引用本文: 馬冉, 張麗, 馬蘭花, 王美燕, 章偉. HIVEP2基因相關以癲癇為主要表型的常染色體顯性智力障礙43型一例并文獻分析. 癲癇雜志, 2025, 11(1): 83-88. doi: 10.7507/2096-0247.202410006 復制
版權信息: ?四川大學華西醫院華西期刊社《癲癇雜志》版權所有,未經授權不得轉載、改編
常染色體顯性智力障礙43型(Mental retardation type 43,MRD43,OMIM 616977)是由HIVEP2基因變異導致的一種罕見常染色體顯性遺傳病,由Ranch于2012年首次報道,本病主要特征為發育遲緩、智力障礙[1,2]。對國內外HIVEP2基因變異案例報道進行文獻回顧,共檢索到20例相關病例[1-10],其中20/20存在發育落后,19/20存在智力障礙,4/20存在癲癇。現報道1例通過全外顯子基因測序確診的HIVEP2基因變異[c.6667C>T (p.Arg2223*)]導致以癲癇為主要表型的MRD43患兒,擴展了該病癲癇表型的藥物治療譜。本研究獲得石河子大學第一附屬醫院醫學倫理委員會審核批準(KJ2022-216-01)及患兒家屬知情同意。
病例資料 患兒 男,7歲10月齡。因“24 h無熱抽搐發作2次”就診石河子大學第一附屬醫院。患兒于當日夜間入睡1 h后無明顯誘因出現嘔吐胃內容物1次,隨后出現四肢強直陣攣,呼之不應,意識喪失,雙眼上翻,牙關緊閉,口唇發紺,喉中發聲,約1~2 min意識恢復,發作后有四肢發軟,自覺乏力;次日晨時9:00患兒于睡眠臨清醒時再次發作,發作前也有嘔吐,發作形式基本同前;隨后急診來院。患兒為G2P1,足月剖宮產,混合喂養,生后體健,Apgar評分不詳,3月齡抬頭、6月齡會坐、12月齡會站、16月齡會走、可喊“爸爸、媽媽”。患兒現為一年級學生,言語欠清晰,運動欠協調,學習成績較差,注意力不集中,平日多動行為。否認窒息等異常出生史、頭顱外傷史、癲癇等家族遺傳史。
體格檢查:體溫36.9℃,脈搏82次/min,呼吸25次/min,血壓:92/54 mmHg,身高136 cm,體質量30 kg,頭圍51 cm。神志清楚,精神反應正常,微笑面容,全身皮膚未見皮疹及出血點,心肺腹查體未見異常;四肢肌力5級,肌張力正常,四肢活動自如,腱反射對稱引出,踝陣攣陰性,病理征陰性,腦膜刺激征未引出;余查體未見異常。
輔助檢查:患兒入院前(2024年2月15日)完善學齡韋氏智力測試:智商58分(具體不詳),未行特殊干預。入院后(2024年3月19日)完善血常規、肝功能[乳酸脫氫酶287.3 U/L,正常(120.0~250.0 U/L)]、腎功能、心肌酶、凝血功能、血氨、乳酸、炎癥指標、血沉、免疫5項、傳染病四項、糞便常規、尿常規、心電圖:未見異常。頭顱核磁共振成像(2024年3月20日)示:透明隔間腔增寬,右側篩竇囊腫(圖1)。4 h 視頻腦電圖(2024年3月21日)示:額、前顳區頻發性棘波、棘慢波放電,左側明顯(圖2)。2024年4月20日行《中國修訂韋氏兒童智力量表(C-WISC)》測試示:智商55,言語智商65,操作智商54;社會生活適應能力檢查:8分;注意力測試:52分;提示智商低下、注意缺陷多動障礙,輕度社會適應能力落后。入院經監護人知情同意各抽取患兒及父母外周靜脈血2 mL,送檢全外顯子組高通量測序、基因組拷貝數變異測序及線粒體全基因組合檢測發現HIVEP2基因出現一處無義變異:c.6667C>T,p.Arg2223*(圖3),HIVEP2基因第6667位C被替換為T,檢測結果經Sanger測序驗證(圖4)。本例變異為雜合無義突變,變異發生于精氨酸,該無義變異會導致編碼蛋白序列提前終止,產生截短蛋白或被降解,可能會對蛋白質的結構和功能產生較大影響。當前該位點在普通東亞人群中的頻率未見報道,既往有文獻報道相關疾病患者檢出該變異且均為新發變異。根據美國醫學遺傳學與基因組學會(American College of Medical Genetics and Genomics,ACMG)指南[11]分析,該變異被判定為致病性變異(PVS1_M+ PM2_P+PM6_S)。該變異高度吻合患兒表型:微笑面容、癲癇發作、發育遲緩、智力低下、注意缺陷多動障礙,最終,該患兒被診斷為MRD43。
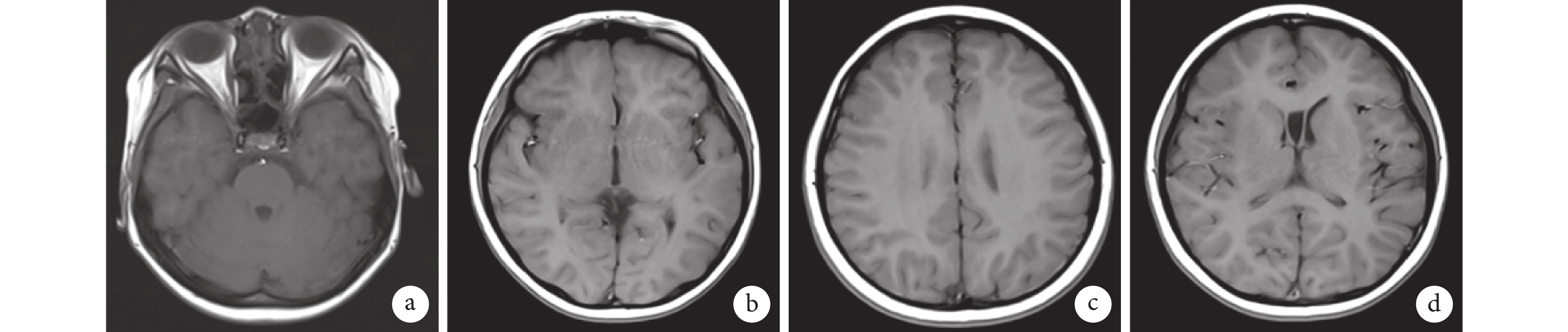
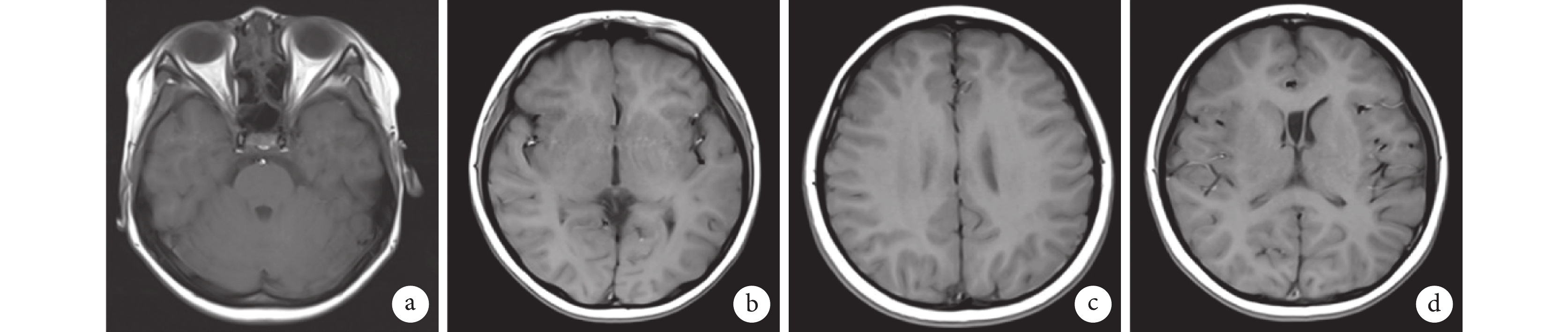 圖1
患兒頭部磁共振成像平掃
圖1
患兒頭部磁共振成像平掃
腦MRI未見異常,透明隔間腔增寬,右側篩竇囊腫
 圖2
患兒視頻腦電圖
圖2
患兒視頻腦電圖
額、前顳區頻發性棘波、棘慢波放電,左側明顯。a. 清醒狀態;b. 睡眠狀態
 圖3
HIVEP2 蛋白致病變異位置示意圖
圖3
HIVEP2 蛋白致病變異位置示意圖
 圖4
HIVEP2突變位點的Sanger測序結果
圖4
HIVEP2突變位點的Sanger測序結果
該患兒的
診斷、治療、隨訪:根據患兒臨床表型(智力低下、發育遲緩、癲癇發作、注意缺陷、多動障礙)、查體(微笑面容、運動欠協調)、輔助檢查及基因檢出結果,診斷“① 癲癇[局灶性發作,伴自主神經癥狀的自限性癲癇(self-limited epilepsy with autonomic seizures,SeLEAS)]?:患兒于7歲起病,頭圍及神經系統體格檢查陰性,自起病患兒癲癇發作少于5次,每次癲癇發作均發生在睡眠過程中,癲癇發作前患兒均有嘔吐癥狀,嘔吐是SeLEAS最常見的自主神經癥狀,患兒的腦電圖示額、前顳區棘波、棘慢波放電,頭顱核磁未提示異常,全外顯子組高通量測序提示患兒存在HIVEP2基因變異,故診斷。予以口服左乙拉西坦片控制癲癇發作,逐漸增量至750 mg(25 mg/kg),每日兩次,近6月未再出現癲癇發作;② MRD43,囑患兒加強社會適應行為訓練;③ 注意缺陷多動障礙,口服鹽酸托莫西汀,20 mg(0.67 mg/kg),每日兩次;④ 智力低下”。直至2024年10月,共隨訪6個月,患兒未再出現癲癇發作,多動行為較前有明顯改善,注意力較前集中,但學習成績差,我科門診隨診。
討論 HIVEP2(OMIM:616977)屬于ZAS蛋白家族,位于6q23-q24位點,作為一種鋅指蛋白參與機體免疫、大腦發育、脂肪及骨形成[10]。HIVEP2在多巴胺神經元細胞質及細胞核中表達,是調節NF-?B和神經發育必需的多種基因的轉錄因子,它通過激活SLC6A3基因調整多巴胺能活性在神經元發育等過程中發揮關鍵作用 [2,12]。有研究報道HIVEP2可通過抑制c-myc等基因的轉錄影響細胞生長、凋亡[13]。
截止2024年10月11日,以“HIVEP2”或“常染色體顯性智力障礙43型”或“Mental retardation type 43”為關鍵詞檢索中國知識基礎設施、維普、萬方、pubmed、WOS、Uptodate等數據庫,共檢索到20例已報道病例(包括18個新發變異:無義、錯義突變、移碼突變,2個遺傳變異來源于家系),其中包括16個HIVEP2基因突變位點,見表1[1-10]。
回顧既往已報道HIVEP2基因變異病例[1-10,13],智力障礙、發育遲緩(包括語言、運動)、肌張力減低為主要臨床特征,但HIVEP2變異患者的智力測評情況不等,基因型與智力障礙表型不具有一致性;少數患兒存在面容異常(如天使綜合征面容、小頭畸形、眼距寬、鼻根寬、小嘴等)、頭顱結構異常、注意缺陷多動障礙、異常的精神行為、癲癇發作、呼吸道及胃腸道癥狀等,存在社交適應、情感及認知行為相關障礙。
癲癇發作是HIVEP2變異患者的一種罕見的臨床表型,當前少有關于癲癇表型的詳細診療記錄,HIVEP2基因變異可能導致的癲癇發作表型仍需進一步探討。Steinfeld等 [3]于2016年報道2例HIVEP2變異病例,1例患者有全面強直發作、肌陣攣、復雜部分癲癇發作類型,腦電圖提示前額雙側尖波和慢波,頭顱核磁共振成像提示存在輕度腦容積減低,無治療記錄;另1例患者生后共出現兩次癲癇發作(1次發熱,1次無發熱),頭顱核磁共振成像及腦電圖未見異常,無治療情況記錄。王星辰等[5]于2022年報道一例男性患兒,3歲起病,癲癇發作形式為全面強直發作,頭顱正電子發射-計算機斷層掃描提示雙側額葉近中線處代謝減低、顳葉代謝減低、海馬萎縮,左側顯著;雙側丘腦、基底節區、皮髓質交界區多發對稱性鈣化灶以及透明間腔擴大,視頻腦電圖提示清醒腦電圖有癲癇波:全導陣發同步發放少量中高幅棘慢波、多棘慢波;雙側額極、額、中央區、前顳極、額中線、中央中線陣發(非)同步出現較多中棘慢波,中顳區偶有出現,雙側前頭部顯著;睡眠期腦電圖同清醒期。就診后規律口服丙戊酸鈉,癲癇發作未能完全控制。王新平等[4]于2023年報道報道一例HIVEP2變異合并癲癇性腦病患者,癲癇發作起病時間不詳,頭顱核磁共振成像未見異常,癲癇發作形式及腦電圖均未描述。本例患兒在睡眠時出現癲癇發作,表現為局灶性發作,其癲癇綜合征考慮為SeLEAS,與既往文獻報道癲癇發作形式有所差異,表明HIVEP2基因變異導致的癲癇表型存在多種發作形式。
研究報道,從HIVEP2基因敲除小鼠的大腦中檢測到升高的炎癥標志物,如GFAP、NADH/NADPH oxidase p22 phox等,小鼠經氟哌啶醇及抗炎藥物治療后,其記憶能力及精神行為較前改善,因此,此類藥物被認為可用于改善具有此類癥狀的人群[14]。此外,也有報道哌醋甲酯可以改善HIVEP2基因變異患者的注意缺陷多動障礙癥狀[8]。Abreu等[6]認為 HIVEP2基因突變導致MRD43患者無特異性治療方案,語言及職業技能訓練、身心療法或騎馬等戶外活動有助于這類患者獲益[14]。當前無明確藥物推薦用于HIVEP2基因變異致癲癇發作患者,本例患兒未使用氟哌啶醇、抗炎藥物或哌醋甲酯,口服左乙拉西坦聯合鹽酸托莫西汀,患兒末次癲癇發作完全控制,且注意力較前集中、多動癥狀較前減輕,表明左乙拉西坦及托莫西汀對于HIVEP2基因變異導致癲癇發作及注意缺陷多動障礙治療效果較好。
本文描述了1例以癲癇為主要表現的HIVEP2基因c.6667C>T(p.Arg2223*)位點突變導致常染色體顯性智力障礙43型兒童病例,于學齡期起病,癲癇發作類型為局灶性發作。Park等[2]在2019年首次報道一例c.6667C>T(p.Arg2223*)位點新發突變病例,該患者為29歲女性,臨床表型表現為輕度語言發育落后,2歲時說第一句話;輕度智商減低,智商測定值為52;輕度運動發育遲緩, 20月齡可獨立行走;肌張力低下,不存在其他精神行為異常;腦電圖正常,無癲癇發作;性格躁動,常出現呼吸道感染及胃腸功能紊亂。2022年Quental等[8]報道了第2例該位點變異病例,11歲男童出現新發突變,臨床表型為智力障礙、言語表達延遲、注意力缺陷、多動障礙、運動協調能力欠佳、自幼喂養困難,無癲癇發作,頭顱核磁未見異常,韋氏智力評分為59分。與此前報道2例HIVEP2基因[c.6667C>T (p.Arg2223*)]變異病例相比,除智力低下、發育遲緩、注意缺陷多動障礙、運動能力欠協調之外,本例患兒還表現有癲癇表型、微笑面容,結合家系全外顯子測序結果,HIVEP2基因變異[c.6667C>T (p.Arg2223*)]被認為是致使該患者癲癇發作的病因。
HIVEP2基因與神經系統發育有關,Park等[2]研究發現HIVEP2基因敲除小鼠表現出智力障礙、活動異常。Takagi等[15]在HIVEP2基因缺陷患者中通常觀察到智力缺陷、多動障礙及焦躁,本例患兒伴有智力障礙、注意力缺陷及多動障礙與此前報道一致。HIVEP2參與2型生長抑素受體的表達,該基因變異可能影響大腦神經元發育成熟[1],這可能是HIVEP2基因突變患者合并癲癇發作的原因之一。部分患者伴有頭顱結構異常,如小腦、胼胝體不完全發育、髓鞘形成不全、額葉萎縮等,但頭顱核磁共振成像異常與癲癇發作之間一致性尚不明確[1,6,8]。本例患兒頭顱核磁共振成像雖然提示顱內透明隔間腔增寬,尚無證據支持癲癇發作與其有關。
綜上,本研究報道了1例HIVEP2基因新發無義變異致使MRD43合并癲癇的病例,該患兒存在微笑面容、發育遲緩、智力低下、注意缺陷多動障礙、癲癇發作,本病例擴展了HIVEP2基因c.6667C>T (p.Arg2223*) 變異位點的臨床表型。當前HIVEP2基因變異所致MRD43無特異性治療方法,應盡早結合患兒臨床特征及相關遺傳學檢測方案明確病因,及早進行對癥及康復訓練。本病例表明左乙拉西坦對于HIVEP2基因變異所致癲癇有效,鹽酸托莫西汀對于治療注意缺陷多動障礙有效,幫助臨床醫師加深對HIVEP2基因變異疾病的認識,同時,也表明全外顯子測序在明確遺傳性疾病患者的致病原因方面是一種重要的診斷工具。
利益沖突聲明 所有作者無利益沖突。
常染色體顯性智力障礙43型(Mental retardation type 43,MRD43,OMIM 616977)是由HIVEP2基因變異導致的一種罕見常染色體顯性遺傳病,由Ranch于2012年首次報道,本病主要特征為發育遲緩、智力障礙[1,2]。對國內外HIVEP2基因變異案例報道進行文獻回顧,共檢索到20例相關病例[1-10],其中20/20存在發育落后,19/20存在智力障礙,4/20存在癲癇。現報道1例通過全外顯子基因測序確診的HIVEP2基因變異[c.6667C>T (p.Arg2223*)]導致以癲癇為主要表型的MRD43患兒,擴展了該病癲癇表型的藥物治療譜。本研究獲得石河子大學第一附屬醫院醫學倫理委員會審核批準(KJ2022-216-01)及患兒家屬知情同意。
病例資料 患兒 男,7歲10月齡。因“24 h無熱抽搐發作2次”就診石河子大學第一附屬醫院。患兒于當日夜間入睡1 h后無明顯誘因出現嘔吐胃內容物1次,隨后出現四肢強直陣攣,呼之不應,意識喪失,雙眼上翻,牙關緊閉,口唇發紺,喉中發聲,約1~2 min意識恢復,發作后有四肢發軟,自覺乏力;次日晨時9:00患兒于睡眠臨清醒時再次發作,發作前也有嘔吐,發作形式基本同前;隨后急診來院。患兒為G2P1,足月剖宮產,混合喂養,生后體健,Apgar評分不詳,3月齡抬頭、6月齡會坐、12月齡會站、16月齡會走、可喊“爸爸、媽媽”。患兒現為一年級學生,言語欠清晰,運動欠協調,學習成績較差,注意力不集中,平日多動行為。否認窒息等異常出生史、頭顱外傷史、癲癇等家族遺傳史。
體格檢查:體溫36.9℃,脈搏82次/min,呼吸25次/min,血壓:92/54 mmHg,身高136 cm,體質量30 kg,頭圍51 cm。神志清楚,精神反應正常,微笑面容,全身皮膚未見皮疹及出血點,心肺腹查體未見異常;四肢肌力5級,肌張力正常,四肢活動自如,腱反射對稱引出,踝陣攣陰性,病理征陰性,腦膜刺激征未引出;余查體未見異常。
輔助檢查:患兒入院前(2024年2月15日)完善學齡韋氏智力測試:智商58分(具體不詳),未行特殊干預。入院后(2024年3月19日)完善血常規、肝功能[乳酸脫氫酶287.3 U/L,正常(120.0~250.0 U/L)]、腎功能、心肌酶、凝血功能、血氨、乳酸、炎癥指標、血沉、免疫5項、傳染病四項、糞便常規、尿常規、心電圖:未見異常。頭顱核磁共振成像(2024年3月20日)示:透明隔間腔增寬,右側篩竇囊腫(圖1)。4 h 視頻腦電圖(2024年3月21日)示:額、前顳區頻發性棘波、棘慢波放電,左側明顯(圖2)。2024年4月20日行《中國修訂韋氏兒童智力量表(C-WISC)》測試示:智商55,言語智商65,操作智商54;社會生活適應能力檢查:8分;注意力測試:52分;提示智商低下、注意缺陷多動障礙,輕度社會適應能力落后。入院經監護人知情同意各抽取患兒及父母外周靜脈血2 mL,送檢全外顯子組高通量測序、基因組拷貝數變異測序及線粒體全基因組合檢測發現HIVEP2基因出現一處無義變異:c.6667C>T,p.Arg2223*(圖3),HIVEP2基因第6667位C被替換為T,檢測結果經Sanger測序驗證(圖4)。本例變異為雜合無義突變,變異發生于精氨酸,該無義變異會導致編碼蛋白序列提前終止,產生截短蛋白或被降解,可能會對蛋白質的結構和功能產生較大影響。當前該位點在普通東亞人群中的頻率未見報道,既往有文獻報道相關疾病患者檢出該變異且均為新發變異。根據美國醫學遺傳學與基因組學會(American College of Medical Genetics and Genomics,ACMG)指南[11]分析,該變異被判定為致病性變異(PVS1_M+ PM2_P+PM6_S)。該變異高度吻合患兒表型:微笑面容、癲癇發作、發育遲緩、智力低下、注意缺陷多動障礙,最終,該患兒被診斷為MRD43。
圖1
患兒頭部磁共振成像平掃
腦MRI未見異常,透明隔間腔增寬,右側篩竇囊腫
圖2
患兒視頻腦電圖
額、前顳區頻發性棘波、棘慢波放電,左側明顯。a. 清醒狀態;b. 睡眠狀態
圖3
HIVEP2 蛋白致病變異位置示意圖
圖4
HIVEP2突變位點的Sanger測序結果
該患兒的
診斷、治療、隨訪:根據患兒臨床表型(智力低下、發育遲緩、癲癇發作、注意缺陷、多動障礙)、查體(微笑面容、運動欠協調)、輔助檢查及基因檢出結果,診斷“① 癲癇[局灶性發作,伴自主神經癥狀的自限性癲癇(self-limited epilepsy with autonomic seizures,SeLEAS)]?:患兒于7歲起病,頭圍及神經系統體格檢查陰性,自起病患兒癲癇發作少于5次,每次癲癇發作均發生在睡眠過程中,癲癇發作前患兒均有嘔吐癥狀,嘔吐是SeLEAS最常見的自主神經癥狀,患兒的腦電圖示額、前顳區棘波、棘慢波放電,頭顱核磁未提示異常,全外顯子組高通量測序提示患兒存在HIVEP2基因變異,故診斷。予以口服左乙拉西坦片控制癲癇發作,逐漸增量至750 mg(25 mg/kg),每日兩次,近6月未再出現癲癇發作;② MRD43,囑患兒加強社會適應行為訓練;③ 注意缺陷多動障礙,口服鹽酸托莫西汀,20 mg(0.67 mg/kg),每日兩次;④ 智力低下”。直至2024年10月,共隨訪6個月,患兒未再出現癲癇發作,多動行為較前有明顯改善,注意力較前集中,但學習成績差,我科門診隨診。
討論 HIVEP2(OMIM:616977)屬于ZAS蛋白家族,位于6q23-q24位點,作為一種鋅指蛋白參與機體免疫、大腦發育、脂肪及骨形成[10]。HIVEP2在多巴胺神經元細胞質及細胞核中表達,是調節NF-?B和神經發育必需的多種基因的轉錄因子,它通過激活SLC6A3基因調整多巴胺能活性在神經元發育等過程中發揮關鍵作用 [2,12]。有研究報道HIVEP2可通過抑制c-myc等基因的轉錄影響細胞生長、凋亡[13]。
截止2024年10月11日,以“HIVEP2”或“常染色體顯性智力障礙43型”或“Mental retardation type 43”為關鍵詞檢索中國知識基礎設施、維普、萬方、pubmed、WOS、Uptodate等數據庫,共檢索到20例已報道病例(包括18個新發變異:無義、錯義突變、移碼突變,2個遺傳變異來源于家系),其中包括16個HIVEP2基因突變位點,見表1[1-10]。
回顧既往已報道HIVEP2基因變異病例[1-10,13],智力障礙、發育遲緩(包括語言、運動)、肌張力減低為主要臨床特征,但HIVEP2變異患者的智力測評情況不等,基因型與智力障礙表型不具有一致性;少數患兒存在面容異常(如天使綜合征面容、小頭畸形、眼距寬、鼻根寬、小嘴等)、頭顱結構異常、注意缺陷多動障礙、異常的精神行為、癲癇發作、呼吸道及胃腸道癥狀等,存在社交適應、情感及認知行為相關障礙。
癲癇發作是HIVEP2變異患者的一種罕見的臨床表型,當前少有關于癲癇表型的詳細診療記錄,HIVEP2基因變異可能導致的癲癇發作表型仍需進一步探討。Steinfeld等 [3]于2016年報道2例HIVEP2變異病例,1例患者有全面強直發作、肌陣攣、復雜部分癲癇發作類型,腦電圖提示前額雙側尖波和慢波,頭顱核磁共振成像提示存在輕度腦容積減低,無治療記錄;另1例患者生后共出現兩次癲癇發作(1次發熱,1次無發熱),頭顱核磁共振成像及腦電圖未見異常,無治療情況記錄。王星辰等[5]于2022年報道一例男性患兒,3歲起病,癲癇發作形式為全面強直發作,頭顱正電子發射-計算機斷層掃描提示雙側額葉近中線處代謝減低、顳葉代謝減低、海馬萎縮,左側顯著;雙側丘腦、基底節區、皮髓質交界區多發對稱性鈣化灶以及透明間腔擴大,視頻腦電圖提示清醒腦電圖有癲癇波:全導陣發同步發放少量中高幅棘慢波、多棘慢波;雙側額極、額、中央區、前顳極、額中線、中央中線陣發(非)同步出現較多中棘慢波,中顳區偶有出現,雙側前頭部顯著;睡眠期腦電圖同清醒期。就診后規律口服丙戊酸鈉,癲癇發作未能完全控制。王新平等[4]于2023年報道報道一例HIVEP2變異合并癲癇性腦病患者,癲癇發作起病時間不詳,頭顱核磁共振成像未見異常,癲癇發作形式及腦電圖均未描述。本例患兒在睡眠時出現癲癇發作,表現為局灶性發作,其癲癇綜合征考慮為SeLEAS,與既往文獻報道癲癇發作形式有所差異,表明HIVEP2基因變異導致的癲癇表型存在多種發作形式。
研究報道,從HIVEP2基因敲除小鼠的大腦中檢測到升高的炎癥標志物,如GFAP、NADH/NADPH oxidase p22 phox等,小鼠經氟哌啶醇及抗炎藥物治療后,其記憶能力及精神行為較前改善,因此,此類藥物被認為可用于改善具有此類癥狀的人群[14]。此外,也有報道哌醋甲酯可以改善HIVEP2基因變異患者的注意缺陷多動障礙癥狀[8]。Abreu等[6]認為 HIVEP2基因突變導致MRD43患者無特異性治療方案,語言及職業技能訓練、身心療法或騎馬等戶外活動有助于這類患者獲益[14]。當前無明確藥物推薦用于HIVEP2基因變異致癲癇發作患者,本例患兒未使用氟哌啶醇、抗炎藥物或哌醋甲酯,口服左乙拉西坦聯合鹽酸托莫西汀,患兒末次癲癇發作完全控制,且注意力較前集中、多動癥狀較前減輕,表明左乙拉西坦及托莫西汀對于HIVEP2基因變異導致癲癇發作及注意缺陷多動障礙治療效果較好。
本文描述了1例以癲癇為主要表現的HIVEP2基因c.6667C>T(p.Arg2223*)位點突變導致常染色體顯性智力障礙43型兒童病例,于學齡期起病,癲癇發作類型為局灶性發作。Park等[2]在2019年首次報道一例c.6667C>T(p.Arg2223*)位點新發突變病例,該患者為29歲女性,臨床表型表現為輕度語言發育落后,2歲時說第一句話;輕度智商減低,智商測定值為52;輕度運動發育遲緩, 20月齡可獨立行走;肌張力低下,不存在其他精神行為異常;腦電圖正常,無癲癇發作;性格躁動,常出現呼吸道感染及胃腸功能紊亂。2022年Quental等[8]報道了第2例該位點變異病例,11歲男童出現新發突變,臨床表型為智力障礙、言語表達延遲、注意力缺陷、多動障礙、運動協調能力欠佳、自幼喂養困難,無癲癇發作,頭顱核磁未見異常,韋氏智力評分為59分。與此前報道2例HIVEP2基因[c.6667C>T (p.Arg2223*)]變異病例相比,除智力低下、發育遲緩、注意缺陷多動障礙、運動能力欠協調之外,本例患兒還表現有癲癇表型、微笑面容,結合家系全外顯子測序結果,HIVEP2基因變異[c.6667C>T (p.Arg2223*)]被認為是致使該患者癲癇發作的病因。
HIVEP2基因與神經系統發育有關,Park等[2]研究發現HIVEP2基因敲除小鼠表現出智力障礙、活動異常。Takagi等[15]在HIVEP2基因缺陷患者中通常觀察到智力缺陷、多動障礙及焦躁,本例患兒伴有智力障礙、注意力缺陷及多動障礙與此前報道一致。HIVEP2參與2型生長抑素受體的表達,該基因變異可能影響大腦神經元發育成熟[1],這可能是HIVEP2基因突變患者合并癲癇發作的原因之一。部分患者伴有頭顱結構異常,如小腦、胼胝體不完全發育、髓鞘形成不全、額葉萎縮等,但頭顱核磁共振成像異常與癲癇發作之間一致性尚不明確[1,6,8]。本例患兒頭顱核磁共振成像雖然提示顱內透明隔間腔增寬,尚無證據支持癲癇發作與其有關。
綜上,本研究報道了1例HIVEP2基因新發無義變異致使MRD43合并癲癇的病例,該患兒存在微笑面容、發育遲緩、智力低下、注意缺陷多動障礙、癲癇發作,本病例擴展了HIVEP2基因c.6667C>T (p.Arg2223*) 變異位點的臨床表型。當前HIVEP2基因變異所致MRD43無特異性治療方法,應盡早結合患兒臨床特征及相關遺傳學檢測方案明確病因,及早進行對癥及康復訓練。本病例表明左乙拉西坦對于HIVEP2基因變異所致癲癇有效,鹽酸托莫西汀對于治療注意缺陷多動障礙有效,幫助臨床醫師加深對HIVEP2基因變異疾病的認識,同時,也表明全外顯子測序在明確遺傳性疾病患者的致病原因方面是一種重要的診斷工具。
利益沖突聲明 所有作者無利益沖突。