引用本文: 張磊, 劉沛洋, 王海燕, 王茹. 首診于眼科的Cohen綜合征1例. 中華眼底病雜志, 2024, 40(3): 222-224. doi: 10.3760/cma.j.cn511434-20220722-00415 復制
版權信息: ?四川大學華西醫院華西期刊社《中華眼底病雜志》版權所有,未經授權不得轉載、改編
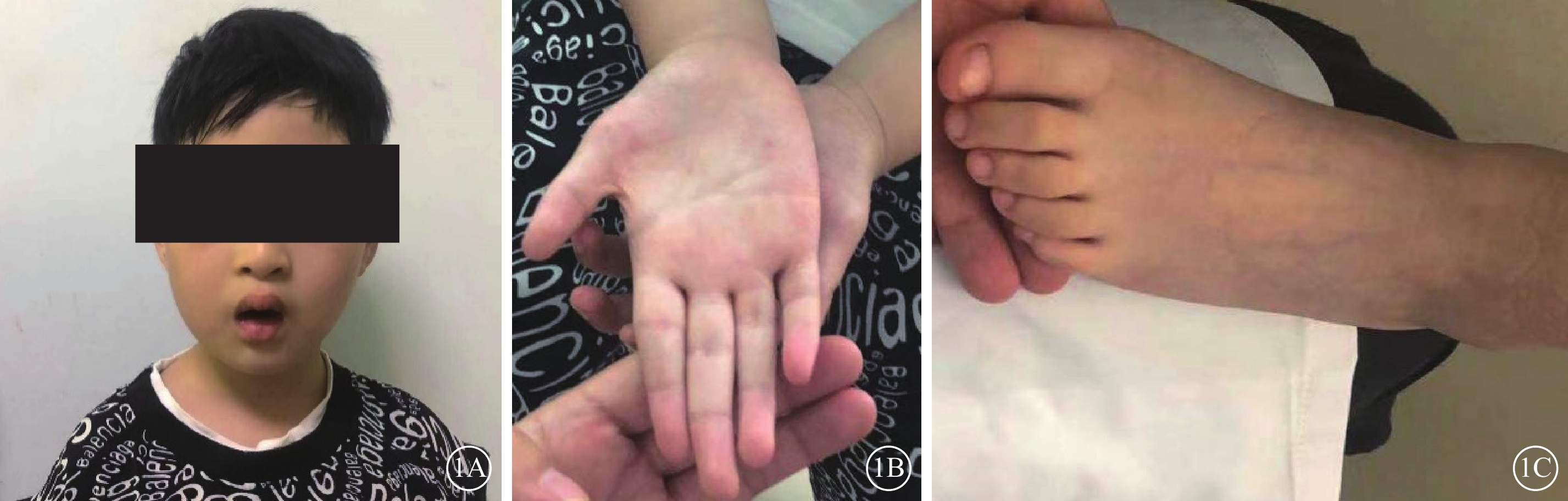
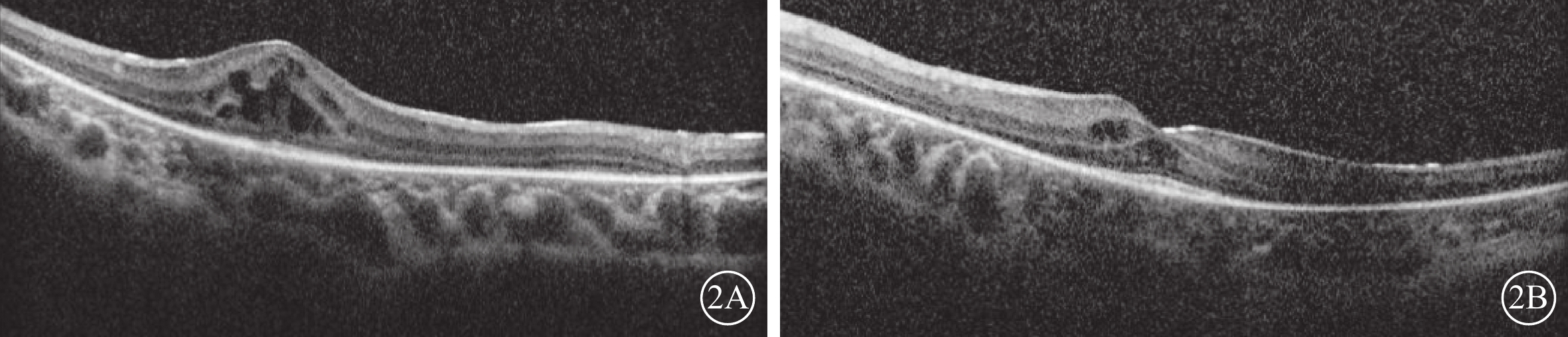
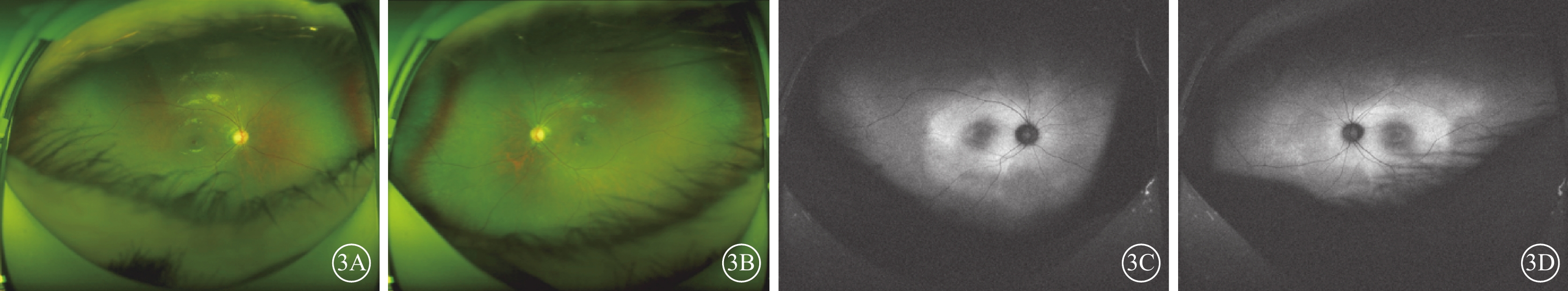
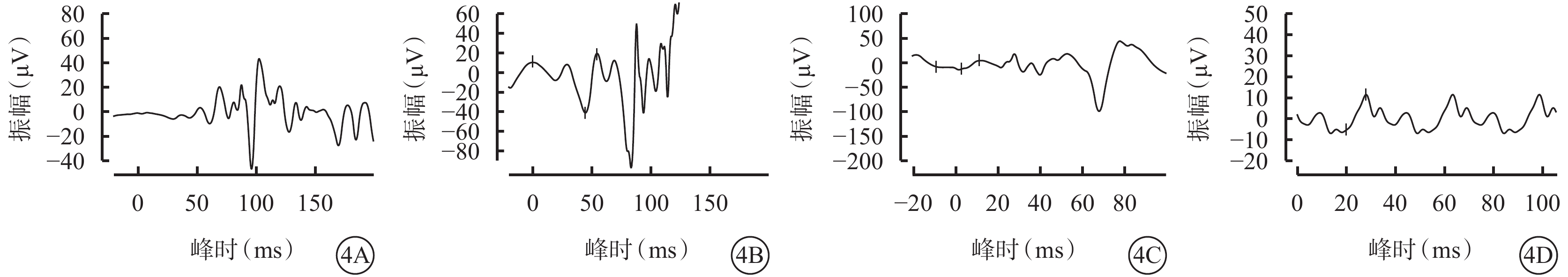
患兒男,6歲。因雙眼畏光2年于2021年3月12日到陜西省眼科醫院就診。患兒為第一胎第一產,足月孕39周+5順產。1歲時外院兒科診斷為“先天發育遲緩”,運動較同齡人差(約2歲時能說完整的詞語,3歲時可行走)。否認夜盲及色盲,否認癲癇病史。父母非近親結婚,無家族遺傳病史。體格檢查:體重24 kg,身高120 cm,頭圍48 cm;神志清晰,反應遲鈍;面容特殊(頭圍小,頭發濃密、發際線低,瞼裂下斜,人中短,上嘴唇凸出)(圖1A);通貫掌紋,手指腳趾較為纖細(圖1B,1C);骨骼發育、心臟彩色超聲及心電圖檢查無異常。實驗室檢查:血常規測得白細胞5.86×109個/L,中性粒細胞1.46×109 個/L,中性粒細胞百分比24.8%。患兒父母表型無異常。眼科檢查:雙眼裸眼視力均為0.2;眼壓正常。眼底檢查,雙眼視盤邊界清楚,顏色略淡,視網膜平伏,黃斑中心凹反光不清。光相干斷層掃描(OCT)檢查,雙眼黃斑囊樣反射,除中心凹外,橢圓體帶和外層視網膜萎縮(圖2)。激光掃描檢眼鏡檢查,雙眼黃斑中心凹旁色素沉著伴“牛眼樣”黃斑病變(圖3A,3B)。眼底自身熒光成像顯示雙眼黃斑環形強自身熒光,周圍環繞弱自身熒光,外圍強自身熒光累及后極部和視盤鼻側(圖3C,3D)。全視野視網膜電圖(ERG)檢查,雙眼ERG各波振幅下降(圖4)。門診初步診斷:雙眼黃斑囊樣水腫(CME)。
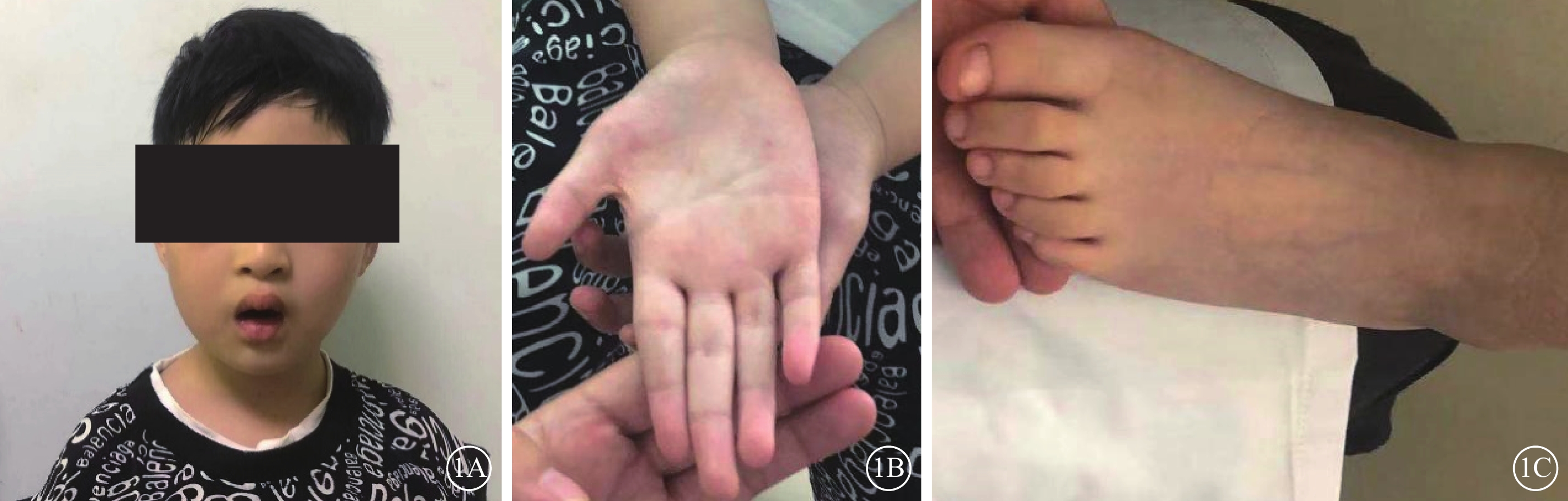 圖1
Cohen綜合征患兒體格檢查像
圖1
Cohen綜合征患兒體格檢查像
1A示面容外觀像,頭圍小,頭發濃密、發際線低,瞼裂下斜,人中短,上嘴唇凸出;1B示手掌外觀像,手指纖細,通貫掌紋;1C示腳掌外觀像,腳趾纖細
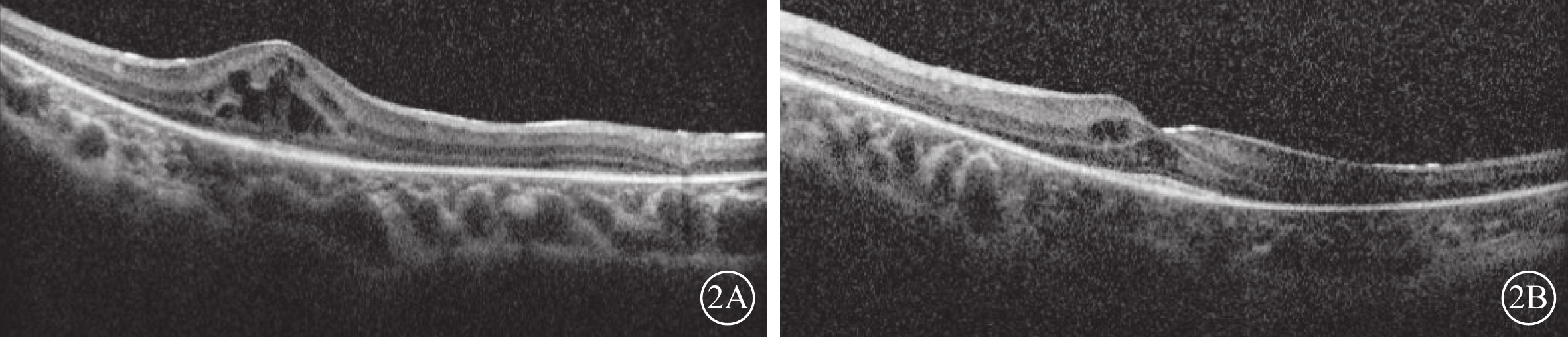 圖2
Cohen綜合征患兒雙眼光相干斷層掃描像
圖2
Cohen綜合征患兒雙眼光相干斷層掃描像
2A、2B分別示右眼、左眼 雙眼黃斑囊樣反射,除中心凹外,橢圓體帶缺失和外層視網膜萎縮
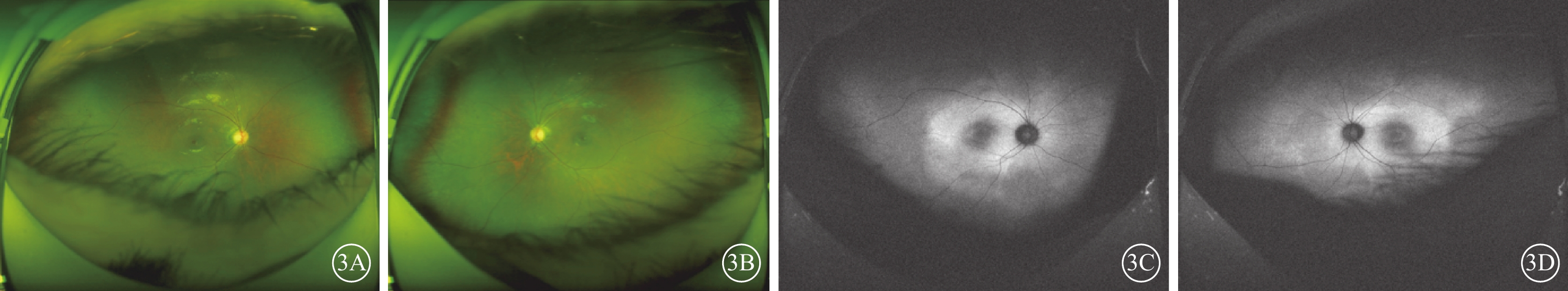 圖3
Cohen綜合征患兒雙眼激光掃描檢眼鏡及眼底自身熒光像
圖3
Cohen綜合征患兒雙眼激光掃描檢眼鏡及眼底自身熒光像
3A、3B分別示右眼、左眼激光掃描檢眼鏡像,雙眼黃斑中心凹旁色素沉著伴“牛眼樣”黃斑病變;3C、3D分別示右眼、左眼眼底自身熒光像,雙眼黃斑環形強自身熒光,周圍環繞弱自身熒光,外圍強自身熒光累及后極部
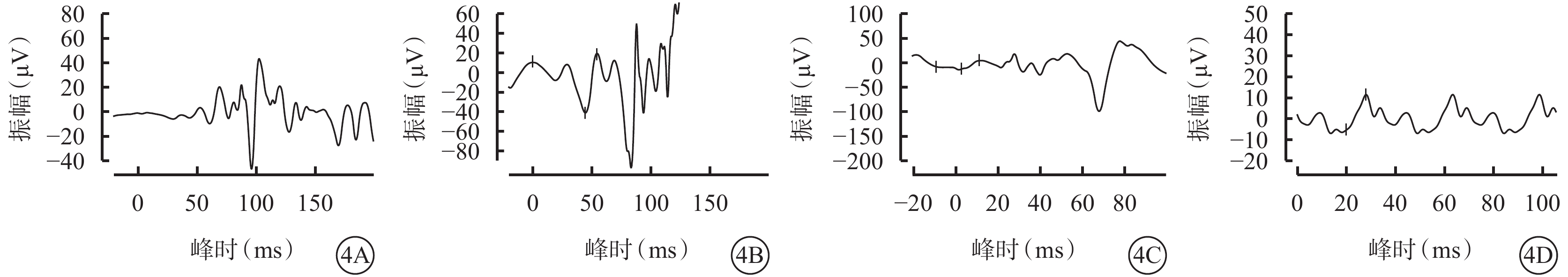 圖4
Cohen綜合征患兒右眼全視野視網膜電圖(ERG)檢查像
圖4
Cohen綜合征患兒右眼全視野視網膜電圖(ERG)檢查像
4A~4D分別示暗適應0.01 ERG、暗適應3.0 ERG、明適應3.0 ERG、明適應30 Hz閃爍光,ERG各波振幅下降
為進一步明確診斷,在遵循《赫爾辛基宣言》原則下并取得患兒家長知情同意后,采集患兒及其父母靜脈血,進行高通量全外顯子組測序。結果顯示,患兒攜帶VPS13B基因復合雜合變異c.6940+1G>T和c.10156dup(p.Thr3386AsnfsTer3),分別來源于父親、母親(圖5)。這兩處變異在人類基因變異數據庫(
 圖5
Cohen綜合征患兒及其父母變異位點測序驗證
圖5
Cohen綜合征患兒及其父母變異位點測序驗證
患兒攜帶
討論 VPS13B基因編碼一種跨膜蛋白,參與囊泡介導的細胞內蛋白質運輸和分類,并在眼睛、血液系統和中樞神經系統的發育和功能中發揮作用。Cohen綜合征是由VPS13B(COH1)基因突變導致的累及全身多器官系統的常染色體隱性遺傳疾病,以軀體特征、智力發育遲緩、中性粒細胞減少和眼部異常為主要特征,又被稱為“腦-肥胖-眼-骨骼”綜合征[1]。
CME在Cohen綜合征患者中發病率較高[2]。本例患兒同樣存在雙眼CME。有研究認為,Cohen綜合征中的黃斑囊腔樣表現可能是由于視網膜結構的異常,而不是血視網膜屏障的破壞和視網膜內液體的積聚,如Cohen綜合征患者可伴有高度近視和后鞏膜葡萄腫,與視網膜劈裂有關,也可能導致相關視網膜病變的CME[3]。Cohen綜合征相關視網膜病變表現為典型的視網膜色素變性,包括夜盲、周邊視力喪失和進行性視網膜變性[1]。Chandler等[4]在22例Cohen綜合征患者中發現,11例患者存在嚴重的晚期視網膜病變,包括血管狹窄、骨細胞樣色素沉著和視盤蒼白。本例患兒視盤顏色略淡,但未發現典型的色素改變,可能由于患兒年齡小,尚未出現這些改變。“牛眼樣”黃斑病變也是Cohen綜合征最常見的視網膜病變。Nasser等[1]報道5例Cohen綜合征患者均存在“牛眼樣”黃斑改變。本例患兒OCT檢查顯示,其黃斑中心保留部分正常的外層結構,而周圍橢圓體帶缺失,雙眼基本對稱;雙眼眼底自身熒光成像呈“牛眼樣”自身熒光改變。
Cohen綜合征患者也常伴有高度近視[5],視力會逐漸惡化,在40歲時有明顯的視力障礙,最終視力可能僅限于數指和光感。ERG在早期可檢測出波形,但隨著年齡增長會顯示出衰減或消失的反應[2]。本例患兒雙眼ERG各波振幅下降,提示視網膜功能下降。因其發育遲緩和配合困難,無法辨認視力表,沒有完成矯正視力檢查;但我們囑其隨訪觀察,注意近視防控,以避免發生高度近視及其并發癥。Cohen綜合征的全身表現包括面部特征:小頭畸形、濃眉、低發際、長而粗的睫毛、下垂或波浪形的眼瞼裂、凸出的中上切牙、耳垂厚而折疊不良等;軀體特征有軀干肥胖、肌張力減退和關節過度伸展;血液學白細胞減少,尤其是中性粒細胞減少。本例患兒除眼部癥狀外,同時存在如發育遲緩、特殊面容和血液學異常表現,符合Cohen綜合征的全身表現。
本例患兒攜帶VPS13B基因復合雜合變異c.6940+1G>T和c.10156dup(p.Thr3386AsnfsTer3),分別遺傳于父親、母親。變異c.6940+1G>T曾在年齡分別為2、3歲的2例無血緣關系的中國男性患兒中被檢出,但這2例患兒均無眼部表型[6]。生物信息學預測該變異可能影響VPS13B基因mRNA的剪接。變異2c.10156dup(p.Thr3386AsnfsTer3)曾在1例2.5歲的意大利男性患兒中被檢出,其眼部存在外斜視伴有眼底異常和先天性黃斑萎縮[7]。該變異為移碼變異,影響閱讀框架,產生一個提前終止密碼子,導致蛋白質截短。VPS13B是高爾基復合體的一種結構膜蛋白,與Rab6(一種參與視網膜運輸的蛋白質)相互作用,VPS13B基因的功能缺失可導致高爾基體完整性紊亂,可能與黃斑囊腔改變有關,但具體機制尚不清楚。有研究認為,該異常可致神經細胞生成減少,最突出的與神經相關表型包括進行性視網膜營養不良、非進行性智力殘疾和出生后發育的小頭畸形[8-9]。本例患兒攜帶的基因變異所致Cohen綜合征表型在中國人群中為首次報道。
Cohen綜合征引起的CME,其經典治療包括局部和口服碳酸酐酶Ⅳ抑制劑(CAI),推測可通過改變視網膜色素上皮和Müller細胞內離子轉運系統的極性排出液體[10]。但Gabrielle等[2]采用CAI治療10例Cohen綜合征引起的CME患者,發現其并不能改善患者水腫程度,且隨著光感受器營養不良的進展,CME可能在成年期自發消失。本例患兒接受乙酰唑胺口服治療,隨訪期間CME沒有明顯變化,還在進一步觀察中。
當兒童出現發育遲緩和視網膜營養不良及其他全身表現時,需考慮 Cohen綜合征。識別其臨床特征并進行確認性基因檢測非常重要,這可以幫助醫生做出準確的診斷,制定個性化的治療方案,根據可能出現的全身問題需重點終身隨訪,預防并發癥,并幫助患兒提高生活質量。
患兒男,6歲。因雙眼畏光2年于2021年3月12日到陜西省眼科醫院就診。患兒為第一胎第一產,足月孕39周+5順產。1歲時外院兒科診斷為“先天發育遲緩”,運動較同齡人差(約2歲時能說完整的詞語,3歲時可行走)。否認夜盲及色盲,否認癲癇病史。父母非近親結婚,無家族遺傳病史。體格檢查:體重24 kg,身高120 cm,頭圍48 cm;神志清晰,反應遲鈍;面容特殊(頭圍小,頭發濃密、發際線低,瞼裂下斜,人中短,上嘴唇凸出)(圖1A);通貫掌紋,手指腳趾較為纖細(圖1B,1C);骨骼發育、心臟彩色超聲及心電圖檢查無異常。實驗室檢查:血常規測得白細胞5.86×109個/L,中性粒細胞1.46×109 個/L,中性粒細胞百分比24.8%。患兒父母表型無異常。眼科檢查:雙眼裸眼視力均為0.2;眼壓正常。眼底檢查,雙眼視盤邊界清楚,顏色略淡,視網膜平伏,黃斑中心凹反光不清。光相干斷層掃描(OCT)檢查,雙眼黃斑囊樣反射,除中心凹外,橢圓體帶和外層視網膜萎縮(圖2)。激光掃描檢眼鏡檢查,雙眼黃斑中心凹旁色素沉著伴“牛眼樣”黃斑病變(圖3A,3B)。眼底自身熒光成像顯示雙眼黃斑環形強自身熒光,周圍環繞弱自身熒光,外圍強自身熒光累及后極部和視盤鼻側(圖3C,3D)。全視野視網膜電圖(ERG)檢查,雙眼ERG各波振幅下降(圖4)。門診初步診斷:雙眼黃斑囊樣水腫(CME)。
圖1
Cohen綜合征患兒體格檢查像
1A示面容外觀像,頭圍小,頭發濃密、發際線低,瞼裂下斜,人中短,上嘴唇凸出;1B示手掌外觀像,手指纖細,通貫掌紋;1C示腳掌外觀像,腳趾纖細
圖2
Cohen綜合征患兒雙眼光相干斷層掃描像
2A、2B分別示右眼、左眼 雙眼黃斑囊樣反射,除中心凹外,橢圓體帶缺失和外層視網膜萎縮
圖3
Cohen綜合征患兒雙眼激光掃描檢眼鏡及眼底自身熒光像
3A、3B分別示右眼、左眼激光掃描檢眼鏡像,雙眼黃斑中心凹旁色素沉著伴“牛眼樣”黃斑病變;3C、3D分別示右眼、左眼眼底自身熒光像,雙眼黃斑環形強自身熒光,周圍環繞弱自身熒光,外圍強自身熒光累及后極部
圖4
Cohen綜合征患兒右眼全視野視網膜電圖(ERG)檢查像
4A~4D分別示暗適應0.01 ERG、暗適應3.0 ERG、明適應3.0 ERG、明適應30 Hz閃爍光,ERG各波振幅下降
為進一步明確診斷,在遵循《赫爾辛基宣言》原則下并取得患兒家長知情同意后,采集患兒及其父母靜脈血,進行高通量全外顯子組測序。結果顯示,患兒攜帶VPS13B基因復合雜合變異c.6940+1G>T和c.10156dup(p.Thr3386AsnfsTer3),分別來源于父親、母親(圖5)。這兩處變異在人類基因變異數據庫(
圖5
Cohen綜合征患兒及其父母變異位點測序驗證
患兒攜帶
討論 VPS13B基因編碼一種跨膜蛋白,參與囊泡介導的細胞內蛋白質運輸和分類,并在眼睛、血液系統和中樞神經系統的發育和功能中發揮作用。Cohen綜合征是由VPS13B(COH1)基因突變導致的累及全身多器官系統的常染色體隱性遺傳疾病,以軀體特征、智力發育遲緩、中性粒細胞減少和眼部異常為主要特征,又被稱為“腦-肥胖-眼-骨骼”綜合征[1]。
CME在Cohen綜合征患者中發病率較高[2]。本例患兒同樣存在雙眼CME。有研究認為,Cohen綜合征中的黃斑囊腔樣表現可能是由于視網膜結構的異常,而不是血視網膜屏障的破壞和視網膜內液體的積聚,如Cohen綜合征患者可伴有高度近視和后鞏膜葡萄腫,與視網膜劈裂有關,也可能導致相關視網膜病變的CME[3]。Cohen綜合征相關視網膜病變表現為典型的視網膜色素變性,包括夜盲、周邊視力喪失和進行性視網膜變性[1]。Chandler等[4]在22例Cohen綜合征患者中發現,11例患者存在嚴重的晚期視網膜病變,包括血管狹窄、骨細胞樣色素沉著和視盤蒼白。本例患兒視盤顏色略淡,但未發現典型的色素改變,可能由于患兒年齡小,尚未出現這些改變。“牛眼樣”黃斑病變也是Cohen綜合征最常見的視網膜病變。Nasser等[1]報道5例Cohen綜合征患者均存在“牛眼樣”黃斑改變。本例患兒OCT檢查顯示,其黃斑中心保留部分正常的外層結構,而周圍橢圓體帶缺失,雙眼基本對稱;雙眼眼底自身熒光成像呈“牛眼樣”自身熒光改變。
Cohen綜合征患者也常伴有高度近視[5],視力會逐漸惡化,在40歲時有明顯的視力障礙,最終視力可能僅限于數指和光感。ERG在早期可檢測出波形,但隨著年齡增長會顯示出衰減或消失的反應[2]。本例患兒雙眼ERG各波振幅下降,提示視網膜功能下降。因其發育遲緩和配合困難,無法辨認視力表,沒有完成矯正視力檢查;但我們囑其隨訪觀察,注意近視防控,以避免發生高度近視及其并發癥。Cohen綜合征的全身表現包括面部特征:小頭畸形、濃眉、低發際、長而粗的睫毛、下垂或波浪形的眼瞼裂、凸出的中上切牙、耳垂厚而折疊不良等;軀體特征有軀干肥胖、肌張力減退和關節過度伸展;血液學白細胞減少,尤其是中性粒細胞減少。本例患兒除眼部癥狀外,同時存在如發育遲緩、特殊面容和血液學異常表現,符合Cohen綜合征的全身表現。
本例患兒攜帶VPS13B基因復合雜合變異c.6940+1G>T和c.10156dup(p.Thr3386AsnfsTer3),分別遺傳于父親、母親。變異c.6940+1G>T曾在年齡分別為2、3歲的2例無血緣關系的中國男性患兒中被檢出,但這2例患兒均無眼部表型[6]。生物信息學預測該變異可能影響VPS13B基因mRNA的剪接。變異2c.10156dup(p.Thr3386AsnfsTer3)曾在1例2.5歲的意大利男性患兒中被檢出,其眼部存在外斜視伴有眼底異常和先天性黃斑萎縮[7]。該變異為移碼變異,影響閱讀框架,產生一個提前終止密碼子,導致蛋白質截短。VPS13B是高爾基復合體的一種結構膜蛋白,與Rab6(一種參與視網膜運輸的蛋白質)相互作用,VPS13B基因的功能缺失可導致高爾基體完整性紊亂,可能與黃斑囊腔改變有關,但具體機制尚不清楚。有研究認為,該異常可致神經細胞生成減少,最突出的與神經相關表型包括進行性視網膜營養不良、非進行性智力殘疾和出生后發育的小頭畸形[8-9]。本例患兒攜帶的基因變異所致Cohen綜合征表型在中國人群中為首次報道。
Cohen綜合征引起的CME,其經典治療包括局部和口服碳酸酐酶Ⅳ抑制劑(CAI),推測可通過改變視網膜色素上皮和Müller細胞內離子轉運系統的極性排出液體[10]。但Gabrielle等[2]采用CAI治療10例Cohen綜合征引起的CME患者,發現其并不能改善患者水腫程度,且隨著光感受器營養不良的進展,CME可能在成年期自發消失。本例患兒接受乙酰唑胺口服治療,隨訪期間CME沒有明顯變化,還在進一步觀察中。
當兒童出現發育遲緩和視網膜營養不良及其他全身表現時,需考慮 Cohen綜合征。識別其臨床特征并進行確認性基因檢測非常重要,這可以幫助醫生做出準確的診斷,制定個性化的治療方案,根據可能出現的全身問題需重點終身隨訪,預防并發癥,并幫助患兒提高生活質量。