引用本文: 張敏, 董照陽, 周昀, 魏偉. Usher綜合征伴黃斑水腫1例. 中華眼底病雜志, 2024, 40(3): 230-232. doi: 10.3760/cma.j.cn511434-20221103-00577 復制
版權信息: ?四川大學華西醫院華西期刊社《中華眼底病雜志》版權所有,未經授權不得轉載、改編
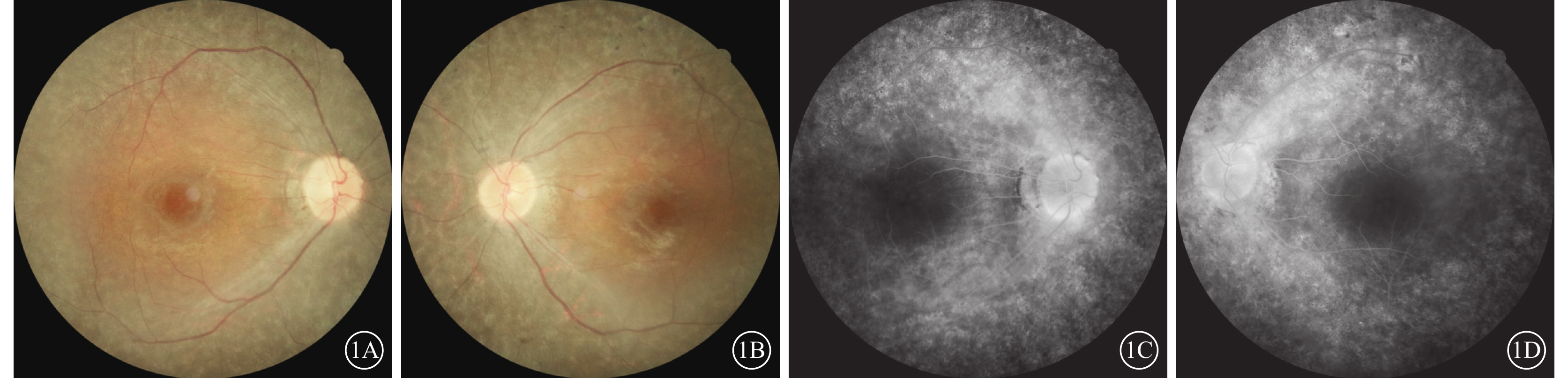
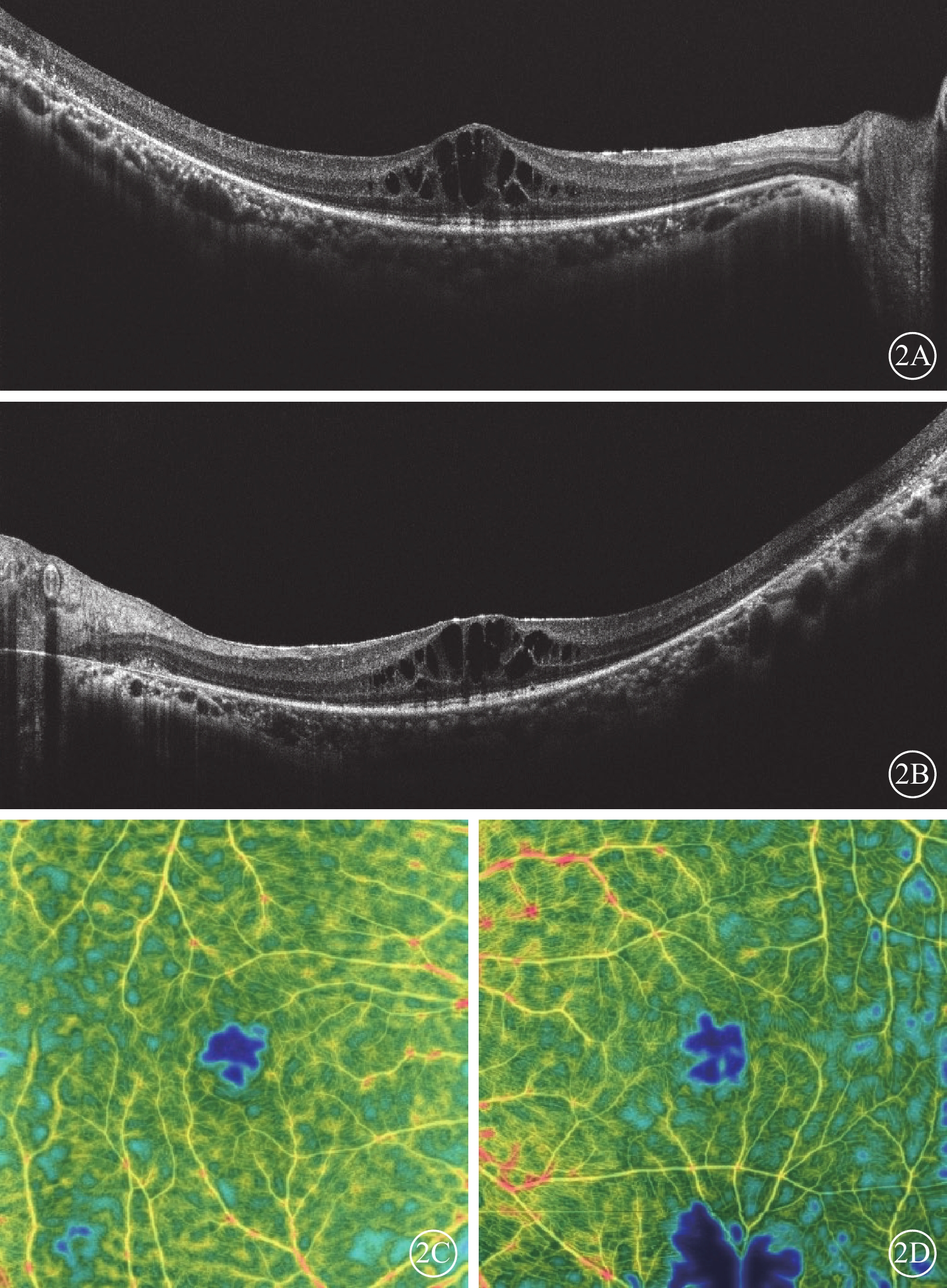
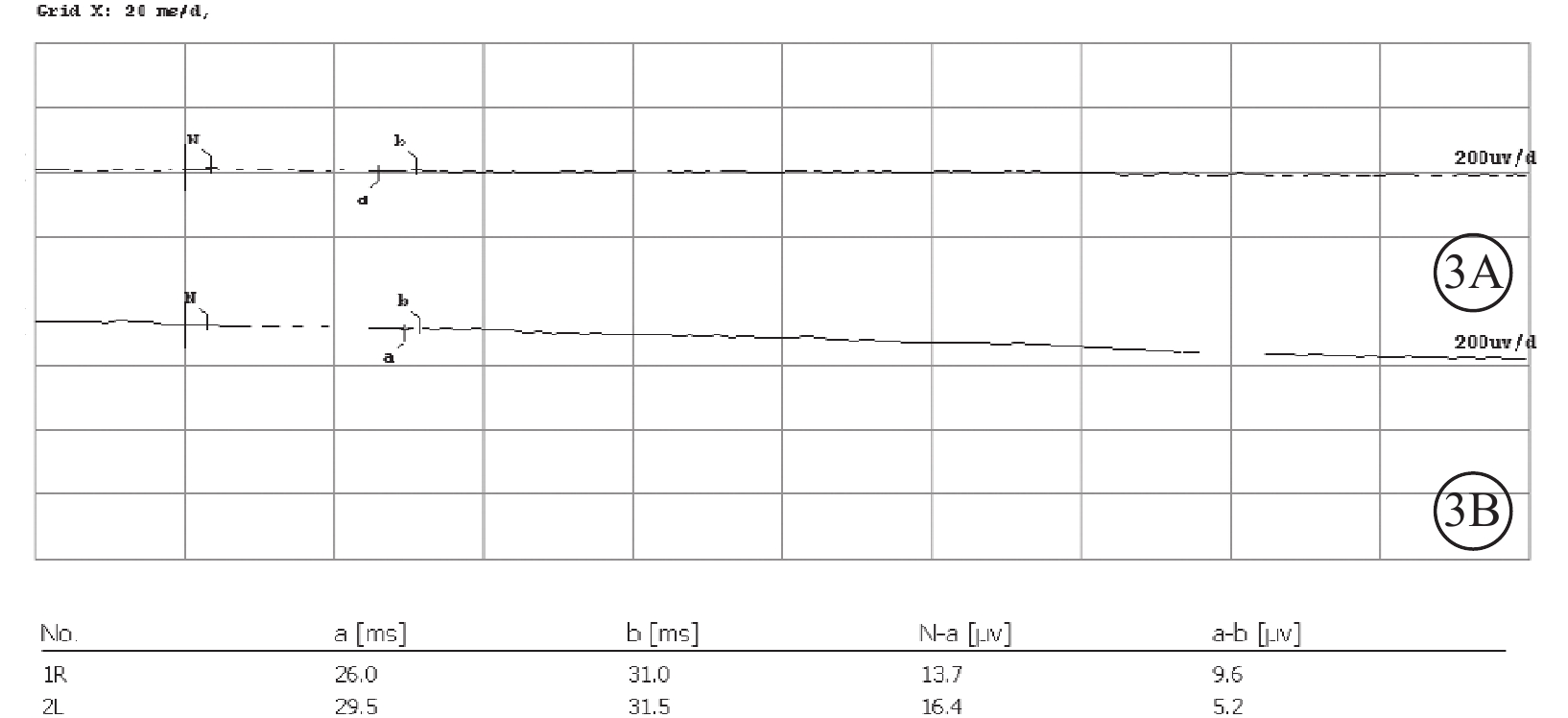
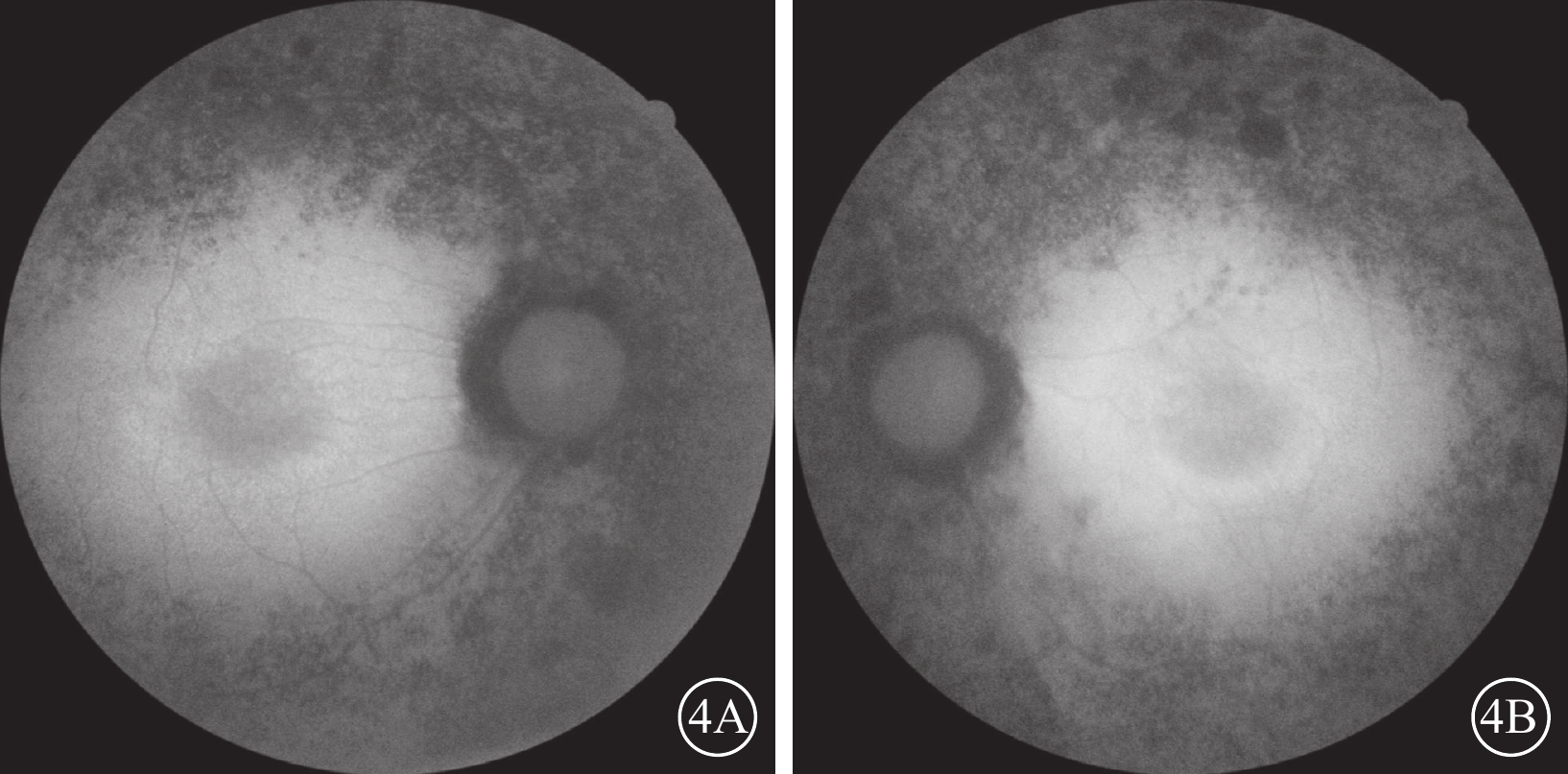
患者女,33歲。因雙眼視物模糊伴行走不穩2年,于2022年6月8日到南京市江寧中醫院眼科就診。患者訴近2年視物模糊明顯,伴上下樓梯行走不穩、行走撞物等;青少年時期初即出現夜盲癥;7歲時確診聽力障礙,配有助聽器,未規律佩戴,聽力無明顯進行性下降。父母非近親結婚,直系親屬無類似病癥。患者閉目直立、快速輪替、指鼻試驗均為陰性。眼科檢查:右眼、左眼最佳矯正視力(BCVA)分別為0.3、0.25。右眼、左眼眼壓分別為15、17 mm Hg(1 mm Hg= 0.133 kPa)。雙眼眼前節未見明顯異常。眼底檢查,雙眼視盤顏色淡紅,視網膜動靜脈細,廣泛青灰色污穢狀改變,骨細胞樣色素沉著,黃斑中心凹反光減弱(圖1A,1B)。熒光素眼底血管造影檢查,雙眼晚期視網膜血管纖細,動靜脈充盈時間延遲,黃斑中心凹周圍至赤道部視網膜斑駁狀強熒光,黃斑區少量熒光素“花瓣樣”積存(圖1C,1D)。光相干斷層掃描(OCT)檢查,雙眼黃斑中心凹視網膜厚度增加,視網膜外核層、外叢狀層及內核層囊樣弱反射(圖2A,2B)。OCT血管成像檢查,雙眼黃斑區旁中心凹視網膜血流密度(PFD)降低(圖2C,2D)。視野檢查,雙眼周邊視野缺損,管狀視野。全視野視網膜電圖檢查,雙眼a波、b波波峰明顯下降,峰時延長,暗適應0.01的振幅近乎呈熄滅型(圖3A,3B)。眼底自發熒光檢查,雙眼黃斑中心凹“花瓣樣”強熒光,中心凹旁強熒光環,環外弱熒光,骨細胞樣色素沉著處無熒光(圖4)。聽力檢查,氣導和骨導呈一致性下降,呈陡降型,低頻聽力輕、中度損失,高頻聽力重度損失。采集患者及其父母外周靜脈血行基因檢測。對患者采用全外顯子組測序,檢測結果通過生物信息學分析后得到候選致病突變位點。運用Sanger測序進行驗證及家系共分離分析,確定致病性突變位點。檢測結果顯示,患者臨床信息相關的1個父源疑似致病性變異,一個母源疑似致病性變異,且均為ADGRV1基因雜合移碼突變(圖5)。診斷:Usher綜合征(USH)2型、視網膜色素變性(RP)伴黃斑囊樣水腫(CME)。予以口服醋甲唑胺25 mg,2次/d治療,持續2個月。OCT檢查,雙眼黃斑水腫明顯改善,視網膜中央黃斑厚度(CMT)顯著下降(圖6)。雙眼BCVA較治療前無明顯變化。停藥后隨訪3個月,CME未見明顯復發。
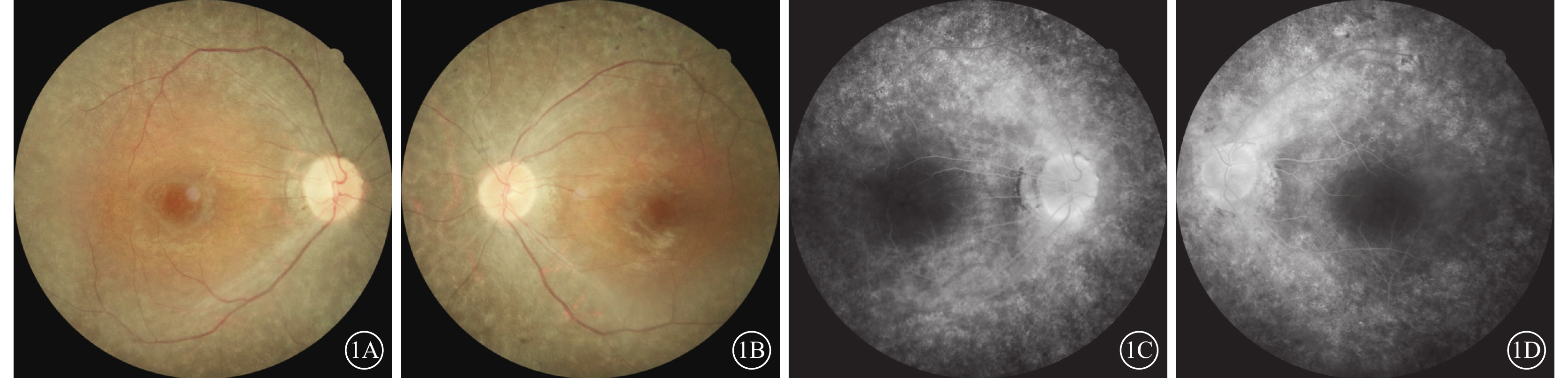 圖1
Usher綜合征伴黃斑水腫患者雙眼彩色眼底像及熒光素眼底血管造影像
圖1
Usher綜合征伴黃斑水腫患者雙眼彩色眼底像及熒光素眼底血管造影像
1A、1B分別示右眼、左眼彩色眼底像,雙眼視盤顏色淡,視網膜動靜脈細,視網膜見廣泛青灰色污穢狀改變,可見骨細胞樣色素沉著,黃斑中心凹反光減弱;1C、1D分別示右眼、左眼熒光素眼底血管造影晚期像,雙眼視網膜血管纖細,動靜脈充盈時間延遲,黃斑中心凹周圍至赤道部視網膜普遍斑駁狀強熒光,黃斑區少量熒光素“花瓣樣”積存
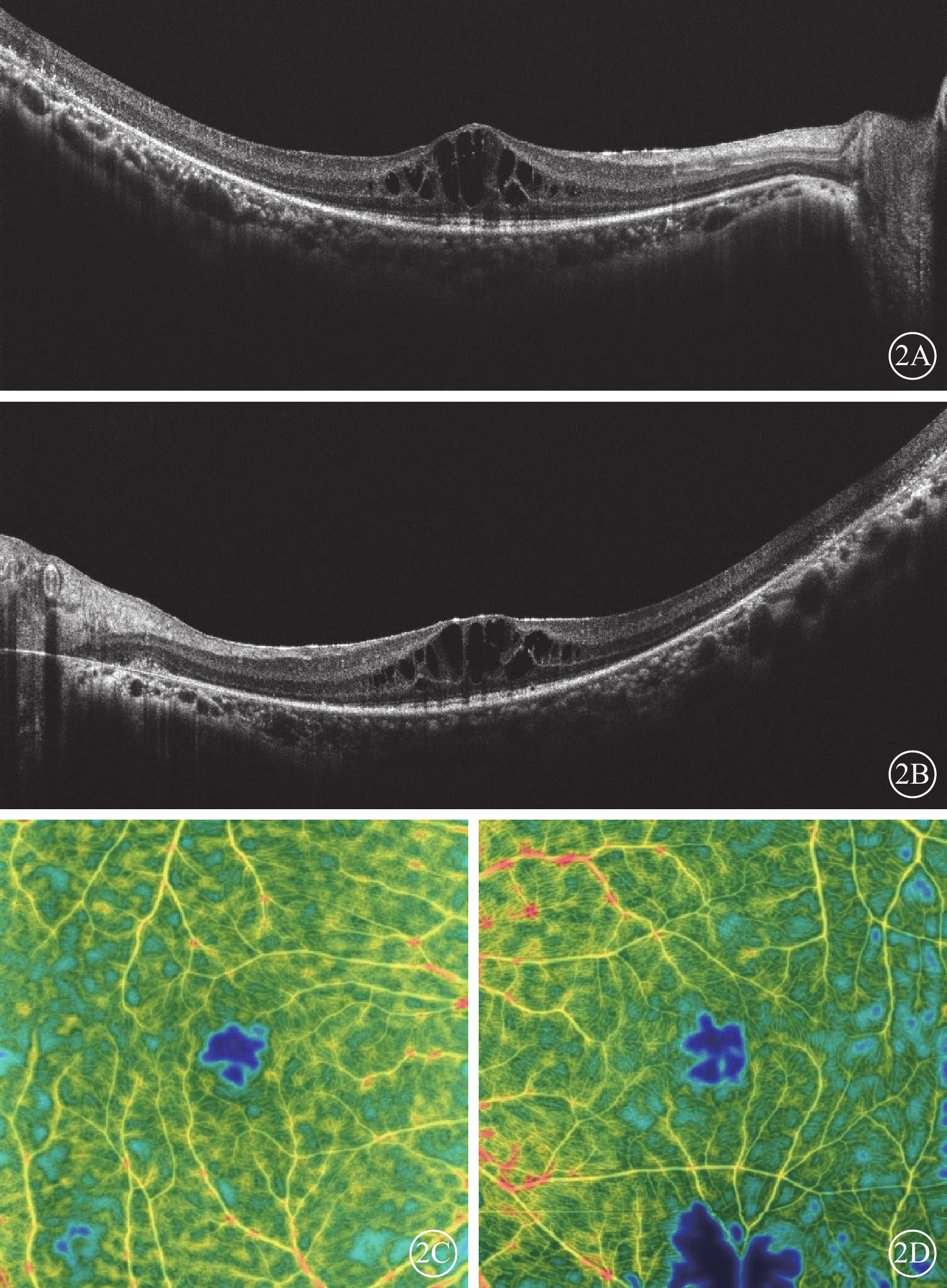 圖2
Usher綜合征伴黃斑水腫患者雙眼OCT及OCTA像
圖2
Usher綜合征伴黃斑水腫患者雙眼OCT及OCTA像
2A、2B分別示右眼、左眼OCT像,雙眼視網膜外核層、外叢狀層及內核層囊樣弱反射;2C、2D分別示右眼、左眼OCTA像,雙眼黃斑區旁中心凹視網膜血流密度降低 OCT:光相干斷層掃描;OCTA:OCT血管成像
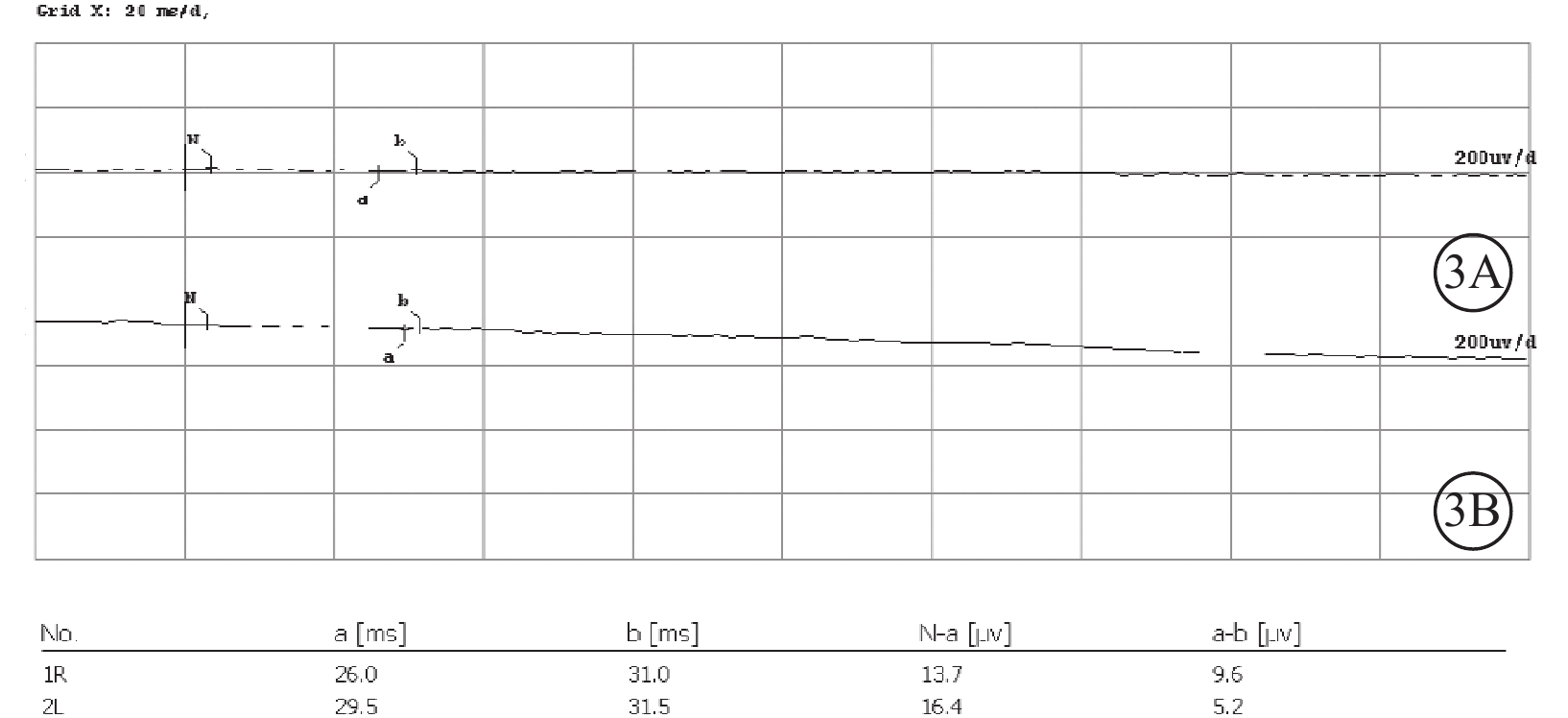 圖3
Usher綜合征伴黃斑水腫患者雙眼全視野視網膜電圖像
圖3
Usher綜合征伴黃斑水腫患者雙眼全視野視網膜電圖像
3A、3B示右眼、左眼暗適應0.01的振幅近乎呈熄滅型
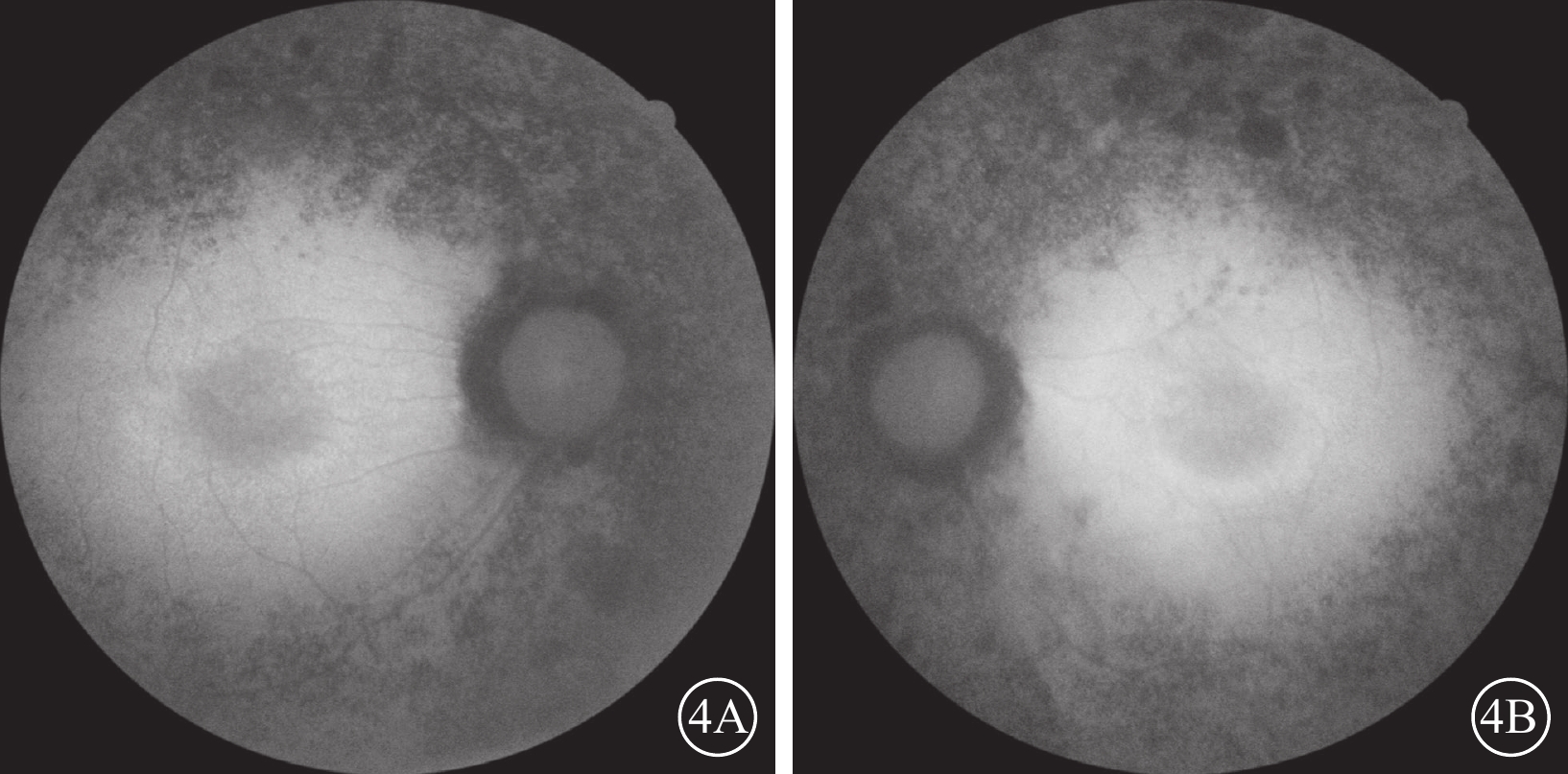 圖4
Usher綜合征伴黃斑水腫患者及眼底自發熒光像
圖4
Usher綜合征伴黃斑水腫患者及眼底自發熒光像
4A、4B分別示右眼、左眼,雙眼黃斑中心凹“花瓣樣”強熒光,黃斑中心凹旁強熒光環,環外弱熒光,骨細胞樣色素沉著處無熒光
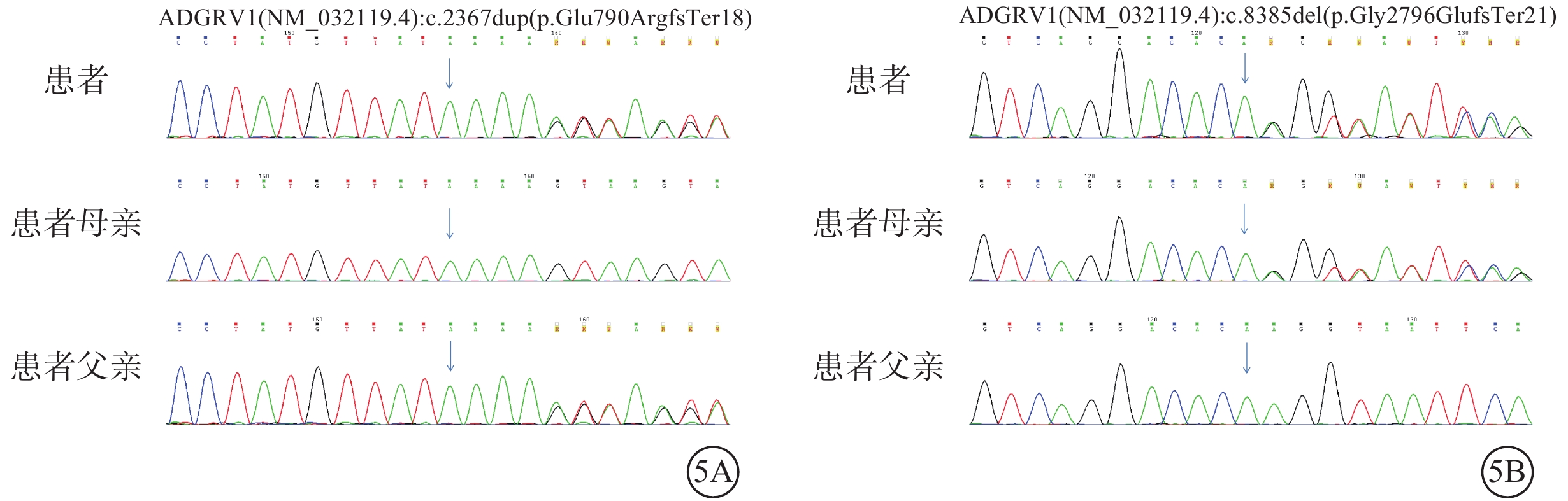
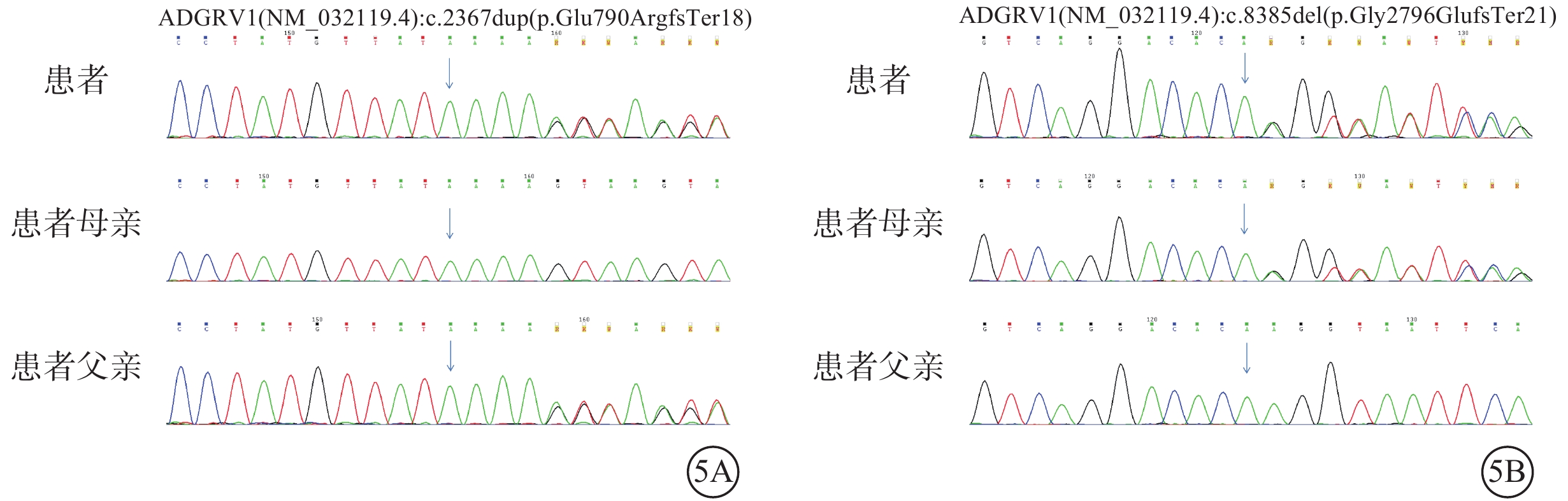 圖5
Usher綜合征伴黃斑水腫患者及其父母基因測序圖
圖5
Usher綜合征伴黃斑水腫患者及其父母基因測序圖
患者臨床信息相關的1個父源疑似致病性變異,一個母源疑似致病性變異,且均為
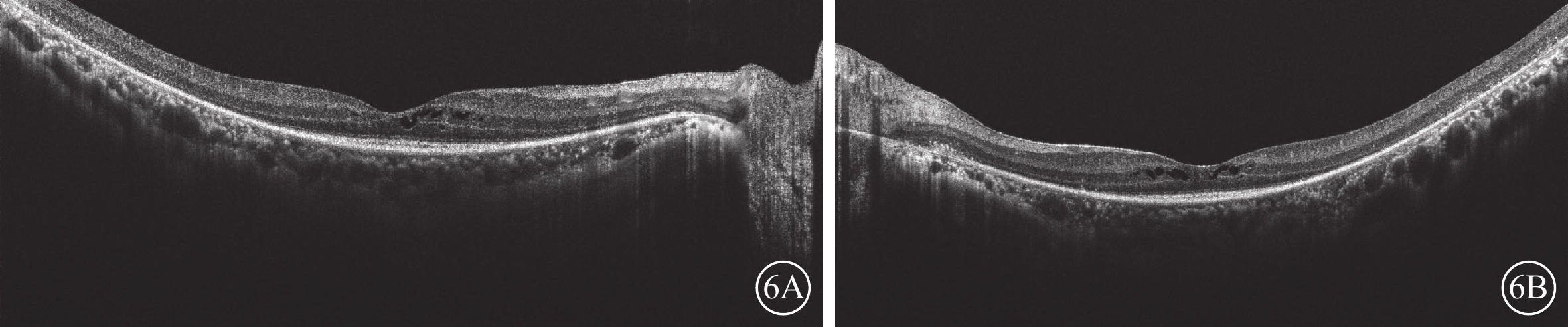
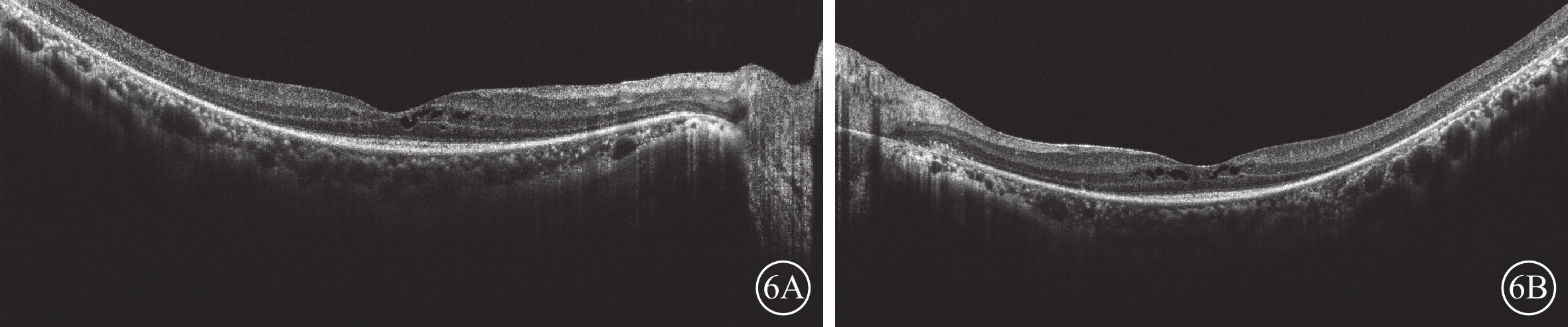 圖6
Usher綜合征伴黃斑水腫患者治療2個月后雙眼光相干斷層掃描像
圖6
Usher綜合征伴黃斑水腫患者治療2個月后雙眼光相干斷層掃描像
6A、6B分別示右眼、左眼,雙眼視網膜中央黃斑厚度顯著下降
討論 USH又稱視網膜色素變性-感音神經性耳聾綜合征,以RP、感音神經性聽力損失(SNHL)伴或不伴前庭功能障礙為特征的常染色體隱性遺傳疾病。依據聽力損傷程度、發病年齡及是否伴有前庭功能障礙,USH分為USH1、USH2、USH3亞型。USH1型RP發病年齡多為10歲前,伴先天性重度或極重度SNHL及明確的前庭功能障礙。USH2型RP發病年齡為10~20歲,伴先天性偏向性中重度SNHL,聽力低頻中度、高頻呈重度損傷,前庭功能可正常。USH3型RP發病年齡不確定或在20歲后,聽力障礙呈語后進行性SNHL,前庭功能發病不確定。
目前臨床以口服及局部使用碳酸酐酶抑制劑(CAI)作為RP-CME一線用藥[1],USH-CME目前參考RP-CME治療[2]。RP-CME發病機制至今尚無明確定論,但目前已存在以下幾種機制學說:(1)Müller細胞水腫和功能障礙;(2)視網膜色素上皮(RPE)泵送機制功能障礙;(3)血視網膜屏障的破壞;(4)炎癥及玻璃體牽引系列反應;(5)抗視網膜抗體作用。RP-CME非單獨某一機制作用結果,而是由多種機制共同參與作用[3]。CAI作用機制復雜,包括減少視網膜血管滲漏并刺激跨血視網膜屏障的主動轉運,加速視網膜下液體吸收并增加視網膜粘連等,其臨床療效得到充分驗證。部分患者在取得療效后的藥物維持階段可能會出現病情不能進一步改善或CME復發,此時停用CAI后1~6個月,再恢復治療,同樣有良好的療效。本例患者在口服醋甲唑胺2個月后,CME明顯改善,停藥后隨訪3個月,未出現CME復發。推測USH2型CME對CAI更敏感,可能是由于USH2型導致的RPE細胞功能損傷具有局限性或可逆性。
此外,基于炎癥反應機制,臨床亦提倡使用糖皮質激素治療RP-CME[4],特別是針對CAI治療不敏感的患者,可以采取玻璃體腔注射糖皮質激素[5],對比口服及局部使用糖皮質激素,其系統性副作用更少[6]。已有臨床研究對比玻璃體內地塞米松植入劑與CAI療效,證明地塞米松植入劑在降低CMT和改善BCVA方面更有效[7-8]。然而考慮到局部注射地塞米松植入劑容易引起青光眼、白內障等風險,且同樣有CME復發風險,需反復多次注射,尚不作為一線用藥。玻璃體腔注射抗血管內皮生長因子藥物、格柵樣激光光凝治療、玻璃體視網膜手術均可用于治療RP-CME,但因研究樣本量較少,療效因研究而異,尚缺乏統計學意義[9-11]。
研究報道,BCVA與RP-CME沒有直接關系,而與橢圓體帶完整性破壞及PFD降低直接相關[12-13]。但CME的出現可能是視網膜逐漸萎縮的信號。因此,橢圓體帶完整性及PFD是判斷患者預后的重要指標。本例USH2型患者PFD降低,中心視力有一定受損,但橢圓體帶結構尚完整,患者此時持續的CME是RP進展的不利信號。故及時治療CME,可有效延緩視力下降。
本例患者提示眼科醫生對診斷RP的患者除了關注其眼部癥狀,還應關注患者全身情況,以進一步明確患者是否患有USH及其他綜合征。因RP-CME在USH患者中發病率高[14],且發病隱匿,建議對USH患者定期復查,以早發現,早治療。
患者女,33歲。因雙眼視物模糊伴行走不穩2年,于2022年6月8日到南京市江寧中醫院眼科就診。患者訴近2年視物模糊明顯,伴上下樓梯行走不穩、行走撞物等;青少年時期初即出現夜盲癥;7歲時確診聽力障礙,配有助聽器,未規律佩戴,聽力無明顯進行性下降。父母非近親結婚,直系親屬無類似病癥。患者閉目直立、快速輪替、指鼻試驗均為陰性。眼科檢查:右眼、左眼最佳矯正視力(BCVA)分別為0.3、0.25。右眼、左眼眼壓分別為15、17 mm Hg(1 mm Hg= 0.133 kPa)。雙眼眼前節未見明顯異常。眼底檢查,雙眼視盤顏色淡紅,視網膜動靜脈細,廣泛青灰色污穢狀改變,骨細胞樣色素沉著,黃斑中心凹反光減弱(圖1A,1B)。熒光素眼底血管造影檢查,雙眼晚期視網膜血管纖細,動靜脈充盈時間延遲,黃斑中心凹周圍至赤道部視網膜斑駁狀強熒光,黃斑區少量熒光素“花瓣樣”積存(圖1C,1D)。光相干斷層掃描(OCT)檢查,雙眼黃斑中心凹視網膜厚度增加,視網膜外核層、外叢狀層及內核層囊樣弱反射(圖2A,2B)。OCT血管成像檢查,雙眼黃斑區旁中心凹視網膜血流密度(PFD)降低(圖2C,2D)。視野檢查,雙眼周邊視野缺損,管狀視野。全視野視網膜電圖檢查,雙眼a波、b波波峰明顯下降,峰時延長,暗適應0.01的振幅近乎呈熄滅型(圖3A,3B)。眼底自發熒光檢查,雙眼黃斑中心凹“花瓣樣”強熒光,中心凹旁強熒光環,環外弱熒光,骨細胞樣色素沉著處無熒光(圖4)。聽力檢查,氣導和骨導呈一致性下降,呈陡降型,低頻聽力輕、中度損失,高頻聽力重度損失。采集患者及其父母外周靜脈血行基因檢測。對患者采用全外顯子組測序,檢測結果通過生物信息學分析后得到候選致病突變位點。運用Sanger測序進行驗證及家系共分離分析,確定致病性突變位點。檢測結果顯示,患者臨床信息相關的1個父源疑似致病性變異,一個母源疑似致病性變異,且均為ADGRV1基因雜合移碼突變(圖5)。診斷:Usher綜合征(USH)2型、視網膜色素變性(RP)伴黃斑囊樣水腫(CME)。予以口服醋甲唑胺25 mg,2次/d治療,持續2個月。OCT檢查,雙眼黃斑水腫明顯改善,視網膜中央黃斑厚度(CMT)顯著下降(圖6)。雙眼BCVA較治療前無明顯變化。停藥后隨訪3個月,CME未見明顯復發。
圖1
Usher綜合征伴黃斑水腫患者雙眼彩色眼底像及熒光素眼底血管造影像
1A、1B分別示右眼、左眼彩色眼底像,雙眼視盤顏色淡,視網膜動靜脈細,視網膜見廣泛青灰色污穢狀改變,可見骨細胞樣色素沉著,黃斑中心凹反光減弱;1C、1D分別示右眼、左眼熒光素眼底血管造影晚期像,雙眼視網膜血管纖細,動靜脈充盈時間延遲,黃斑中心凹周圍至赤道部視網膜普遍斑駁狀強熒光,黃斑區少量熒光素“花瓣樣”積存
圖2
Usher綜合征伴黃斑水腫患者雙眼OCT及OCTA像
2A、2B分別示右眼、左眼OCT像,雙眼視網膜外核層、外叢狀層及內核層囊樣弱反射;2C、2D分別示右眼、左眼OCTA像,雙眼黃斑區旁中心凹視網膜血流密度降低 OCT:光相干斷層掃描;OCTA:OCT血管成像
圖3
Usher綜合征伴黃斑水腫患者雙眼全視野視網膜電圖像
3A、3B示右眼、左眼暗適應0.01的振幅近乎呈熄滅型
圖4
Usher綜合征伴黃斑水腫患者及眼底自發熒光像
4A、4B分別示右眼、左眼,雙眼黃斑中心凹“花瓣樣”強熒光,黃斑中心凹旁強熒光環,環外弱熒光,骨細胞樣色素沉著處無熒光
圖5
Usher綜合征伴黃斑水腫患者及其父母基因測序圖
患者臨床信息相關的1個父源疑似致病性變異,一個母源疑似致病性變異,且均為
圖6
Usher綜合征伴黃斑水腫患者治療2個月后雙眼光相干斷層掃描像
6A、6B分別示右眼、左眼,雙眼視網膜中央黃斑厚度顯著下降
討論 USH又稱視網膜色素變性-感音神經性耳聾綜合征,以RP、感音神經性聽力損失(SNHL)伴或不伴前庭功能障礙為特征的常染色體隱性遺傳疾病。依據聽力損傷程度、發病年齡及是否伴有前庭功能障礙,USH分為USH1、USH2、USH3亞型。USH1型RP發病年齡多為10歲前,伴先天性重度或極重度SNHL及明確的前庭功能障礙。USH2型RP發病年齡為10~20歲,伴先天性偏向性中重度SNHL,聽力低頻中度、高頻呈重度損傷,前庭功能可正常。USH3型RP發病年齡不確定或在20歲后,聽力障礙呈語后進行性SNHL,前庭功能發病不確定。
目前臨床以口服及局部使用碳酸酐酶抑制劑(CAI)作為RP-CME一線用藥[1],USH-CME目前參考RP-CME治療[2]。RP-CME發病機制至今尚無明確定論,但目前已存在以下幾種機制學說:(1)Müller細胞水腫和功能障礙;(2)視網膜色素上皮(RPE)泵送機制功能障礙;(3)血視網膜屏障的破壞;(4)炎癥及玻璃體牽引系列反應;(5)抗視網膜抗體作用。RP-CME非單獨某一機制作用結果,而是由多種機制共同參與作用[3]。CAI作用機制復雜,包括減少視網膜血管滲漏并刺激跨血視網膜屏障的主動轉運,加速視網膜下液體吸收并增加視網膜粘連等,其臨床療效得到充分驗證。部分患者在取得療效后的藥物維持階段可能會出現病情不能進一步改善或CME復發,此時停用CAI后1~6個月,再恢復治療,同樣有良好的療效。本例患者在口服醋甲唑胺2個月后,CME明顯改善,停藥后隨訪3個月,未出現CME復發。推測USH2型CME對CAI更敏感,可能是由于USH2型導致的RPE細胞功能損傷具有局限性或可逆性。
此外,基于炎癥反應機制,臨床亦提倡使用糖皮質激素治療RP-CME[4],特別是針對CAI治療不敏感的患者,可以采取玻璃體腔注射糖皮質激素[5],對比口服及局部使用糖皮質激素,其系統性副作用更少[6]。已有臨床研究對比玻璃體內地塞米松植入劑與CAI療效,證明地塞米松植入劑在降低CMT和改善BCVA方面更有效[7-8]。然而考慮到局部注射地塞米松植入劑容易引起青光眼、白內障等風險,且同樣有CME復發風險,需反復多次注射,尚不作為一線用藥。玻璃體腔注射抗血管內皮生長因子藥物、格柵樣激光光凝治療、玻璃體視網膜手術均可用于治療RP-CME,但因研究樣本量較少,療效因研究而異,尚缺乏統計學意義[9-11]。
研究報道,BCVA與RP-CME沒有直接關系,而與橢圓體帶完整性破壞及PFD降低直接相關[12-13]。但CME的出現可能是視網膜逐漸萎縮的信號。因此,橢圓體帶完整性及PFD是判斷患者預后的重要指標。本例USH2型患者PFD降低,中心視力有一定受損,但橢圓體帶結構尚完整,患者此時持續的CME是RP進展的不利信號。故及時治療CME,可有效延緩視力下降。
本例患者提示眼科醫生對診斷RP的患者除了關注其眼部癥狀,還應關注患者全身情況,以進一步明確患者是否患有USH及其他綜合征。因RP-CME在USH患者中發病率高[14],且發病隱匿,建議對USH患者定期復查,以早發現,早治療。