引用本文: 高杰, 孫建, 孫雷, 李艷秋, 劉同同, 曹靜, 王濤. COL9A3基因新變異致Stickler綜合征一家系2例. 中華眼底病雜志, 2024, 40(4): 313-314. doi: 10.3760/cma.j.cn511434-20230425-00188 復制
版權信息: ?四川大學華西醫院華西期刊社《中華眼底病雜志》版權所有,未經授權不得轉載、改編
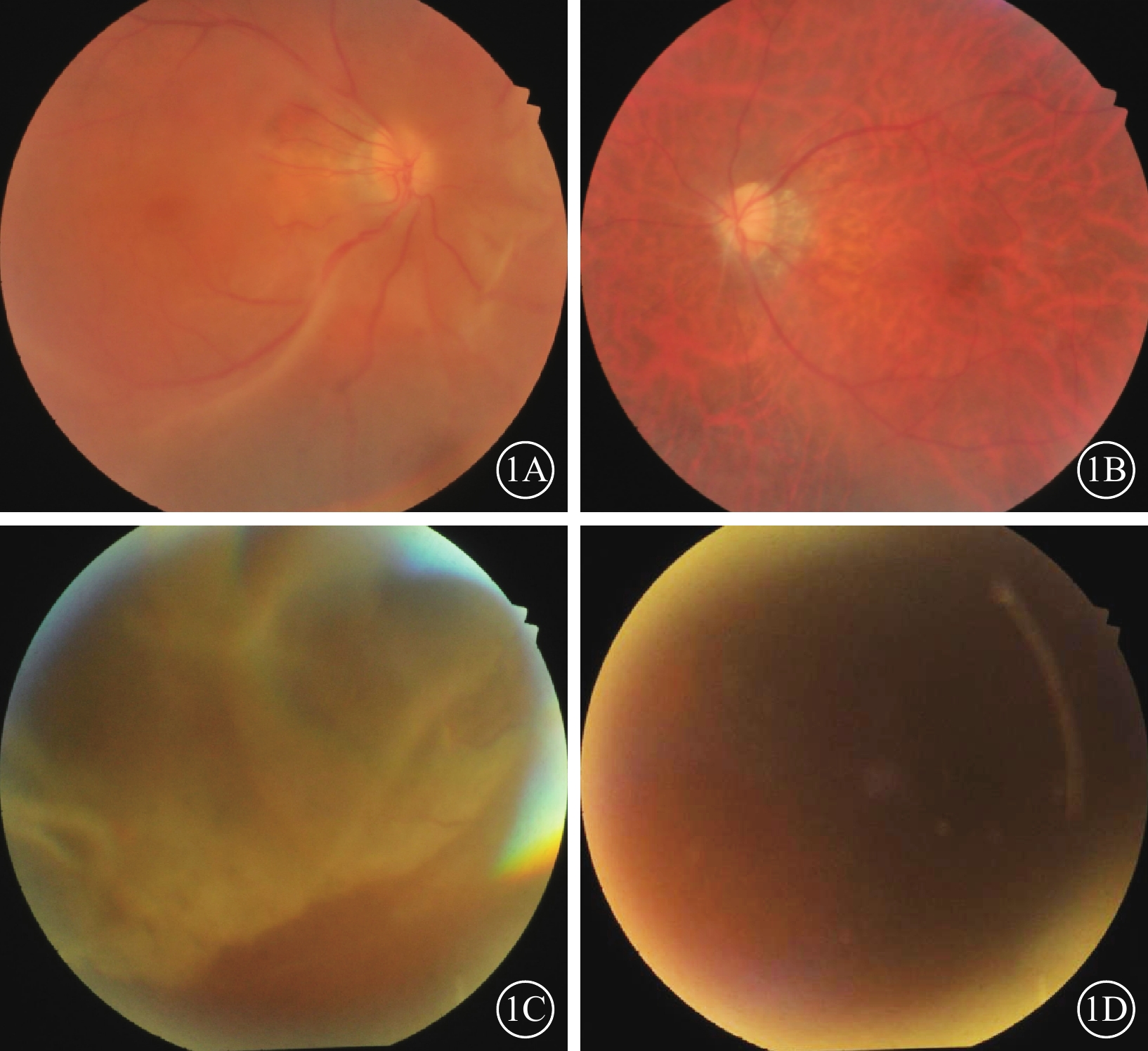
例1 患者男,47歲。因無明顯誘因突感右眼視物不清,于2019年3月7日到山東省臨沂市人民醫院眼科就診。既往雙眼高度近視病史30余年。眼科檢查:右眼、左眼最佳矯正視力(BCVA)分別為0.10、0.15。右眼、左眼眼壓分別為13.0、22.3 mm Hg(1 mm Hg=0.133 kPa)。雙眼后囊膜混濁。眼底檢查:右眼玻璃體腔內重度混濁,可見大量色素顆粒,鼻下方視網膜脫離明顯,黃斑區受累,鼻上方中緯部約1個視盤直徑大小的視網膜撕裂孔,變性區明顯;左眼玻璃體腔輕度混濁,視網膜平伏,視盤顏色淡紅,顳側萎縮弧,黃斑區中心凹反光消失,視網膜血管走形正常,“豹紋狀”眼底(圖1A,1B)。全身情況:體態臃腫,鼻梁塌陷,面中部后縮,感音性耳聾。診斷:雙眼高度近視、右眼視網膜脫離。
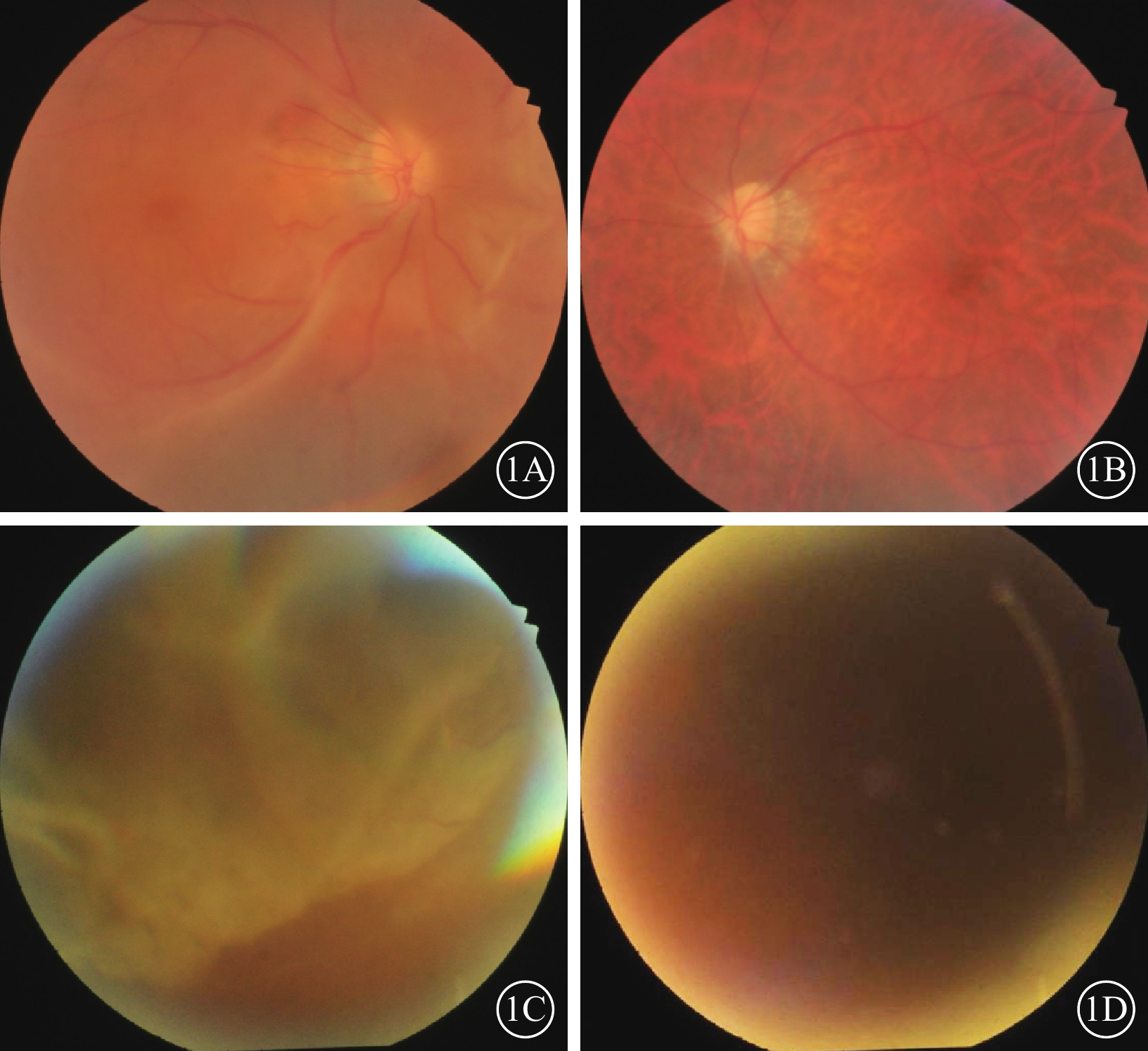 圖1
COL9A3基因新變異致Stickler綜合征一家系2例患者雙眼彩色眼底像
圖1
COL9A3基因新變異致Stickler綜合征一家系2例患者雙眼彩色眼底像
1A、1B分別示例1患者右眼、左眼彩色眼底像,右眼玻璃體腔內可見大量色素顆粒,鼻下方視網膜脫離明顯,黃斑區受累,鼻上方中緯部約1個視盤直徑大小的視網膜撕裂孔,變性區明顯。左眼玻璃體腔輕度混濁,視網膜平伏,視盤顏色淡紅,顳側萎縮弧,黃斑區中心凹反光消失,視網膜血管走形正常,豹紋狀眼底。1C、1D分別示例2患者右眼、左眼彩色眼底像,右眼視網膜大范圍脫落,其余窺不清。左眼晶狀體嚴重混濁眼底無法窺入
例2 患者女,45歲,例1患者胞妹。因無明顯誘因突感右眼視物遮擋感,于2022年10月2日到山東省臨沂市人民醫院眼科就診。既往雙眼高度近視病史30余年。眼科檢查:右眼、左眼BCVA分別為0.2、手動/20 cm。右眼、左眼眼壓分別為19.0、21.0 mm Hg(1 mm Hg=0.133 kPa)。雙眼后囊膜混濁,左眼晶狀體混濁。眼底檢查:右眼視網膜大范圍脫落,其余窺不清;左眼晶狀體嚴重混濁眼底無法窺入(圖1C,1D)。全身情況:體態臃腫,鼻梁塌陷,面中部后縮,感音性耳聾。診斷:雙眼高度近視、右眼視網膜脫離、左眼白內障。
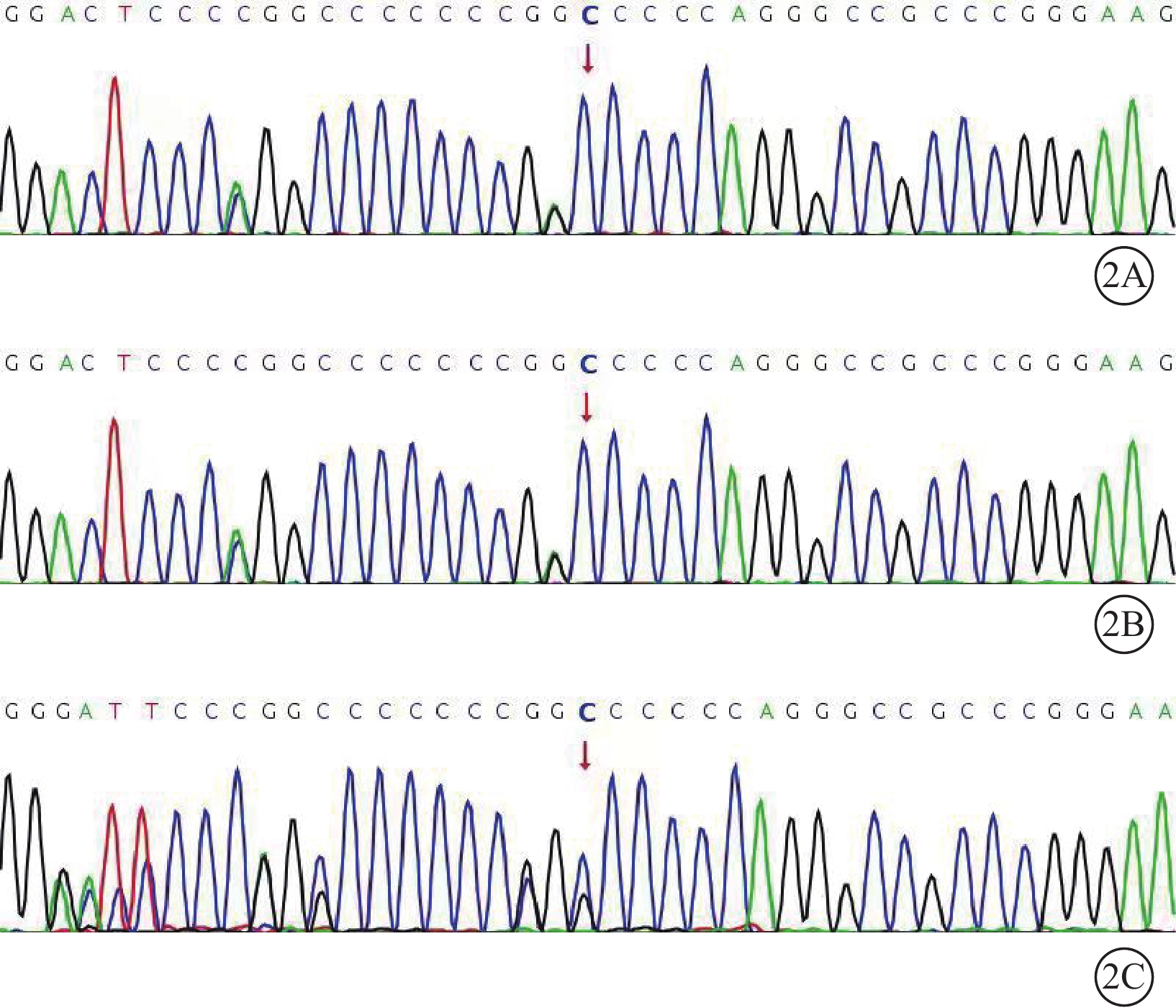
例1患者自述其父有視網膜脫離病史,玻璃體切割手術聯合硅油注入手術史,因已去世所以無法追蹤。綜合2例患者的臨床表現,考慮Stickler綜合征的可能性大。征求患者同意后行高通量測序基因檢測,均行Sanger驗證。結果顯示,2例患者均攜帶COL9A3基因的2個雜合突變M1:c.104_105delinsA:p.G35Dfs*53和M2:c.110del:p.P37Qfs*51。其母親僅攜帶M2的雜合變異(圖2)。通過二代測序BAM文件圖可推斷,在例1患者染色體中M1、M2分別位于20號兩條染色體q13.3位置,且互為反式位置構成復合雜合變異(圖3)。M1和M2變異在基因組聚合數據庫中未被收錄,根據美國醫學遺傳學與基因組學學會指南,該變異判定為疑似致病性突變。綜合臨床表現及基因檢測結果,2例患者確診Stickler綜合征Ⅵ型。
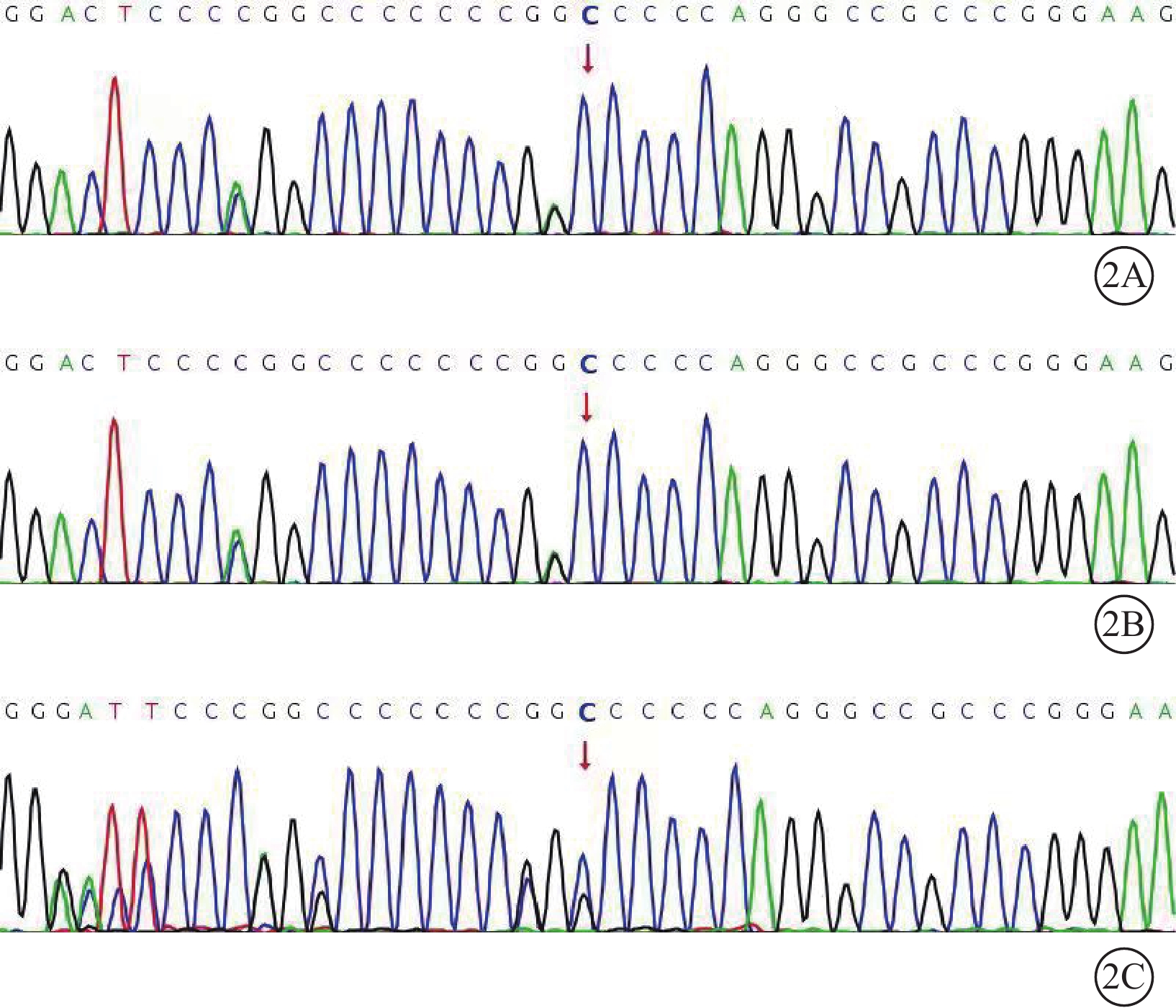 圖2
COL2A3基因新變異致的Stickler綜合征一家系2例基因測序結果
圖2
COL2A3基因新變異致的Stickler綜合征一家系2例基因測序結果
2A~2C分別示例1、例2患者及其母親。2例患者均攜帶
 圖3
COL2A3基因新變異致Stickler綜合征患者(例1患者)二代測序BAM文件圖
圖3
COL2A3基因新變異致Stickler綜合征患者(例1患者)二代測序BAM文件圖
藍色表示變異M1,紅色表示變異M2
討論 Stickler綜合征是一種結締組織病,其主要表現為:(1)眼部表現:先天性高度近視、青年時期的白內障、眼軸增長、玻璃體變性和液化、視網膜格子樣變性、視網膜裂孔、視網膜脫離以及極少數的青光眼。(2)顱面部面中部發育不良:顴骨發育不全、鼻梁扁平、腭裂。(3)聽覺障礙:即幼年時期漸進性耳聾,包括感音性耳聾、傳導性耳聾、混合性耳聾。(4)骨關節異常:骨關節炎、身材矮小、髖關節病變、步態蹣跚。眼底病變與Stickler綜合征的視力預后尤為相關,其膠原蛋白的發育不良導致患者發育期嚴重的玻璃體腔內變性液化,從而容易發生巨大視網膜裂孔并視網膜脫離,巨大裂孔常可導致脈絡膜脫離、眼球萎縮等繼發性嚴重眼部病變。與特發性視網膜裂孔的干孔相比,因Stickler綜合征患者的玻璃體變性導致的對裂孔邊緣的牽引,液化的玻璃體極易通過經裂孔進入玻璃體下形成較大范圍的視網膜脫離甚至可達鋸齒緣。通常根據患者眼部表現并結合患者全身情況即可做出初步診斷,明確分型診斷還需進一步基因檢測[1]。
Stickler綜合征目前被分為六型[1]。因分型不同主要臨床表現特點也不盡相同[2],Ⅰ型由COL2A1基因突變引起,導致視網膜脫落的風險最高。Ⅱ型由COL11A1基因突變引起,包括眼部畸形。Ⅲ型由COL11A2基因突變引起,不包括眼部畸形。Ⅱ和Ⅲ型比Ⅰ型更有可能出現聽力喪失。Ⅳ型和Ⅴ型分別由COL9A1或COL9A2基因突變引起,是Stickler綜合征中的常染色體隱性遺傳類型[3-6]。Ⅵ型由COL9A3基因突變引起,因報道罕見遺傳類型不詳。
Stickler綜合征基因突變位點眾多,臨床表型復雜。以往國內報道大多是由COL2A1基因突變引起的Stickler綜合征Ⅰ型,此病例家系發病突變基因位點位于染色體20q13.3的COL9A3基因,既往關于COL9A3基因突變患者在全球范圍內僅報道過7例患者[3-6],其中6例患者為純合突變,1例患者為復合雜合變異[6],國內暫無相關病例報道。Stickler綜合征的視網膜脫離更復雜,也更難管理,患者視網膜脫離修復的成功率非常低[7]。目前可以通過遺傳、生物信息學分析準確預測高危人群來判斷視網膜脫離的可能性,并對高危人群提供預防性手術,可降低失明風險。
本文2例患者所在家系具有明顯遺傳病遺傳特征,先證者及其胞妹的臨床表現高度相似,主要表現為先天性高度近視、青年時期發病的白內障、視網膜脫離、玻璃體混濁、漸進性聽力喪失,體態臃腫、鼻梁塌陷、面中部后縮。針對此類患者我們眼科治療過程中主要采用對癥治療如配戴眼鏡矯正視力、玻璃體切割手術、硅油注入手術、玻璃體腔注射抗血管內皮生長因子藥物,白內障超聲乳化聯合工晶狀體植入手術等,短期內達到了良好的治療效果。但因患者病情復雜需要密切隨訪,持續觀察眼部情況的變化。
本文在一個Stickler綜合征家系中檢測到COL9A3基因新的致病性變異,豐富了COL9A3基因的突變譜和臨床表型譜,并結合循證醫學證據對此家系其余親屬建議行預防性診療。Stickler綜合征的表型具有復雜多樣化,基因檢測為Stickler綜合征的診斷、分型及治療提供了參考價值。Stickler綜合征預后普遍較差,早期臨床干預可為患者提供相應收益。未來需要更大樣本量、更長隨訪時間的研究來進一步驗證各類預防Stickler綜合征視網膜脫離的方式及其療效及安全性。
例1 患者男,47歲。因無明顯誘因突感右眼視物不清,于2019年3月7日到山東省臨沂市人民醫院眼科就診。既往雙眼高度近視病史30余年。眼科檢查:右眼、左眼最佳矯正視力(BCVA)分別為0.10、0.15。右眼、左眼眼壓分別為13.0、22.3 mm Hg(1 mm Hg=0.133 kPa)。雙眼后囊膜混濁。眼底檢查:右眼玻璃體腔內重度混濁,可見大量色素顆粒,鼻下方視網膜脫離明顯,黃斑區受累,鼻上方中緯部約1個視盤直徑大小的視網膜撕裂孔,變性區明顯;左眼玻璃體腔輕度混濁,視網膜平伏,視盤顏色淡紅,顳側萎縮弧,黃斑區中心凹反光消失,視網膜血管走形正常,“豹紋狀”眼底(圖1A,1B)。全身情況:體態臃腫,鼻梁塌陷,面中部后縮,感音性耳聾。診斷:雙眼高度近視、右眼視網膜脫離。
圖1
COL9A3基因新變異致Stickler綜合征一家系2例患者雙眼彩色眼底像
1A、1B分別示例1患者右眼、左眼彩色眼底像,右眼玻璃體腔內可見大量色素顆粒,鼻下方視網膜脫離明顯,黃斑區受累,鼻上方中緯部約1個視盤直徑大小的視網膜撕裂孔,變性區明顯。左眼玻璃體腔輕度混濁,視網膜平伏,視盤顏色淡紅,顳側萎縮弧,黃斑區中心凹反光消失,視網膜血管走形正常,豹紋狀眼底。1C、1D分別示例2患者右眼、左眼彩色眼底像,右眼視網膜大范圍脫落,其余窺不清。左眼晶狀體嚴重混濁眼底無法窺入
例2 患者女,45歲,例1患者胞妹。因無明顯誘因突感右眼視物遮擋感,于2022年10月2日到山東省臨沂市人民醫院眼科就診。既往雙眼高度近視病史30余年。眼科檢查:右眼、左眼BCVA分別為0.2、手動/20 cm。右眼、左眼眼壓分別為19.0、21.0 mm Hg(1 mm Hg=0.133 kPa)。雙眼后囊膜混濁,左眼晶狀體混濁。眼底檢查:右眼視網膜大范圍脫落,其余窺不清;左眼晶狀體嚴重混濁眼底無法窺入(圖1C,1D)。全身情況:體態臃腫,鼻梁塌陷,面中部后縮,感音性耳聾。診斷:雙眼高度近視、右眼視網膜脫離、左眼白內障。
例1患者自述其父有視網膜脫離病史,玻璃體切割手術聯合硅油注入手術史,因已去世所以無法追蹤。綜合2例患者的臨床表現,考慮Stickler綜合征的可能性大。征求患者同意后行高通量測序基因檢測,均行Sanger驗證。結果顯示,2例患者均攜帶COL9A3基因的2個雜合突變M1:c.104_105delinsA:p.G35Dfs*53和M2:c.110del:p.P37Qfs*51。其母親僅攜帶M2的雜合變異(圖2)。通過二代測序BAM文件圖可推斷,在例1患者染色體中M1、M2分別位于20號兩條染色體q13.3位置,且互為反式位置構成復合雜合變異(圖3)。M1和M2變異在基因組聚合數據庫中未被收錄,根據美國醫學遺傳學與基因組學學會指南,該變異判定為疑似致病性突變。綜合臨床表現及基因檢測結果,2例患者確診Stickler綜合征Ⅵ型。
圖2
COL2A3基因新變異致的Stickler綜合征一家系2例基因測序結果
2A~2C分別示例1、例2患者及其母親。2例患者均攜帶
圖3
COL2A3基因新變異致Stickler綜合征患者(例1患者)二代測序BAM文件圖
藍色表示變異M1,紅色表示變異M2
討論 Stickler綜合征是一種結締組織病,其主要表現為:(1)眼部表現:先天性高度近視、青年時期的白內障、眼軸增長、玻璃體變性和液化、視網膜格子樣變性、視網膜裂孔、視網膜脫離以及極少數的青光眼。(2)顱面部面中部發育不良:顴骨發育不全、鼻梁扁平、腭裂。(3)聽覺障礙:即幼年時期漸進性耳聾,包括感音性耳聾、傳導性耳聾、混合性耳聾。(4)骨關節異常:骨關節炎、身材矮小、髖關節病變、步態蹣跚。眼底病變與Stickler綜合征的視力預后尤為相關,其膠原蛋白的發育不良導致患者發育期嚴重的玻璃體腔內變性液化,從而容易發生巨大視網膜裂孔并視網膜脫離,巨大裂孔常可導致脈絡膜脫離、眼球萎縮等繼發性嚴重眼部病變。與特發性視網膜裂孔的干孔相比,因Stickler綜合征患者的玻璃體變性導致的對裂孔邊緣的牽引,液化的玻璃體極易通過經裂孔進入玻璃體下形成較大范圍的視網膜脫離甚至可達鋸齒緣。通常根據患者眼部表現并結合患者全身情況即可做出初步診斷,明確分型診斷還需進一步基因檢測[1]。
Stickler綜合征目前被分為六型[1]。因分型不同主要臨床表現特點也不盡相同[2],Ⅰ型由COL2A1基因突變引起,導致視網膜脫落的風險最高。Ⅱ型由COL11A1基因突變引起,包括眼部畸形。Ⅲ型由COL11A2基因突變引起,不包括眼部畸形。Ⅱ和Ⅲ型比Ⅰ型更有可能出現聽力喪失。Ⅳ型和Ⅴ型分別由COL9A1或COL9A2基因突變引起,是Stickler綜合征中的常染色體隱性遺傳類型[3-6]。Ⅵ型由COL9A3基因突變引起,因報道罕見遺傳類型不詳。
Stickler綜合征基因突變位點眾多,臨床表型復雜。以往國內報道大多是由COL2A1基因突變引起的Stickler綜合征Ⅰ型,此病例家系發病突變基因位點位于染色體20q13.3的COL9A3基因,既往關于COL9A3基因突變患者在全球范圍內僅報道過7例患者[3-6],其中6例患者為純合突變,1例患者為復合雜合變異[6],國內暫無相關病例報道。Stickler綜合征的視網膜脫離更復雜,也更難管理,患者視網膜脫離修復的成功率非常低[7]。目前可以通過遺傳、生物信息學分析準確預測高危人群來判斷視網膜脫離的可能性,并對高危人群提供預防性手術,可降低失明風險。
本文2例患者所在家系具有明顯遺傳病遺傳特征,先證者及其胞妹的臨床表現高度相似,主要表現為先天性高度近視、青年時期發病的白內障、視網膜脫離、玻璃體混濁、漸進性聽力喪失,體態臃腫、鼻梁塌陷、面中部后縮。針對此類患者我們眼科治療過程中主要采用對癥治療如配戴眼鏡矯正視力、玻璃體切割手術、硅油注入手術、玻璃體腔注射抗血管內皮生長因子藥物,白內障超聲乳化聯合工晶狀體植入手術等,短期內達到了良好的治療效果。但因患者病情復雜需要密切隨訪,持續觀察眼部情況的變化。
本文在一個Stickler綜合征家系中檢測到COL9A3基因新的致病性變異,豐富了COL9A3基因的突變譜和臨床表型譜,并結合循證醫學證據對此家系其余親屬建議行預防性診療。Stickler綜合征的表型具有復雜多樣化,基因檢測為Stickler綜合征的診斷、分型及治療提供了參考價值。Stickler綜合征預后普遍較差,早期臨床干預可為患者提供相應收益。未來需要更大樣本量、更長隨訪時間的研究來進一步驗證各類預防Stickler綜合征視網膜脫離的方式及其療效及安全性。