引用本文: 中國抗癲癇協會創新與轉化專業委員會, 中華醫學會兒科學分會罕見病學組, 中華醫學會兒科學分會神經學組. 類細胞周期蛋白依賴性蛋白激酶5缺乏癥診斷與治療的中國專家共識. 癲癇雜志, 2024, 10(6): 467-477. doi: 10.7507/2096-0247.202408008 復制
版權信息: ?四川大學華西醫院華西期刊社《癲癇雜志》版權所有,未經授權不得轉載、改編
類細胞周期蛋白依賴性蛋白激酶5(cyclin-dependent kinase like 5,CDKL5,亦稱為細胞周期蛋白依賴性激酶樣5)缺乏癥(CDKL5 deficiency disorder,CDD)是一種嬰兒期起病的罕見X-連鎖顯性遺傳病,表現為難以控制的癲癇發作、嚴重全面性發育遲緩以及視覺、睡眠、消化等多方面障礙[1,2],嚴重影響患者及其家庭的生活質量。CDD表型復雜多樣,需結合臨床表現和基因檢測結果進行診斷,并與其他常見發育性癲癇性腦病(developmental and epileptic encephalopathy,DEE)鑒別[3]。目前尚無針對CDD病因的治療方法,臨床以綜合管理及對癥治療為主[4]。近年歐美地區專家先后發布《國際共識:CDKL5缺乏癥患者的評估和管理建議》[1]以及《為CDKL5缺乏癥患者提供高質量照護:歐洲專家小組關于患者病程的意見》[4],為CDD的診療提供了初步參考,但目前仍缺乏符合我國國情的規范化指導。中國抗癲癇協會創新與轉化專業委員會、中華醫學會兒科學分會神經學組及中華醫學會兒科學分會罕見病學組共同組織領域內專家,結合權威共識推薦、現有研究證據及臨床實踐經驗,制定了《類細胞周期蛋白依賴性蛋白激酶5缺乏癥診斷與治療的中國專家共識》(后簡稱“本共識”),旨在為我國CDD的診斷及管理提供具體建議和指導。
1 共識制定方法
本共識編寫組由我國兒童神經及罕見病領域資深專家組成,具有廣泛代表性。編寫組首先就共識的目的、主題范圍、框架達成一致,內容涉及CDD的疾病概述、篩查診斷與臨床管理。檢索數據庫包括MEDLINE(PubMed)、Embase、The Cochrane Library、中國知網數據庫、萬方知識數據服務平臺等。納入文章類型包括指南、共識、薈萃分析、隨機對照臨床試驗、非隨機對照臨床試驗、隊列研究、病例報道等。檢索詞包括“CDKL5缺乏癥/CDKL5 deficiency disorder/CDD”,“流行病學/epidemiology”,“疾病機制/pathology”,“臨床表現/clinical presentation”,“診斷/diagnosis”,“基因檢測/genetic testing”,“抗癲癇發作藥物/anti-seizure medications/ASMs”,“生酮飲食/ketogenic diet”,“癲癇外科手術/epilepsy surgery” 等。檢索語言限定為中文或英文,檢索時間截至2024年6月15日,納入標準為與CDD或兒童難治性癲癇治療相關且可獲取全文。由于罕見病領域文獻有限,除科研數據庫以外,編寫組亦檢索了權威機構平臺,包括國際CDKL5研究基金會[5]、CDKL5 UK[6]、中國CDKL5互助聯盟[7]等。
因探討CDD治療的研究有限,制定抗癲癇發作治療推薦意見時,除臨床數據外,編寫組亦借鑒了國際抗癲癇聯盟(International League Against Epilepsy,ILAE)、中國抗癲癇協會(China Association Against Epilepsy,CAAE)、中華醫學會(Chinese Medical Association,CMA)、美國神經病學學會(American Academy of Neurology,AAN)、美國癲癇協會(American Epilepsy Society,AES)、英國國家衛生與臨床優化研究所(National Institute for Health and Care Excellence,NICE)等權威機構就兒科的抗癲癇治療作出的推薦意見,并參考了編寫組專家治療兒童癲癇的豐富臨床經驗。編寫組根據資料檢索結果撰寫共識初稿,經深入討論、反復推敲對關鍵問題形成8條推薦意見,并依據英國牛津大學循證醫學中心的證據分級和推薦標準(表1)[8]對其進行分級。
隨后,編寫組進行了線上德爾菲調查,將德爾菲問卷和證據綜述發送給支持《共識》編寫的專家組成員,專家組成員結合文獻證據及臨床經驗填寫問卷。德爾菲問卷的每個問題結果分5級,包括完全同意、同意、不確定、不同意及完全不同意,每個問題后專家均可填寫補充意見。選擇強烈同意及同意的專家比例>75%表示就該條目達成共識;如存在未達成共識的條目,編寫組將根據反饋意見修改相關推薦意見后再次進行調查。首輪調查于2024年7月12日進行,共收回有效問卷41份,專家組對所有推薦意見均達成共識,共識率為82.9%~100%,故未再次進行調查。
2 疾病概述
2.1 流行病學
CDD在新生兒中的發病率為1:60 000~1:40 000,在女性中更為常見,與男性發病率比值為4:1~12:1[1]。盡管CDD相對罕見,但早發性DEE患者中伴CDKL5變異的比例達4.4%~37.5% [9-13]。我國多項研究亦顯示CDKL5是嬰兒期起病DEE患者中常見的致病基因[13-15]。
CDD嚴重影響患者的生活質量,雖然目前尚無高質量研究給出CDD患者的預期壽命[16],但臨床不乏在兒童或青少年時期死亡病例報道[5,16]。CDD致殘率高,許多患者終身無法達到普通兒童早期發展的發育里程碑,如獨坐、獨站等[3,17]。CDD患者預后與其疾病嚴重程度、臨床管理(尤其是癲癇發作控制)及照護條件等多種因素相關。研究顯示,與獨坐及獨立行走相關的因素包括有利的基因型、癲癇發作起病較晚、1歲內接受相應治療等[18]。一項在嬰兒癲癇性痙攣綜合征(infantile epileptic spasms syndrome,IESS)患者中進行的試驗顯示,起病至開始給與治療的間期短、對治療的早期應答,與較好的發育及癲癇發作預后相關[19]。同時,有研究顯示,相較于其他病因所致的IESS患者,CDD患者自起病至首次接受治療的間隔往往較長[20]。因此,臨床需及早診斷并治療CDD,以期改善患者預后。
2.2 發病機制
CDKL5是1998年Montini等[21]應用外顯子捕獲技術在Xp22區域克隆到的一個新基因,于2003年首次在伴智力障礙的女性IESS患者中發現該基因變異[22-24]。CDKL5有多種經選擇性剪接產生的異構體,有些廣泛表達,有些則主要在大腦中表達[16,21,25,26]。在mRNA層面,CDKL5在大腦皮層、海馬體、紋狀體和嗅球等結構中含量最高,在包括谷氨酸能神經元和γ -氨基丁酸(γ-aminobutyric acid,GABA)能神經元在內的前腦神經元中含量尤其豐富[16]。CDKL5水平在產前最低,在神經系統快速發育的圍產期和產后水平最高(尤其是在大腦皮層和海馬體),對神經元的形成和成熟極為重要[27]。研究已證實,CDKL5參與神經元的遷移、形成和生長,突觸的發育和正常功能的行使[27],對維持正常的興奮-抑制平衡和記憶至關重要[28]。缺乏CDKL5會導致樹突分支減少、神經元回路連接受損等多種問題,嚴重影響神經系統的發育及正常功能[16]。研究發現敲除前腦谷氨酸能神經元中的Cdkl5基因可導致小鼠自發性癲癇發作[29,30]。這些研究將有助于構建CDD動物模型、深入了解其病理機制以及研發臨床治療藥物。
2.3 臨床表現
CDD是一種以癲癇發作為主要表現的多系統受累疾病,起病早,癥狀重,表型復雜多樣,男性患者往往比女性患者病情更為嚴重[31]。國外數據顯示,CDD中位起病年齡為6周,90%的患者在3月齡內起病[31]。我國病例報道中,CDD患者多于出生后1~12個月起病[32-38],就診年齡多在3月齡~3歲之間[34,38]。
2.3.1 癲癇發作
ILAE將CDD患者的癲癇綜合征歸為病因特異性發育性癲癇性腦病(CDKL5-DEE)[39],其癲癇通常在嬰兒期起病,發作頻繁(多數患者每日均有發作),且難以控制[3,31]。癲癇發作類型隨年齡增長而變化,在約25%的個體中,癲癇性痙攣發作是初始發作類型,其他發作類型包括強直、局灶、肌陣攣、全面性強直-陣攣及序貫發作[3]。根據癲癇發作類型及腦電圖特點,臨床上將CDD相關癲癇分為3期,包括嬰兒期起病的運動性發作(Ⅰ期,1~10周)、伴癲癇性痙攣發作的癲癇性腦病(Ⅱ期,6~36月齡)以及難治性癲癇(Ⅲ期,2~11歲)(表2)[40-42]。不同分期癲癇發作間期的腦電圖存在差異。I期發作間期腦電圖可能正常,或表現出彌漫性慢波或局灶性癲癇樣放電[1,3]。隨著疾病發展,Ⅱ、Ⅲ期患者在發作間期腦電圖多表現為高度失律以及彌漫性慢波伴多灶癲癇樣放電[41]。
過度運動-強直-痙攣序貫發作是CDD特征性的癲癇發作類型,見于多數病例。其第一階段以過度運動起始,伴有搖擺、蹬踢和發聲,持續10~60 s。隨后是強直期,四肢伸展或是上肢伸直及下肢屈曲,持續20~45 s。繼而演變為痙攣發作,持續 1~15 min。另外,部分CDD患者會經歷無發作“蜜月期”,即癲癇發作減少或消失一段時間,通常發生在2月齡~11歲(中位年齡為2歲),持續時間為2.5個月~6年(中位時間為6個月)[41]。值得注意的是,雖然難治性癲癇是CDD最常見的臨床表現之一,但臨床上亦觀察到極少數(<1%)CDD患者無癲癇發作[43-45]。
2.3.2 嚴重全面性發育遲緩、腦性視覺損傷及其他共患病
盡管CDD患者達到同一發育里程碑的時間存在較大差異,但所有患者均存在嚴重的全面性發育遲緩,包括運動、溝通(言語和語言)以及認知等多方面發育落后[3,31,46],約60%的患者能獨坐,20%左右的患者可獨行,40%~70%的患者無法抓取及持有物體[3]。許多患者存在異常動作,包括刻板動作(如將手反復放入口中、手臂拍動或揮動、交叉雙腿等)、舞蹈癥等[3]。約50%的患者在6歲時可咿呀學語,僅約25%的患者在7歲時可說出單個單詞[31]。
值得注意的是,約80%的CDD患者存在腦性視覺損傷,其表現為異常眼動,包括內斜視、外斜視、水平性或旋轉性眼震等,以及對強光的異常注視和反應等,常伴有視線接觸不良和視覺追蹤障礙[3,31,42,47-49]。此外,約90%的CDD患者存在睡眠障礙,主要體現為無法維持正常睡眠狀態,如連續幾晚缺乏睡眠及后續的過度睡眠,睡眠障礙通常終身存在[3,31]。約70%的患者存在胃腸道功能障礙,常見便秘和胃食管反流[3]。約30%的患者存在進食障礙,部分患者需要胃造口管等方式喂養。CDD患者可能存在的其他臨床表現還包括行為問題(如自閉癥樣表現)、肌肉骨骼異常、反復感染(以呼吸道感染最為常見)、面容異常(如眼距寬、鼻梁塌)等[1,3,4,32,35]。
2.4 基因型與表型的相關性
CDD患者中存在多種CDKL5變異,國際CDD數據庫中285例CDD患者攜帶了200多種CDKL5變異,分析每種基因型與表型的相關性非常困難[2,50]。根據這些變異在基因上的位置及對CDKL5功能可能產生的影響進行基因型-表型分析,研究發現具有第172~781個氨基酸之間的截斷變異的患者癲癇發作率最低,在第781個氨基酸后存在截斷變異的患者比存在其他變異的患者發育相對好,更早達到粗大運動、精細運動和溝通發育里程碑[2,43,46,51]。亦有研究發現變異似乎與癲癇發作類型、高度失律、無發作“蜜月期”和腦性視覺損傷無相關性[42]。MacKay等[50]發現存在p.Arg178Trp、p.Arg559*和p.Arg178Gln變異的患者較為嚴重,而p.Arg134*、p.Arg550*以及p.Glu55Argfs*20所致的CDD相對較輕。未來需要更多系統性研究進一步探索CDD患者中基因型與表型的相關性。
推薦意見1:CDD起病早,癥狀重,表型復雜多樣(證據級別2b)。臨床應重視CDD的早期識別,及早診斷、及時干預,從而改善患者的預后(專家意見,共識率100%)。
3 CDD的篩查與診斷
CDD的診斷應該遵循ILAE的診斷標準(表3)[52]。盡管CDD臨床表現具有一定的特異性,但需基因檢測確診,且需與其他常見DEE鑒別[3,31]。
3.1 篩查和輔助檢查
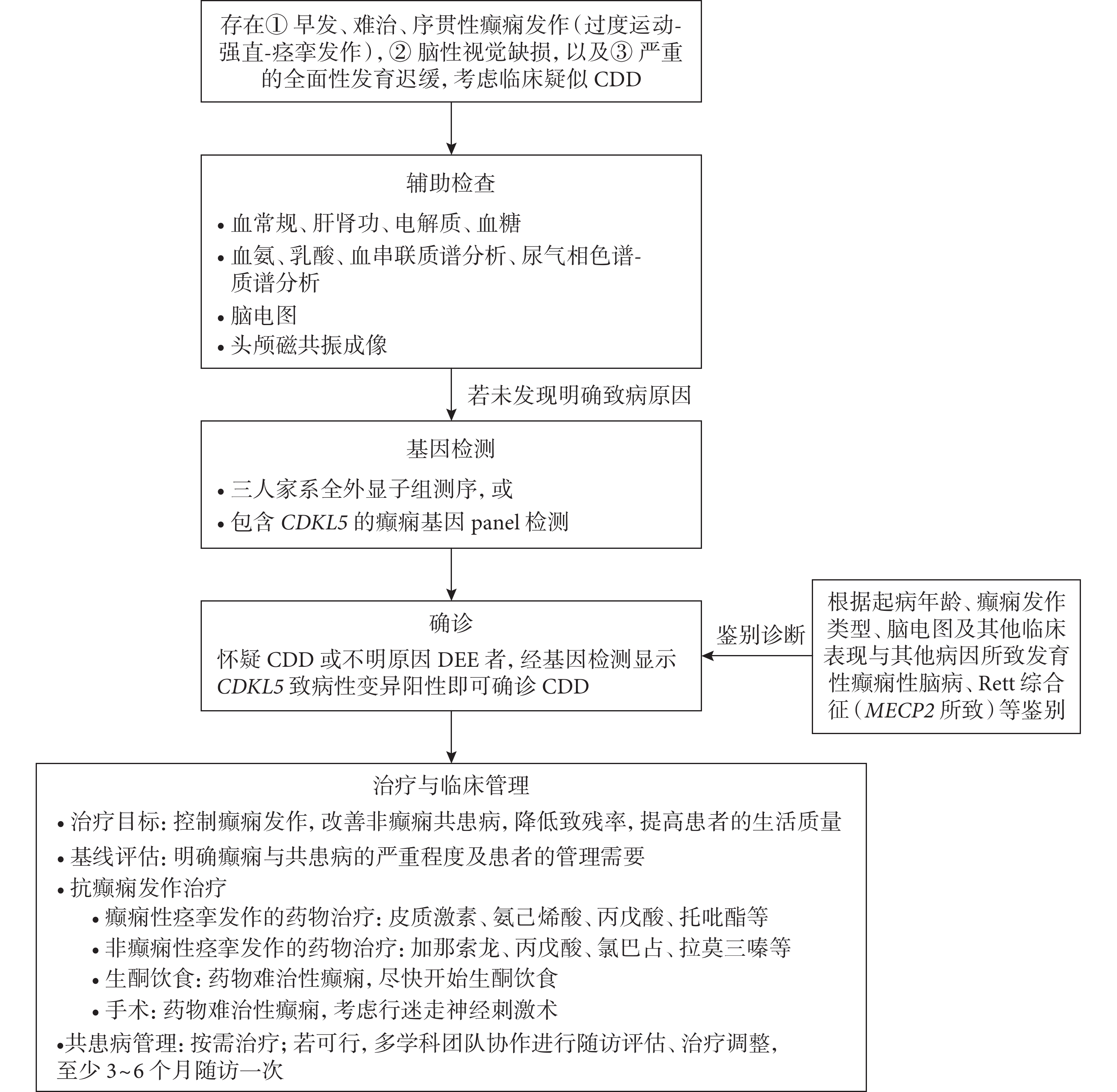
若患者存在① 早發、難治、過度運動-強直-痙攣序貫發作,② 腦性視覺缺損,以及③ 嚴重的全面性發育遲緩,則考慮臨床疑似CDD。疑似CDD的患者可通過輔助檢查排查及辨別可能的病因。對血、尿和腦脊液進行相應的檢查有助于排查代謝性疾病[53]。癲癇發作間期的腦電圖特征可幫助與其他DEE鑒別。CDD患者的磁共振成像(magnetic resonance imaging,MRI)缺乏特異性,可能正常或異常,可表現為大腦皮層萎縮、腦外間隙增寬等情況[3,33,34,38]。雖然血常規、生化、血氨、乳酸、血串聯質譜分析、尿氣相色譜-質譜分析、腦電圖或者MRI結果并非確診CDD的必要條件,但建議進行這些輔助檢查,以排除其他病因。圖1概括了CDD患者臨床診療的步驟和基本原則。
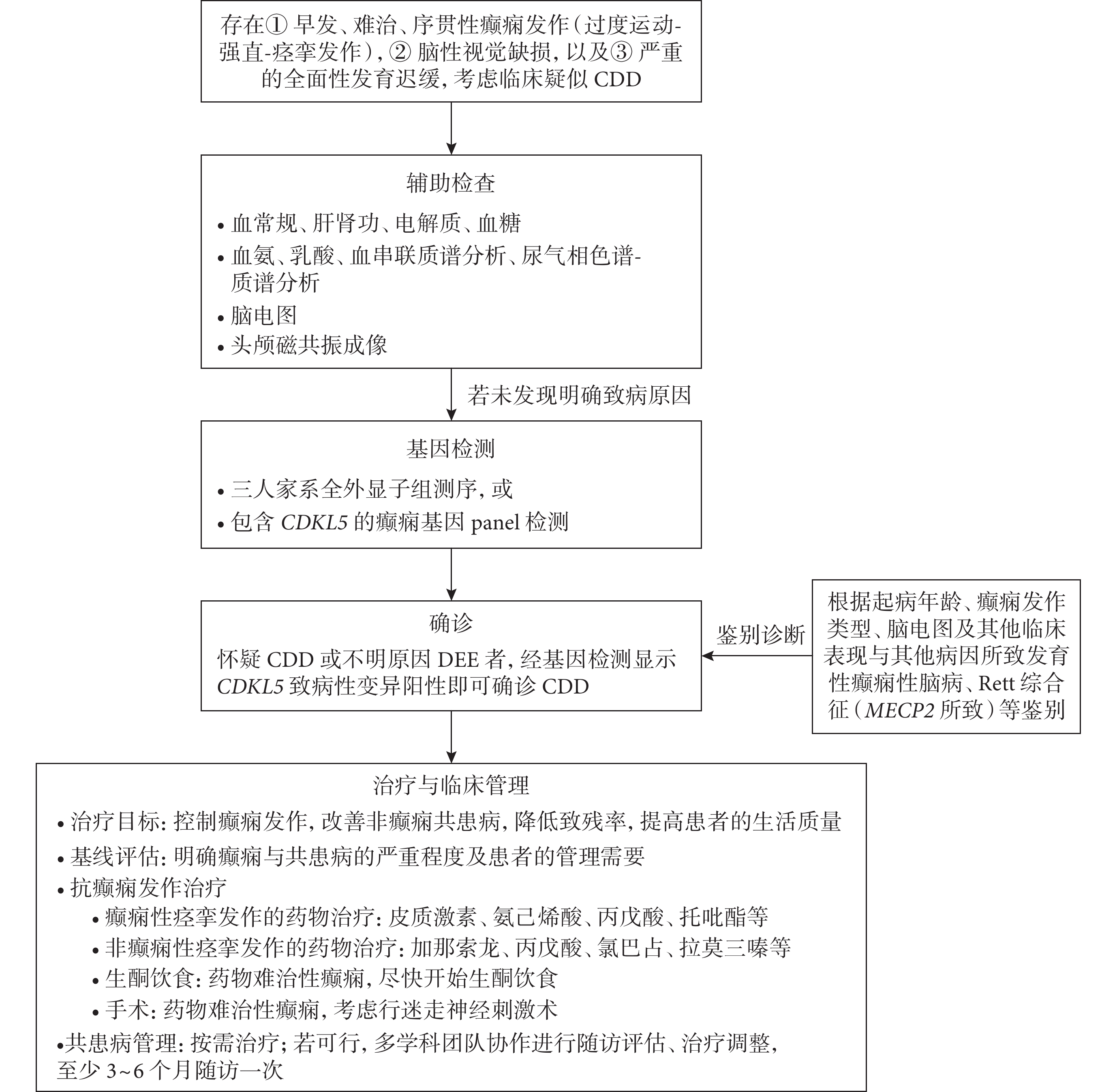 圖1
CDD診治流程圖
圖1
CDD診治流程圖
推薦意見2:當患者出現① 早發、難治、序貫性癲癇發作(過度運動-強直-痙攣發作),② 腦性視覺缺損,以及③ 嚴重的全面性發育遲緩,考慮臨床疑似CDD(專家意見,共識率100%)。
3.2 確診CDD
基因檢測陽性是確診CDD的必要條件[3,52]。對于懷疑CDD或不明原因DEE者,建議行患者及其父母的三人核心家系全外顯子組測序(trio-based whole exome sequencing,Trio-WES)或包括CDKL5基因的癲癇基因panel檢測。
推薦意見3:當患者臨床疑似CDD,為明確CDD診斷,建議進行如下基因檢測:① 三人核心家系全外顯子組測序,或 ② 包含CDKL5基因的癲癇基因panel檢測 (專家意見,共識率100%)。
3.3 鑒別診斷
CDD起病早、多種癲癇發作形式、發育遲緩及行為異常等特征,需要與其他病因所致的DEE,MECP2相關的Rett綜合征等相鑒別。
4 CDD的治療與臨床管理
4.1 治療目標與原則
CDD的治療目標為控制癲癇發作,改善各種非癲癇共患病,降低致殘率,提高患者的生活質量。CDD的治療原則包括:① 首先采用抗癲癇發作藥物(anti-seizure medications,ASMs)治療癲癇發作,根據患者具體情況個體化選擇藥物、調整劑量,以期達到最佳的癲癇控制效果;② 出現藥物難治性癲癇的患者,可盡快開始生酮飲食治療;③ 在一些藥物難治性癲癇病例中,可考慮神經調控手術治療,如迷走神經刺激術(vagus nerve stimulation,VNS);④ 評估并治療非癲癇共患病,加強訓練干預,按需使用康復治療,以減少總體疾病負擔;⑤ 如果條件允許,根據患者情況制定個體化的、多學科綜合管理方案,并定期隨訪評估,以期最大程度提高患者的生活質量。
4.2 抗CDD相關癲癇發作的藥物治療
CDD相關的癲癇多為藥物難治性癲癇,目前癲癇發作的藥物治療,療效有限,且隨著治療時間的延長,療效逐漸降低[54,55],并存在安全性挑戰[54-57]。加那索龍(ganaxolone)是全球首個獲批用于CDD癲癇發作的藥物治療[58-60],目前國內尚無其他藥物獲批用于該患者人群的治療,臨床亟需更多療效明確、安全性良好的創新治療方案,持續控制癲癇發作,提高患者的生活質量。
4.2.1 嬰兒癲癇性痙攣綜合征的藥物治療
癲癇性痙攣發作是CDD患者的初始癲癇發作類型。研究顯示,促腎上腺皮質激素(adrenocorticotropic hormone,ACTH)或口服大劑量潑尼松龍治療IESS的效果,優于包括氨己烯酸在內的ASMs[61,62]。ILAE指南[63]和《中國發育性癲癇性腦病激素治療臨床實踐指南(2022版)》(以下簡稱“中國DEE激素治療指南”)[64]均推薦優先使用激素治療IESS。激素的選擇及使用方法可參照《中國DEE激素治療指南》。
同時,研究顯示激素治療CDD相關癲癇發作(包括癲癇性痙攣發作)時的初期應答率可能較高,但隨著治療時間延長,應答率逐漸下降[20,54,55,65]。如,Olson等[54]2021年的研究顯示,ACTH、類固醇治療CDD相關癲癇(包括IESS)的2周治療應答率(應答定義為≥1種類型的癲癇發作頻率降低≥50%)分別為42%(8/19)、33%(7/21),而兩者3個月的應答率均下降為0。若激素治療癲癇性痙攣發作無效或者患者不能耐受,可考慮開始使用其他ASMs,綜合國內外指南推薦,可考慮使用的ASMs包括氨己烯酸、丙戊酸和托吡酯等[63,66]。
4.2.2 其他類型癲癇發作的藥物治療
研究顯示近3/4的CDD患者同時服用≥2種的ASMs[41,54],然而,隨著服藥時間的增加,療效逐漸降低[54,55]。基于明確報道應答率、評估方法以及樣本量等標準,下文概述4項探討ASMs治療CDD相關癲癇發作療效的研究。
這4項研究中,Olson等[54]2021年的北美研究納入了177例CDD患者,回顧患者的治療記錄,確定了最常用的ASMs并評估其在14天和3個月時的應答率。Muller等[55]2016年的研究,通過歐洲和美國21個中心收集數據,回顧性評估了39例CDD患者在使用ASMs和生酮飲食時的長期療效,重點關注患者在3、6和12個月時的應答情況(應答定義為與基線前4周相比,過去4周內癲癇發作頻率降低>50%)。Kobayashi 等[65]2021年的研究調查了29例日本CDD患者的ASMs使用情況,通過回顧病歷評估藥物的應答率和長期反應。Amin等[67] 2017年的研究通過CDKL5-UK慈善機構數據庫調查了44例來自美國、歐洲和英國的CDD患者,使用在線問卷的方法收集家長或照護人員提供的患者信息,評估ASMs、VNS和生酮飲食的效果。
基于以上研究,CDD患者最常使用的ASMs包括左乙拉西坦、托吡酯、苯巴比妥、丙戊酸、氨己烯酸、氯巴占、拉莫三嗪等[54,55,65,67];基于2項有明確ASMs應答率定義的縱向研究,丙戊酸、氨己烯酸、氯巴占和拉莫三嗪為有效ASMs (使用者≥20例,3個月應答率≥20%)[54,55]。Olson等[54]研究中,有效ASMs(3個月應答率)為氯巴占(36%,9/25)、氨己烯酸(33%,9/27)和丙戊酸(28%,7/25)。Muller等[55]研究中,有效ASMs(3個月應答率)為氨己烯酸(32%,8/25)、拉莫三嗪(22%,5/23)和丙戊酸(21%,7/34)。另外,Muller等[55]研究中氯巴占3個月應答率為24%(4/17),Olson等[54]研究中拉莫三嗪3個月應答率為13%(2/15)。這四項研究均未根據癲癇發作類型區分療效[54,55,65,67],此外,兩項縱向研究均顯示患者對ASMs的應答率隨著時間而降低[54,55]。Muller等[55]研究中,氨己烯酸、拉莫三嗪、丙戊酸和氯巴占在12個月時的應答率分別降至4%(1/25)、9%(2/23)、9%(3/34)、和0(0/17)。吡侖帕奈作為一種新型ASM,只有個別研究報道了其在極少數CDD患者中的使用,目前缺乏其直接用于治療CDD相關癲癇的高質量循證醫學依據[68]。因此,本共識未能對其治療CDD相關癲癇提供推薦意見。
4.2.3 抗CDD相關癲癇發作新藥進展
加那索龍是一種具有神經活性的類固醇,可正向別構調節中樞神經系統γ -氨基丁酸A型(γ-aminobutyric acid type A,GABAA)受體,加那索龍與GABAA受體結合后,放大GABA與GABAA受體結合后的氯離子通道的激活作用,增強氯離子流,從而抑制過度興奮的神經活動。另外,加那索龍同時作用于介導間歇性抑制的突觸內GABAA受體以及持續性抑制的突觸外的GABAA受體,實現對于間歇性及持續性過度興奮神經活動的抑制[69]。
一項名為MARIGOLD的三期、隨機、雙盲、安慰劑對照試驗,入組了101例經基因檢測確診CDKL5變異、伴早發性且難以控制的癲癇發作和神經發育障礙的患者,其中2~11歲患者占82.2%,青少年患者占16.8%[59,70,71]。試驗證實加那索龍可有效、持續地改善CDD相關癲癇發作且耐受性良好[70,71]。在為期17周的雙盲期,加那索龍組主要運動性癲癇發作頻率較基線下降百分比(30.7%)顯著高于安慰劑組(6.9%)(P=0.0 036)[70],在開放標簽階段中持續使用加那索龍2年的受試者中,主要運動性癲癇發作相對于基線的降幅達到48.2%(證據等級 1b)[71]。在MARIGOLD研究中,加那索龍最常見不良反應為嗜睡、發熱、上呼吸道感染、鎮靜,加那索龍組因不良反應導致停藥的發生率低于安慰劑組,2年長期擴展研究未見新發不良反應,顯示了長期使用的安全性[70,71]。加那索龍是全球首個獲批用于CDD相關癲癇治療的藥物,已獲美國藥品食品監督局(Food and Drug Administration,FDA)和歐洲藥監局(European Medicines Agency,EMA)批準用于治療CDD相關的癲癇發作[58,59],于2024年獲得中國藥品監督管理局批準用于在≥2歲患者中治療CDD相關的癲癇發作[60]。
CDD患者需要長期甚至終身用藥,因此ASMs的安全性對患者至關重要。為有效管理ASMs的不良反應,臨床上需采取定期監測、個性化調整劑量、患者/照護者教育等措施。管理ASMs不良反應的更多措施,可參考我國于2023年發布的《抗癲癇發作藥物不良反應管理指南》[56]。
推薦意見4:確診CDD后,優先使用皮質激素治療CDD相關癲癇性痙攣發作(證據等級2b,推薦強度B;共識率97.6%);如皮質激素治療無效或不耐受,推薦開始使用其他ASMs(包括氨己烯酸、丙戊酸、托吡酯)進行針對癲癇性痙攣發作的治療(專家意見,共識率100%)。
推薦意見5:在控制CDD相關的其他癲癇發作時,建議選用加那索龍(證據等級1b,推薦強度A;共識率100%),丙戊酸、氯巴占或拉莫三嗪(證據等級2b,推薦強度B;共識率100%)進行抗癲癇發作治療。
4.3 抗癲癇發作的非藥物治療
4.3.1 生酮飲食
生酮飲食是一種高脂肪、低碳水化合物的飲食方式,有助于穩定神經元的興奮性而減少癲癇發作[72]。生酮飲食對部分難治性癲癇有顯著療效[73,74],但也可能帶來一些副作用,如便秘、嘔吐、腎結石、高脂血癥、低血糖癥、低蛋白血癥等[75]。超過半數的 CDD 患者曾采用過生酮飲食[54,76,77]。現有研究對CDD患者能否從生酮飲食中獲益結論不一,較大型的研究支持CDD患者采用生酮飲食[76,78],而較小型的研究則顯示CDD患者不能從生酮飲食中獲益[41]。
盡管生酮飲食治療CDD相關癲癇發作時療效有限、持續時間短[67,76],且有一定副作用,鑒于CDD患者的治療選擇有限,編寫小組仍建議把生酮飲食作為治療CDD相關癲癇發作的常規手段之一。生酮飲食治療各環節需注意的事項,可參考我國2019年發布的《生酮飲食治療兒童癲癇性腦病循證指南》[75]。該指南推薦,在排除不適合生酮飲食治療的禁忌證或存在針對病因的其他更有效治療措施后,且通常在≥2種ASMs 治療失敗后,可考慮生酮飲食治療[75]。該指南同時建議,對CDD可進行生酮飲食治療[75]。在癲癇患者中,生酮飲食一般在啟動后1~2周起效,3個月內效果穩定,因此建議CDD患者啟動生酮飲食后堅持至少3個月[79]。有效者堅持生酮飲食2年后,若經醫生評估后可終止,逐漸過渡到普通飲食[79]。
推薦意見6:針對CDD相關的藥物難治性癲癇患者,應盡快開始生酮飲食,作為二線抗癲癇發作治療的補充(證據等級2b,推薦強度B;共識率100%);如果生酮飲食持續治療3個月無效 ,考慮停用生酮飲食(專家意見,共識率92.7%);如果生酮飲食有效,可持續治療2年,再酌情逐漸停用(專家意見,共識率97.6%)。
4.3.2 手術治療
根據我國專家于2021年制定的《迷走神經刺激治療藥物難治性癲癇的中國專家共識》[80],如果藥物治療無法有效控制患者癲癇發作、且患者符合ILAE于2010年發布的藥物難治性癲癇的診斷標準[81],可考慮對其進行VNS治療。VNS是一種可有效控制兒童癲癇發作的手段[82,83]。目前僅有數項小型回顧性研究探討了VNS在CDD患者中的療效[65,84-86]。一項國際回顧性研究顯示,69%(25/36)的CDD患者在接受VNS治療后癲癇發作控制得以改善,其中68%(17/25)發作頻率降低、72%(18/25)發作時間縮短、60%(15/25)發作強度減弱[84]。這項研究提示,VNS可作為CDD患者抗癲癇發作治療的一個選擇。VNS治療的適應證、禁忌癥、注意事項可參照我國的專家共識,其術前評估應由神經內科和/或兒科醫師、癲癇外科醫師及電生理醫師共同參與[80]。
鑒于胼胝體切開術治療 CDD 的臨床數據非常有限[54,65],國內已很少應用且無相關指導意見[87],故編寫小組不建議把胼胝體切開術作為治療 CDD 的常規手段。
推薦意見7:針對CDD相關的藥物難治性癲癇患者,考慮迷走神經刺激術(證據等級2b,推薦強度B;共識率95.1%)。
4.4 共患病的多學科管理
CDD患者,除癲癇外,常同時伴有多種共患病,嚴重影響CDD患者的生活質量,需接受多學科治療[1,3,4,88]。盡管目前尚無研究直接比較CDD、Rett綜合征、唐氏綜合征對患者生活質量的影響,但CDD患者的總生活質量得分似乎低于Rett綜合征患者與唐氏綜合征患者[2,89]。另外,無論是與普通人群相比,還是與Rett綜合征或唐氏綜合征患者的父母相比,CDD患者父母的心理/情感健康均較差[2]。可見,CDD給患者及其家庭帶了沉重的精神負擔。因此,有效的多學科照護對改善患者及其家庭的生活質量不可或缺[1,3,4]。
確診CDD后,如可行,建議由多學科團隊協作對患者進行全面評估,以明確癲癇與共患病的嚴重程度以及患者的管理需要。評估的內容除癲癇特征外,還需包括日常活動及睡眠情況、發育情況(包括體格、運動、認知等)、胃腸道功能及喂養情況、肌肉骨骼、視力、聽力、呼吸、心臟、皮膚、泌尿、牙科等[1,3,5]。根據評估結果,制定多學科管理策略,并對其進行長期隨訪、定期評估。患者的基線檢測結果應妥善存檔,以便后續評估隨訪。除醫療層面的評估及管理之外,還可包括解釋CDD的遺傳學原理,介紹相應的社區/線上資源等。
因CDD患者的運動障礙、認識障礙、溝通障礙突出。患者確診后應給與綜合評估,需評估運動障礙及其對粗大運動技能和精細運動技能的影響,進而考慮適應性設備以及物理治療/康復治療的需求,按需對CDD患者進行治療[1,3]。按照運動障礙的具體情況,也可考慮使用巴氯芬、肉毒桿菌毒素或其他治療運動障礙的藥物[3]。同時,對認知障礙和語言障礙進行專業評估,必要情況下考慮早期干預和支持,例如,考慮是否需要使用輔助溝通工具,如開關、觸摸板或眼控輔助設備等[1]。
推薦意見8:CDD是多系統受累疾病,如果條件允許,建議確診后由多學科團隊協作進行多學科隨訪評估、治療調整,且至少3~6個月隨訪一次(專家意見,共識率100%)。
5 結論與展望
本共識結合國內外最新診治研究進展、相關指南共識和編寫專家的臨床經驗,從CDD的篩查診斷、抗癲癇發作治療與共患病的管理三方面制定了推薦意見。CDD的臨床表現具有一定的特異性,但基因檢測陽性是確診的必要條件。CDD相關癲癇為難治性癲癇,可通過調整ASMs、采用生酮飲食、進行VNS手術等方式,盡量控制癲癇發作。CDD患者伴多種共患病,應根據患者具體情況采取個性化的多學科照護模式,以期盡量降低致殘率,提高患者的生活質量。CDD對患者家庭造成的心理及生活負擔不容忽視,因此除醫學干預,社會支持系統的完善同樣至關重要。在有條件的情況下,可為患者家庭提供包括心理輔導、經濟援助和社會融入等多方面的支持,幫助改善其生活質量,減輕精神負擔。
值得注意的是,ASMs雖可減輕癲癇負擔,但大多療效短暫或僅能產生部分效果,并且有可能造成多方面的副作用[54,55]。在某些情況下,ASMs產生的負面影響,甚至超過了控制癲癇發作帶來的積極影響[77,90]。因此,使用ASMs時要謹慎權衡治療獲益和其對生活質量的影響。目前可用于治療CDD相關癲癇的藥物有限,除本共識推薦的藥物外,大麻二酚和芬氟拉明對CDD相關癲癇也顯示出一定療效[54,91-93],但二者均未在我國獲批。目前數種針對CDD的療法正在研發中[3,41],這或將為CDD患者提供更多、更有效的治療選擇。另外,在現有基礎上進一步探究CDD病理機制,也將有助于研發有效的CDD針對性法療法。
現有的CDD臨床研究主要以回顧性研究、問卷調查和病例系列/報告為主,高質量的研究稀缺,來自中國的研究尤其少見。因此,本共識的局限性在于大多數推薦意見的證據級別較低。未來需要更多高質量的臨床研究結果,為制定CDD的診療共識提供高級別證據。隨著臨床數據的增加和對該疾病了解的深入,本共識將持續更新,以期進一步促進我國CDD診治水平的提升。
中國抗癲癇協會創新與轉化專業委員會(按作者姓氏拼音排序,排名不分先后)
操德智、陳蕾、崔寧、翟瑄、丁晶、杜麗君、樊紅彬、高峰、郭崇倫、郭強、郭燕舞、韓蘊麗、洪波、胡杰、賈天明、姜玉武、蔣莉、康德智、李海峰、李玲、李路明、李其富、李文玲、李云、林衛紅、凌至培、劉仕勇、劉翔宇、劉曉鳴、劉智勝、龍莉莉、歐紹武、錢若兵、孫巖、湯春輝、田茂強、田梅、王慧芳、王菊莉、王群、王天成、王新軍、王雄飛、鄔巍、吳迪、謝涵、徐紀文、閆宇翔、楊光路、楊麗白、楊小楓、于曉莉、張建昭、張赟健、趙國光、趙婷婷、趙秀鶴、周文靜、朱丹、朱遂強
中華醫學會兒科學分會罕見病學組(按作者姓氏拼音排序,排名不分先后)
陳倩、陳淑麗、陳穎、程亞穎、方昕、胡瑞梅、黃艷智、姜麗紅、金潤銘、蔣莉、李聯僑、李曉華、劉麗、劉小紅、劉曉軍、劉曉梅、劉艷明、邱正慶、單小鷗、田執梁、湯繼宏、王華、王劍、王藝、吳迪、吳瑾、虞雄鷹、徐家麗、徐迎軍、余永國、于永慧、姚寶珍、應艷琴、張改秀、張鋒、張靜、張月華、張一寧、朱海燕、鄒朝春、鄒麗萍
中華醫學會兒科學分會神經學組(按作者姓氏拼音排序,排名不分先后)
卞廣波、陳永前、方方、甘靖、高峰、高麗、郭靜竹、韓虹、韓金芬、韓蘊麗、洪思琦、胡君、季濤云、姜玉武、李保敏、李玲、梁建民、廖建湘、劉艷、彭鏡、孫丹、孫巖、湯春輝、田茂強、王春雨、王紀文、王榮、王守磊、吳德、吳華平、楊光、楊光路、楊健、楊思達、楊志仙、張俊梅、張利亞、張玉琴、趙蓉、鄭幗、周戩平、周水珍、禚志紅
利益沖突聲明 所有作者無利益沖突。
類細胞周期蛋白依賴性蛋白激酶5(cyclin-dependent kinase like 5,CDKL5,亦稱為細胞周期蛋白依賴性激酶樣5)缺乏癥(CDKL5 deficiency disorder,CDD)是一種嬰兒期起病的罕見X-連鎖顯性遺傳病,表現為難以控制的癲癇發作、嚴重全面性發育遲緩以及視覺、睡眠、消化等多方面障礙[1,2],嚴重影響患者及其家庭的生活質量。CDD表型復雜多樣,需結合臨床表現和基因檢測結果進行診斷,并與其他常見發育性癲癇性腦病(developmental and epileptic encephalopathy,DEE)鑒別[3]。目前尚無針對CDD病因的治療方法,臨床以綜合管理及對癥治療為主[4]。近年歐美地區專家先后發布《國際共識:CDKL5缺乏癥患者的評估和管理建議》[1]以及《為CDKL5缺乏癥患者提供高質量照護:歐洲專家小組關于患者病程的意見》[4],為CDD的診療提供了初步參考,但目前仍缺乏符合我國國情的規范化指導。中國抗癲癇協會創新與轉化專業委員會、中華醫學會兒科學分會神經學組及中華醫學會兒科學分會罕見病學組共同組織領域內專家,結合權威共識推薦、現有研究證據及臨床實踐經驗,制定了《類細胞周期蛋白依賴性蛋白激酶5缺乏癥診斷與治療的中國專家共識》(后簡稱“本共識”),旨在為我國CDD的診斷及管理提供具體建議和指導。
1 共識制定方法
本共識編寫組由我國兒童神經及罕見病領域資深專家組成,具有廣泛代表性。編寫組首先就共識的目的、主題范圍、框架達成一致,內容涉及CDD的疾病概述、篩查診斷與臨床管理。檢索數據庫包括MEDLINE(PubMed)、Embase、The Cochrane Library、中國知網數據庫、萬方知識數據服務平臺等。納入文章類型包括指南、共識、薈萃分析、隨機對照臨床試驗、非隨機對照臨床試驗、隊列研究、病例報道等。檢索詞包括“CDKL5缺乏癥/CDKL5 deficiency disorder/CDD”,“流行病學/epidemiology”,“疾病機制/pathology”,“臨床表現/clinical presentation”,“診斷/diagnosis”,“基因檢測/genetic testing”,“抗癲癇發作藥物/anti-seizure medications/ASMs”,“生酮飲食/ketogenic diet”,“癲癇外科手術/epilepsy surgery” 等。檢索語言限定為中文或英文,檢索時間截至2024年6月15日,納入標準為與CDD或兒童難治性癲癇治療相關且可獲取全文。由于罕見病領域文獻有限,除科研數據庫以外,編寫組亦檢索了權威機構平臺,包括國際CDKL5研究基金會[5]、CDKL5 UK[6]、中國CDKL5互助聯盟[7]等。
因探討CDD治療的研究有限,制定抗癲癇發作治療推薦意見時,除臨床數據外,編寫組亦借鑒了國際抗癲癇聯盟(International League Against Epilepsy,ILAE)、中國抗癲癇協會(China Association Against Epilepsy,CAAE)、中華醫學會(Chinese Medical Association,CMA)、美國神經病學學會(American Academy of Neurology,AAN)、美國癲癇協會(American Epilepsy Society,AES)、英國國家衛生與臨床優化研究所(National Institute for Health and Care Excellence,NICE)等權威機構就兒科的抗癲癇治療作出的推薦意見,并參考了編寫組專家治療兒童癲癇的豐富臨床經驗。編寫組根據資料檢索結果撰寫共識初稿,經深入討論、反復推敲對關鍵問題形成8條推薦意見,并依據英國牛津大學循證醫學中心的證據分級和推薦標準(表1)[8]對其進行分級。
隨后,編寫組進行了線上德爾菲調查,將德爾菲問卷和證據綜述發送給支持《共識》編寫的專家組成員,專家組成員結合文獻證據及臨床經驗填寫問卷。德爾菲問卷的每個問題結果分5級,包括完全同意、同意、不確定、不同意及完全不同意,每個問題后專家均可填寫補充意見。選擇強烈同意及同意的專家比例>75%表示就該條目達成共識;如存在未達成共識的條目,編寫組將根據反饋意見修改相關推薦意見后再次進行調查。首輪調查于2024年7月12日進行,共收回有效問卷41份,專家組對所有推薦意見均達成共識,共識率為82.9%~100%,故未再次進行調查。
2 疾病概述
2.1 流行病學
CDD在新生兒中的發病率為1:60 000~1:40 000,在女性中更為常見,與男性發病率比值為4:1~12:1[1]。盡管CDD相對罕見,但早發性DEE患者中伴CDKL5變異的比例達4.4%~37.5% [9-13]。我國多項研究亦顯示CDKL5是嬰兒期起病DEE患者中常見的致病基因[13-15]。
CDD嚴重影響患者的生活質量,雖然目前尚無高質量研究給出CDD患者的預期壽命[16],但臨床不乏在兒童或青少年時期死亡病例報道[5,16]。CDD致殘率高,許多患者終身無法達到普通兒童早期發展的發育里程碑,如獨坐、獨站等[3,17]。CDD患者預后與其疾病嚴重程度、臨床管理(尤其是癲癇發作控制)及照護條件等多種因素相關。研究顯示,與獨坐及獨立行走相關的因素包括有利的基因型、癲癇發作起病較晚、1歲內接受相應治療等[18]。一項在嬰兒癲癇性痙攣綜合征(infantile epileptic spasms syndrome,IESS)患者中進行的試驗顯示,起病至開始給與治療的間期短、對治療的早期應答,與較好的發育及癲癇發作預后相關[19]。同時,有研究顯示,相較于其他病因所致的IESS患者,CDD患者自起病至首次接受治療的間隔往往較長[20]。因此,臨床需及早診斷并治療CDD,以期改善患者預后。
2.2 發病機制
CDKL5是1998年Montini等[21]應用外顯子捕獲技術在Xp22區域克隆到的一個新基因,于2003年首次在伴智力障礙的女性IESS患者中發現該基因變異[22-24]。CDKL5有多種經選擇性剪接產生的異構體,有些廣泛表達,有些則主要在大腦中表達[16,21,25,26]。在mRNA層面,CDKL5在大腦皮層、海馬體、紋狀體和嗅球等結構中含量最高,在包括谷氨酸能神經元和γ -氨基丁酸(γ-aminobutyric acid,GABA)能神經元在內的前腦神經元中含量尤其豐富[16]。CDKL5水平在產前最低,在神經系統快速發育的圍產期和產后水平最高(尤其是在大腦皮層和海馬體),對神經元的形成和成熟極為重要[27]。研究已證實,CDKL5參與神經元的遷移、形成和生長,突觸的發育和正常功能的行使[27],對維持正常的興奮-抑制平衡和記憶至關重要[28]。缺乏CDKL5會導致樹突分支減少、神經元回路連接受損等多種問題,嚴重影響神經系統的發育及正常功能[16]。研究發現敲除前腦谷氨酸能神經元中的Cdkl5基因可導致小鼠自發性癲癇發作[29,30]。這些研究將有助于構建CDD動物模型、深入了解其病理機制以及研發臨床治療藥物。
2.3 臨床表現
CDD是一種以癲癇發作為主要表現的多系統受累疾病,起病早,癥狀重,表型復雜多樣,男性患者往往比女性患者病情更為嚴重[31]。國外數據顯示,CDD中位起病年齡為6周,90%的患者在3月齡內起病[31]。我國病例報道中,CDD患者多于出生后1~12個月起病[32-38],就診年齡多在3月齡~3歲之間[34,38]。
2.3.1 癲癇發作
ILAE將CDD患者的癲癇綜合征歸為病因特異性發育性癲癇性腦病(CDKL5-DEE)[39],其癲癇通常在嬰兒期起病,發作頻繁(多數患者每日均有發作),且難以控制[3,31]。癲癇發作類型隨年齡增長而變化,在約25%的個體中,癲癇性痙攣發作是初始發作類型,其他發作類型包括強直、局灶、肌陣攣、全面性強直-陣攣及序貫發作[3]。根據癲癇發作類型及腦電圖特點,臨床上將CDD相關癲癇分為3期,包括嬰兒期起病的運動性發作(Ⅰ期,1~10周)、伴癲癇性痙攣發作的癲癇性腦病(Ⅱ期,6~36月齡)以及難治性癲癇(Ⅲ期,2~11歲)(表2)[40-42]。不同分期癲癇發作間期的腦電圖存在差異。I期發作間期腦電圖可能正常,或表現出彌漫性慢波或局灶性癲癇樣放電[1,3]。隨著疾病發展,Ⅱ、Ⅲ期患者在發作間期腦電圖多表現為高度失律以及彌漫性慢波伴多灶癲癇樣放電[41]。
過度運動-強直-痙攣序貫發作是CDD特征性的癲癇發作類型,見于多數病例。其第一階段以過度運動起始,伴有搖擺、蹬踢和發聲,持續10~60 s。隨后是強直期,四肢伸展或是上肢伸直及下肢屈曲,持續20~45 s。繼而演變為痙攣發作,持續 1~15 min。另外,部分CDD患者會經歷無發作“蜜月期”,即癲癇發作減少或消失一段時間,通常發生在2月齡~11歲(中位年齡為2歲),持續時間為2.5個月~6年(中位時間為6個月)[41]。值得注意的是,雖然難治性癲癇是CDD最常見的臨床表現之一,但臨床上亦觀察到極少數(<1%)CDD患者無癲癇發作[43-45]。
2.3.2 嚴重全面性發育遲緩、腦性視覺損傷及其他共患病
盡管CDD患者達到同一發育里程碑的時間存在較大差異,但所有患者均存在嚴重的全面性發育遲緩,包括運動、溝通(言語和語言)以及認知等多方面發育落后[3,31,46],約60%的患者能獨坐,20%左右的患者可獨行,40%~70%的患者無法抓取及持有物體[3]。許多患者存在異常動作,包括刻板動作(如將手反復放入口中、手臂拍動或揮動、交叉雙腿等)、舞蹈癥等[3]。約50%的患者在6歲時可咿呀學語,僅約25%的患者在7歲時可說出單個單詞[31]。
值得注意的是,約80%的CDD患者存在腦性視覺損傷,其表現為異常眼動,包括內斜視、外斜視、水平性或旋轉性眼震等,以及對強光的異常注視和反應等,常伴有視線接觸不良和視覺追蹤障礙[3,31,42,47-49]。此外,約90%的CDD患者存在睡眠障礙,主要體現為無法維持正常睡眠狀態,如連續幾晚缺乏睡眠及后續的過度睡眠,睡眠障礙通常終身存在[3,31]。約70%的患者存在胃腸道功能障礙,常見便秘和胃食管反流[3]。約30%的患者存在進食障礙,部分患者需要胃造口管等方式喂養。CDD患者可能存在的其他臨床表現還包括行為問題(如自閉癥樣表現)、肌肉骨骼異常、反復感染(以呼吸道感染最為常見)、面容異常(如眼距寬、鼻梁塌)等[1,3,4,32,35]。
2.4 基因型與表型的相關性
CDD患者中存在多種CDKL5變異,國際CDD數據庫中285例CDD患者攜帶了200多種CDKL5變異,分析每種基因型與表型的相關性非常困難[2,50]。根據這些變異在基因上的位置及對CDKL5功能可能產生的影響進行基因型-表型分析,研究發現具有第172~781個氨基酸之間的截斷變異的患者癲癇發作率最低,在第781個氨基酸后存在截斷變異的患者比存在其他變異的患者發育相對好,更早達到粗大運動、精細運動和溝通發育里程碑[2,43,46,51]。亦有研究發現變異似乎與癲癇發作類型、高度失律、無發作“蜜月期”和腦性視覺損傷無相關性[42]。MacKay等[50]發現存在p.Arg178Trp、p.Arg559*和p.Arg178Gln變異的患者較為嚴重,而p.Arg134*、p.Arg550*以及p.Glu55Argfs*20所致的CDD相對較輕。未來需要更多系統性研究進一步探索CDD患者中基因型與表型的相關性。
推薦意見1:CDD起病早,癥狀重,表型復雜多樣(證據級別2b)。臨床應重視CDD的早期識別,及早診斷、及時干預,從而改善患者的預后(專家意見,共識率100%)。
3 CDD的篩查與診斷
CDD的診斷應該遵循ILAE的診斷標準(表3)[52]。盡管CDD臨床表現具有一定的特異性,但需基因檢測確診,且需與其他常見DEE鑒別[3,31]。
3.1 篩查和輔助檢查
若患者存在① 早發、難治、過度運動-強直-痙攣序貫發作,② 腦性視覺缺損,以及③ 嚴重的全面性發育遲緩,則考慮臨床疑似CDD。疑似CDD的患者可通過輔助檢查排查及辨別可能的病因。對血、尿和腦脊液進行相應的檢查有助于排查代謝性疾病[53]。癲癇發作間期的腦電圖特征可幫助與其他DEE鑒別。CDD患者的磁共振成像(magnetic resonance imaging,MRI)缺乏特異性,可能正常或異常,可表現為大腦皮層萎縮、腦外間隙增寬等情況[3,33,34,38]。雖然血常規、生化、血氨、乳酸、血串聯質譜分析、尿氣相色譜-質譜分析、腦電圖或者MRI結果并非確診CDD的必要條件,但建議進行這些輔助檢查,以排除其他病因。圖1概括了CDD患者臨床診療的步驟和基本原則。
圖1
CDD診治流程圖
推薦意見2:當患者出現① 早發、難治、序貫性癲癇發作(過度運動-強直-痙攣發作),② 腦性視覺缺損,以及③ 嚴重的全面性發育遲緩,考慮臨床疑似CDD(專家意見,共識率100%)。
3.2 確診CDD
基因檢測陽性是確診CDD的必要條件[3,52]。對于懷疑CDD或不明原因DEE者,建議行患者及其父母的三人核心家系全外顯子組測序(trio-based whole exome sequencing,Trio-WES)或包括CDKL5基因的癲癇基因panel檢測。
推薦意見3:當患者臨床疑似CDD,為明確CDD診斷,建議進行如下基因檢測:① 三人核心家系全外顯子組測序,或 ② 包含CDKL5基因的癲癇基因panel檢測 (專家意見,共識率100%)。
3.3 鑒別診斷
CDD起病早、多種癲癇發作形式、發育遲緩及行為異常等特征,需要與其他病因所致的DEE,MECP2相關的Rett綜合征等相鑒別。
4 CDD的治療與臨床管理
4.1 治療目標與原則
CDD的治療目標為控制癲癇發作,改善各種非癲癇共患病,降低致殘率,提高患者的生活質量。CDD的治療原則包括:① 首先采用抗癲癇發作藥物(anti-seizure medications,ASMs)治療癲癇發作,根據患者具體情況個體化選擇藥物、調整劑量,以期達到最佳的癲癇控制效果;② 出現藥物難治性癲癇的患者,可盡快開始生酮飲食治療;③ 在一些藥物難治性癲癇病例中,可考慮神經調控手術治療,如迷走神經刺激術(vagus nerve stimulation,VNS);④ 評估并治療非癲癇共患病,加強訓練干預,按需使用康復治療,以減少總體疾病負擔;⑤ 如果條件允許,根據患者情況制定個體化的、多學科綜合管理方案,并定期隨訪評估,以期最大程度提高患者的生活質量。
4.2 抗CDD相關癲癇發作的藥物治療
CDD相關的癲癇多為藥物難治性癲癇,目前癲癇發作的藥物治療,療效有限,且隨著治療時間的延長,療效逐漸降低[54,55],并存在安全性挑戰[54-57]。加那索龍(ganaxolone)是全球首個獲批用于CDD癲癇發作的藥物治療[58-60],目前國內尚無其他藥物獲批用于該患者人群的治療,臨床亟需更多療效明確、安全性良好的創新治療方案,持續控制癲癇發作,提高患者的生活質量。
4.2.1 嬰兒癲癇性痙攣綜合征的藥物治療
癲癇性痙攣發作是CDD患者的初始癲癇發作類型。研究顯示,促腎上腺皮質激素(adrenocorticotropic hormone,ACTH)或口服大劑量潑尼松龍治療IESS的效果,優于包括氨己烯酸在內的ASMs[61,62]。ILAE指南[63]和《中國發育性癲癇性腦病激素治療臨床實踐指南(2022版)》(以下簡稱“中國DEE激素治療指南”)[64]均推薦優先使用激素治療IESS。激素的選擇及使用方法可參照《中國DEE激素治療指南》。
同時,研究顯示激素治療CDD相關癲癇發作(包括癲癇性痙攣發作)時的初期應答率可能較高,但隨著治療時間延長,應答率逐漸下降[20,54,55,65]。如,Olson等[54]2021年的研究顯示,ACTH、類固醇治療CDD相關癲癇(包括IESS)的2周治療應答率(應答定義為≥1種類型的癲癇發作頻率降低≥50%)分別為42%(8/19)、33%(7/21),而兩者3個月的應答率均下降為0。若激素治療癲癇性痙攣發作無效或者患者不能耐受,可考慮開始使用其他ASMs,綜合國內外指南推薦,可考慮使用的ASMs包括氨己烯酸、丙戊酸和托吡酯等[63,66]。
4.2.2 其他類型癲癇發作的藥物治療
研究顯示近3/4的CDD患者同時服用≥2種的ASMs[41,54],然而,隨著服藥時間的增加,療效逐漸降低[54,55]。基于明確報道應答率、評估方法以及樣本量等標準,下文概述4項探討ASMs治療CDD相關癲癇發作療效的研究。
這4項研究中,Olson等[54]2021年的北美研究納入了177例CDD患者,回顧患者的治療記錄,確定了最常用的ASMs并評估其在14天和3個月時的應答率。Muller等[55]2016年的研究,通過歐洲和美國21個中心收集數據,回顧性評估了39例CDD患者在使用ASMs和生酮飲食時的長期療效,重點關注患者在3、6和12個月時的應答情況(應答定義為與基線前4周相比,過去4周內癲癇發作頻率降低>50%)。Kobayashi 等[65]2021年的研究調查了29例日本CDD患者的ASMs使用情況,通過回顧病歷評估藥物的應答率和長期反應。Amin等[67] 2017年的研究通過CDKL5-UK慈善機構數據庫調查了44例來自美國、歐洲和英國的CDD患者,使用在線問卷的方法收集家長或照護人員提供的患者信息,評估ASMs、VNS和生酮飲食的效果。
基于以上研究,CDD患者最常使用的ASMs包括左乙拉西坦、托吡酯、苯巴比妥、丙戊酸、氨己烯酸、氯巴占、拉莫三嗪等[54,55,65,67];基于2項有明確ASMs應答率定義的縱向研究,丙戊酸、氨己烯酸、氯巴占和拉莫三嗪為有效ASMs (使用者≥20例,3個月應答率≥20%)[54,55]。Olson等[54]研究中,有效ASMs(3個月應答率)為氯巴占(36%,9/25)、氨己烯酸(33%,9/27)和丙戊酸(28%,7/25)。Muller等[55]研究中,有效ASMs(3個月應答率)為氨己烯酸(32%,8/25)、拉莫三嗪(22%,5/23)和丙戊酸(21%,7/34)。另外,Muller等[55]研究中氯巴占3個月應答率為24%(4/17),Olson等[54]研究中拉莫三嗪3個月應答率為13%(2/15)。這四項研究均未根據癲癇發作類型區分療效[54,55,65,67],此外,兩項縱向研究均顯示患者對ASMs的應答率隨著時間而降低[54,55]。Muller等[55]研究中,氨己烯酸、拉莫三嗪、丙戊酸和氯巴占在12個月時的應答率分別降至4%(1/25)、9%(2/23)、9%(3/34)、和0(0/17)。吡侖帕奈作為一種新型ASM,只有個別研究報道了其在極少數CDD患者中的使用,目前缺乏其直接用于治療CDD相關癲癇的高質量循證醫學依據[68]。因此,本共識未能對其治療CDD相關癲癇提供推薦意見。
4.2.3 抗CDD相關癲癇發作新藥進展
加那索龍是一種具有神經活性的類固醇,可正向別構調節中樞神經系統γ -氨基丁酸A型(γ-aminobutyric acid type A,GABAA)受體,加那索龍與GABAA受體結合后,放大GABA與GABAA受體結合后的氯離子通道的激活作用,增強氯離子流,從而抑制過度興奮的神經活動。另外,加那索龍同時作用于介導間歇性抑制的突觸內GABAA受體以及持續性抑制的突觸外的GABAA受體,實現對于間歇性及持續性過度興奮神經活動的抑制[69]。
一項名為MARIGOLD的三期、隨機、雙盲、安慰劑對照試驗,入組了101例經基因檢測確診CDKL5變異、伴早發性且難以控制的癲癇發作和神經發育障礙的患者,其中2~11歲患者占82.2%,青少年患者占16.8%[59,70,71]。試驗證實加那索龍可有效、持續地改善CDD相關癲癇發作且耐受性良好[70,71]。在為期17周的雙盲期,加那索龍組主要運動性癲癇發作頻率較基線下降百分比(30.7%)顯著高于安慰劑組(6.9%)(P=0.0 036)[70],在開放標簽階段中持續使用加那索龍2年的受試者中,主要運動性癲癇發作相對于基線的降幅達到48.2%(證據等級 1b)[71]。在MARIGOLD研究中,加那索龍最常見不良反應為嗜睡、發熱、上呼吸道感染、鎮靜,加那索龍組因不良反應導致停藥的發生率低于安慰劑組,2年長期擴展研究未見新發不良反應,顯示了長期使用的安全性[70,71]。加那索龍是全球首個獲批用于CDD相關癲癇治療的藥物,已獲美國藥品食品監督局(Food and Drug Administration,FDA)和歐洲藥監局(European Medicines Agency,EMA)批準用于治療CDD相關的癲癇發作[58,59],于2024年獲得中國藥品監督管理局批準用于在≥2歲患者中治療CDD相關的癲癇發作[60]。
CDD患者需要長期甚至終身用藥,因此ASMs的安全性對患者至關重要。為有效管理ASMs的不良反應,臨床上需采取定期監測、個性化調整劑量、患者/照護者教育等措施。管理ASMs不良反應的更多措施,可參考我國于2023年發布的《抗癲癇發作藥物不良反應管理指南》[56]。
推薦意見4:確診CDD后,優先使用皮質激素治療CDD相關癲癇性痙攣發作(證據等級2b,推薦強度B;共識率97.6%);如皮質激素治療無效或不耐受,推薦開始使用其他ASMs(包括氨己烯酸、丙戊酸、托吡酯)進行針對癲癇性痙攣發作的治療(專家意見,共識率100%)。
推薦意見5:在控制CDD相關的其他癲癇發作時,建議選用加那索龍(證據等級1b,推薦強度A;共識率100%),丙戊酸、氯巴占或拉莫三嗪(證據等級2b,推薦強度B;共識率100%)進行抗癲癇發作治療。
4.3 抗癲癇發作的非藥物治療
4.3.1 生酮飲食
生酮飲食是一種高脂肪、低碳水化合物的飲食方式,有助于穩定神經元的興奮性而減少癲癇發作[72]。生酮飲食對部分難治性癲癇有顯著療效[73,74],但也可能帶來一些副作用,如便秘、嘔吐、腎結石、高脂血癥、低血糖癥、低蛋白血癥等[75]。超過半數的 CDD 患者曾采用過生酮飲食[54,76,77]。現有研究對CDD患者能否從生酮飲食中獲益結論不一,較大型的研究支持CDD患者采用生酮飲食[76,78],而較小型的研究則顯示CDD患者不能從生酮飲食中獲益[41]。
盡管生酮飲食治療CDD相關癲癇發作時療效有限、持續時間短[67,76],且有一定副作用,鑒于CDD患者的治療選擇有限,編寫小組仍建議把生酮飲食作為治療CDD相關癲癇發作的常規手段之一。生酮飲食治療各環節需注意的事項,可參考我國2019年發布的《生酮飲食治療兒童癲癇性腦病循證指南》[75]。該指南推薦,在排除不適合生酮飲食治療的禁忌證或存在針對病因的其他更有效治療措施后,且通常在≥2種ASMs 治療失敗后,可考慮生酮飲食治療[75]。該指南同時建議,對CDD可進行生酮飲食治療[75]。在癲癇患者中,生酮飲食一般在啟動后1~2周起效,3個月內效果穩定,因此建議CDD患者啟動生酮飲食后堅持至少3個月[79]。有效者堅持生酮飲食2年后,若經醫生評估后可終止,逐漸過渡到普通飲食[79]。
推薦意見6:針對CDD相關的藥物難治性癲癇患者,應盡快開始生酮飲食,作為二線抗癲癇發作治療的補充(證據等級2b,推薦強度B;共識率100%);如果生酮飲食持續治療3個月無效 ,考慮停用生酮飲食(專家意見,共識率92.7%);如果生酮飲食有效,可持續治療2年,再酌情逐漸停用(專家意見,共識率97.6%)。
4.3.2 手術治療
根據我國專家于2021年制定的《迷走神經刺激治療藥物難治性癲癇的中國專家共識》[80],如果藥物治療無法有效控制患者癲癇發作、且患者符合ILAE于2010年發布的藥物難治性癲癇的診斷標準[81],可考慮對其進行VNS治療。VNS是一種可有效控制兒童癲癇發作的手段[82,83]。目前僅有數項小型回顧性研究探討了VNS在CDD患者中的療效[65,84-86]。一項國際回顧性研究顯示,69%(25/36)的CDD患者在接受VNS治療后癲癇發作控制得以改善,其中68%(17/25)發作頻率降低、72%(18/25)發作時間縮短、60%(15/25)發作強度減弱[84]。這項研究提示,VNS可作為CDD患者抗癲癇發作治療的一個選擇。VNS治療的適應證、禁忌癥、注意事項可參照我國的專家共識,其術前評估應由神經內科和/或兒科醫師、癲癇外科醫師及電生理醫師共同參與[80]。
鑒于胼胝體切開術治療 CDD 的臨床數據非常有限[54,65],國內已很少應用且無相關指導意見[87],故編寫小組不建議把胼胝體切開術作為治療 CDD 的常規手段。
推薦意見7:針對CDD相關的藥物難治性癲癇患者,考慮迷走神經刺激術(證據等級2b,推薦強度B;共識率95.1%)。
4.4 共患病的多學科管理
CDD患者,除癲癇外,常同時伴有多種共患病,嚴重影響CDD患者的生活質量,需接受多學科治療[1,3,4,88]。盡管目前尚無研究直接比較CDD、Rett綜合征、唐氏綜合征對患者生活質量的影響,但CDD患者的總生活質量得分似乎低于Rett綜合征患者與唐氏綜合征患者[2,89]。另外,無論是與普通人群相比,還是與Rett綜合征或唐氏綜合征患者的父母相比,CDD患者父母的心理/情感健康均較差[2]。可見,CDD給患者及其家庭帶了沉重的精神負擔。因此,有效的多學科照護對改善患者及其家庭的生活質量不可或缺[1,3,4]。
確診CDD后,如可行,建議由多學科團隊協作對患者進行全面評估,以明確癲癇與共患病的嚴重程度以及患者的管理需要。評估的內容除癲癇特征外,還需包括日常活動及睡眠情況、發育情況(包括體格、運動、認知等)、胃腸道功能及喂養情況、肌肉骨骼、視力、聽力、呼吸、心臟、皮膚、泌尿、牙科等[1,3,5]。根據評估結果,制定多學科管理策略,并對其進行長期隨訪、定期評估。患者的基線檢測結果應妥善存檔,以便后續評估隨訪。除醫療層面的評估及管理之外,還可包括解釋CDD的遺傳學原理,介紹相應的社區/線上資源等。
因CDD患者的運動障礙、認識障礙、溝通障礙突出。患者確診后應給與綜合評估,需評估運動障礙及其對粗大運動技能和精細運動技能的影響,進而考慮適應性設備以及物理治療/康復治療的需求,按需對CDD患者進行治療[1,3]。按照運動障礙的具體情況,也可考慮使用巴氯芬、肉毒桿菌毒素或其他治療運動障礙的藥物[3]。同時,對認知障礙和語言障礙進行專業評估,必要情況下考慮早期干預和支持,例如,考慮是否需要使用輔助溝通工具,如開關、觸摸板或眼控輔助設備等[1]。
推薦意見8:CDD是多系統受累疾病,如果條件允許,建議確診后由多學科團隊協作進行多學科隨訪評估、治療調整,且至少3~6個月隨訪一次(專家意見,共識率100%)。
5 結論與展望
本共識結合國內外最新診治研究進展、相關指南共識和編寫專家的臨床經驗,從CDD的篩查診斷、抗癲癇發作治療與共患病的管理三方面制定了推薦意見。CDD的臨床表現具有一定的特異性,但基因檢測陽性是確診的必要條件。CDD相關癲癇為難治性癲癇,可通過調整ASMs、采用生酮飲食、進行VNS手術等方式,盡量控制癲癇發作。CDD患者伴多種共患病,應根據患者具體情況采取個性化的多學科照護模式,以期盡量降低致殘率,提高患者的生活質量。CDD對患者家庭造成的心理及生活負擔不容忽視,因此除醫學干預,社會支持系統的完善同樣至關重要。在有條件的情況下,可為患者家庭提供包括心理輔導、經濟援助和社會融入等多方面的支持,幫助改善其生活質量,減輕精神負擔。
值得注意的是,ASMs雖可減輕癲癇負擔,但大多療效短暫或僅能產生部分效果,并且有可能造成多方面的副作用[54,55]。在某些情況下,ASMs產生的負面影響,甚至超過了控制癲癇發作帶來的積極影響[77,90]。因此,使用ASMs時要謹慎權衡治療獲益和其對生活質量的影響。目前可用于治療CDD相關癲癇的藥物有限,除本共識推薦的藥物外,大麻二酚和芬氟拉明對CDD相關癲癇也顯示出一定療效[54,91-93],但二者均未在我國獲批。目前數種針對CDD的療法正在研發中[3,41],這或將為CDD患者提供更多、更有效的治療選擇。另外,在現有基礎上進一步探究CDD病理機制,也將有助于研發有效的CDD針對性法療法。
現有的CDD臨床研究主要以回顧性研究、問卷調查和病例系列/報告為主,高質量的研究稀缺,來自中國的研究尤其少見。因此,本共識的局限性在于大多數推薦意見的證據級別較低。未來需要更多高質量的臨床研究結果,為制定CDD的診療共識提供高級別證據。隨著臨床數據的增加和對該疾病了解的深入,本共識將持續更新,以期進一步促進我國CDD診治水平的提升。
中國抗癲癇協會創新與轉化專業委員會(按作者姓氏拼音排序,排名不分先后)
操德智、陳蕾、崔寧、翟瑄、丁晶、杜麗君、樊紅彬、高峰、郭崇倫、郭強、郭燕舞、韓蘊麗、洪波、胡杰、賈天明、姜玉武、蔣莉、康德智、李海峰、李玲、李路明、李其富、李文玲、李云、林衛紅、凌至培、劉仕勇、劉翔宇、劉曉鳴、劉智勝、龍莉莉、歐紹武、錢若兵、孫巖、湯春輝、田茂強、田梅、王慧芳、王菊莉、王群、王天成、王新軍、王雄飛、鄔巍、吳迪、謝涵、徐紀文、閆宇翔、楊光路、楊麗白、楊小楓、于曉莉、張建昭、張赟健、趙國光、趙婷婷、趙秀鶴、周文靜、朱丹、朱遂強
中華醫學會兒科學分會罕見病學組(按作者姓氏拼音排序,排名不分先后)
陳倩、陳淑麗、陳穎、程亞穎、方昕、胡瑞梅、黃艷智、姜麗紅、金潤銘、蔣莉、李聯僑、李曉華、劉麗、劉小紅、劉曉軍、劉曉梅、劉艷明、邱正慶、單小鷗、田執梁、湯繼宏、王華、王劍、王藝、吳迪、吳瑾、虞雄鷹、徐家麗、徐迎軍、余永國、于永慧、姚寶珍、應艷琴、張改秀、張鋒、張靜、張月華、張一寧、朱海燕、鄒朝春、鄒麗萍
中華醫學會兒科學分會神經學組(按作者姓氏拼音排序,排名不分先后)
卞廣波、陳永前、方方、甘靖、高峰、高麗、郭靜竹、韓虹、韓金芬、韓蘊麗、洪思琦、胡君、季濤云、姜玉武、李保敏、李玲、梁建民、廖建湘、劉艷、彭鏡、孫丹、孫巖、湯春輝、田茂強、王春雨、王紀文、王榮、王守磊、吳德、吳華平、楊光、楊光路、楊健、楊思達、楊志仙、張俊梅、張利亞、張玉琴、趙蓉、鄭幗、周戩平、周水珍、禚志紅
利益沖突聲明 所有作者無利益沖突。