最常見疾病之一,目前尚無明確的用藥指南。國外一項大樣本研究結果顯示,苯巴比妥為新生兒驚厥一線用藥[12]。國內學者也有報道,苯巴比妥治療KCNQ2基因突變致新生兒驚厥有一定療效[13]。本例患兒新生兒期起病,在未明確具體病因之前,首選苯巴比妥治療。明確為KCNQ3-BFNE后,若苯巴比妥治療效果不理想,或僅部分控制,可換用鈉通道阻滯劑。
根據美國醫學遺傳學與基因組學學會指南,KCNQ3基因c.1018G>C(p.G340R)為可能致病性變異。頻率證據PM2方面,該變異在千人基因組(1 000G)、人類外顯子數據庫(ExAC)和人群基因組突變頻率數據庫(gnomAD)中等多個人群頻率庫中頻率為0。
KCNQ3基因p.G340V在良性新生驚厥中有報道[14],符合PM5證據。該變異在多種哺乳動物之間均為高度保守(圖3),有害性預測軟件REVEL為有害0.987分,SIFT為有害0分,CADD為有害32分,符合PP3條件。患兒臨床表型高度符合自限性家族性新生兒癲癇,母親成年癲癇并對鈉離子阻滯劑高度敏感,符合KCNQ3相關癲癇表現,符合PP4條件,但關于成人KCNQ3相關癲癇暫未見文獻報告,或關于良性新生兒驚厥的患兒成年后是否仍有癲癇發作方面還未有相關文獻報道。有研究提示KCNQ2的S6節段內高度保守的殘基Gly-301(相關KCNQ3的Gly-340位點)對KCNQ通道門控至關重要[15],也支持本位點變異為致病性變異證據。
KCNQ3基因編碼鉀離子電壓門控通道超家族亞基Kv7.3,位于神經細胞軸突后段,在大腦皮層、海馬、尾狀核、杏仁核和丘腦中廣泛表達,主要與KCNQ2基因編碼的同源Kv7.2組裝成M通道,M通道在保持細胞膜靜息電位的穩定性中發揮重要作用,并調節神經元興奮性[16,17]。KCNQ3基因功能喪失性變異通常會改變鉀離子通道的門控作用、運輸及亞細胞定位功能,鉀通道功能喪失性變異可致神經興奮性增高。KCNQ3變異體(c.719T>G;p.M240R)倉鼠細胞中的細胞膜片鉗記錄顯示,M240R的Kv7.3通道無功能改變,而包含M240R突變亞基的Kv7.3與Kv7.2結合的異聚通道顯示激活門控中約10 mV的去極化位移,與體外LOF效應一致[18]。KCNQ鉀通道與電壓門控鈉通道共同定位于神經元細胞膜,二者相互作用并調節[19],因此鉀通道功能障礙者可能對SCB有反應[20]。奧卡西平通過靶向鈉通道阻斷鈉離子在動作電位過程中的移動從而防止癲癇活動的發生[21],拉考沙胺通過增強鈉通道的慢失活起到抗癲癇作用[22]。因此,對于GOF鈉通道變異和LOF鉀通道變異的患者,SCB是對發病機制的一種精準治療[23]。
GOF KCNQ3亞基改變導致神經發育性障礙相關表型及LOF改變造成新生兒期的良性驚厥的機制不明確,大部分研究基于KCNQ2和KCNQ3形成異聚體(KCNQ2/3)模型進行探討。部分研究示,在癲癇性腦病和自閉癥譜系障礙患者中發現了多個KCNQ3的GOF變異。Kv7.3亞基包含6個跨膜片段(S1-S6)和胞質N端和C端,在S1-S4電壓傳感域中,S4跨膜段包含一系列帶正電荷的精氨酸殘基,這些殘基使該通道能夠隨著膜電位的變化而進行開合[18]。KCNQ3基因R227和R230是S4段最外的2個精氨酸,使其產生的鉀離子電流減少,減慢細胞的復極化或產生復極化不完全,導致細胞的興奮性增加或動作電位的自發放。Tristan等[9]對11例神經發育障礙患者進行研究,發現KCNQ3基因多個個體出現新生雜合子變異R230(R230C、R230H、R230S)和R227(R227Q),所有患者在出生2年內都表現出全面發育遲緩,大多數(8/11,73%)表現為語言障礙,所有患者都有自閉癥特征,5例(5/11,45%)被診斷為自閉癥譜系障礙,11例中有8例(73%)經常出現睡眠激活的多灶性癲癇樣放電,大多數患者(9/11,82%)未出現癲癇發作,在新生兒期也無患者出現癲癇發作,突變體KCNQ3通道的電壓鉗記錄顯示這些變異為GOF效應。多個研究示KCNQ3的P574位點異常與自閉癥一定相關[24-26],其機制與Kv7.3/Kv7.2異聚體模型有明顯的區別。Miceli等[26]在3例無驚厥史的自閉癥譜系障礙兒童個體中發現c.1720C>T(p.P574S)核苷酸變化,p.P574S的Kv7.3變異與Kv7.5共同表達時,顯著降低了非洲爪蛙卵母細胞中鉀電流的幅度,但與Kv7.2或Kv7.4無電流改變,異聚體Kv7.3/Kv7.5通道的功能障礙與某些形式的自閉癥譜系障礙、癲癇和可能的其他精神疾病的發病機制有關。
大部分KCNQ2或KCNQ3相關BFNE兒童的癲癇發作可通過常規抗癲癇發作藥物(尤其鈉通道阻滯劑)控制[27]。相反,對于KCNQ2或KCNQ3造成的嚴重發育性癲癇腦病,還未有很好的抗癲癇方案[28],依佐加濱(ezogabine)及瑞替加賓(retigabine)是一種選擇性Kv7通道激活劑,已被證明對功能喪失性Kv7.2變異患者的癲癇發作控制及發育起積極作用[29,30],但由于其不利的風險/效益比限制了其臨床應用。在非藥物療法中,生酮飲食被證明對KCNQ2變異引起的神經發育障礙兒童特別有效,但作用機制尚未完全了解,也暫無數據表明對KCNQ3相關疾病有影響[31]。
利益沖突聲明 所有作者無利益沖突。