引用本文: 左華欣, 周曉芳, 任曉暾, 施維, 李莉, 余繼鋒, 彭春霞. 兒童Ⅰ型唾液酸沉積癥并發黃斑“櫻桃紅斑”1例. 中華眼底病雜志, 2024, 40(1): 64-66. doi: 10.3760/cma.j.cn511434-20231113-00455 復制
版權信息: ?四川大學華西醫院華西期刊社《中華眼底病雜志》版權所有,未經授權不得轉載、改編
患者男,15歲。因走路易跌倒3年至北京兒童醫院神經內科就診,因雙眼進行性視力下降轉診于眼科。患兒1個月前無發熱性抽搐2次,表現為雙眼上翻,口唇發紺,牙關緊閉,約1~2 min緩解。無全身病史及家族遺傳病史。眼科檢查:右眼、左眼最佳矯正視力均為20/100;屈光狀態:右眼-4.50 DS/-2.75 DC×180°、 左眼-2.00 DS/-2.75 DC×5°。眼壓:右眼、左眼分別為19、16 mm Hg(1 mm Hg=0.133 kPa)。雙眼眼球運動正常;瞳孔圓,直徑2.5 mm,對光反射存在;晶狀體可見少量白色點狀混濁。眼底檢查,雙眼視盤顏色淡、邊界清楚,杯盤比約0.3~0.4,黃斑呈“櫻桃紅斑”改變(圖1)。視覺誘發電位(VEP)檢查,雙眼在低、中、高空間頻率刺激下P100波振幅明顯降低(圖2)。視網膜電圖(ERG)檢查,雙眼分別在明、暗適應刺激下a、b波振幅大致正常。光相干斷層掃描(OCT)檢查,雙眼視盤周圍神經纖維層厚度降低,右眼、左眼分別為76、79 μm;雙眼黃斑中心凹形態大致正常,神經節細胞層因唾液酸沉積反射增強,與神經纖維層反射強度相同,界限不清,融合為一層,兩層厚度正常(圖3)。神經系統檢查,四肢肌力及肌張力正常,腱反射正常,雙側巴賓斯基征(+),踝陣攣(-)。共濟運動失調檢測,指鼻試驗不準,走直線不穩。顱腦核磁共振成像(MRI)檢查及腦電圖檢查未見異常。眼科初步診斷:雙眼視神經病變、雙眼黃斑病變。神經內科建議進一步完善外周血基因檢查明確診斷。
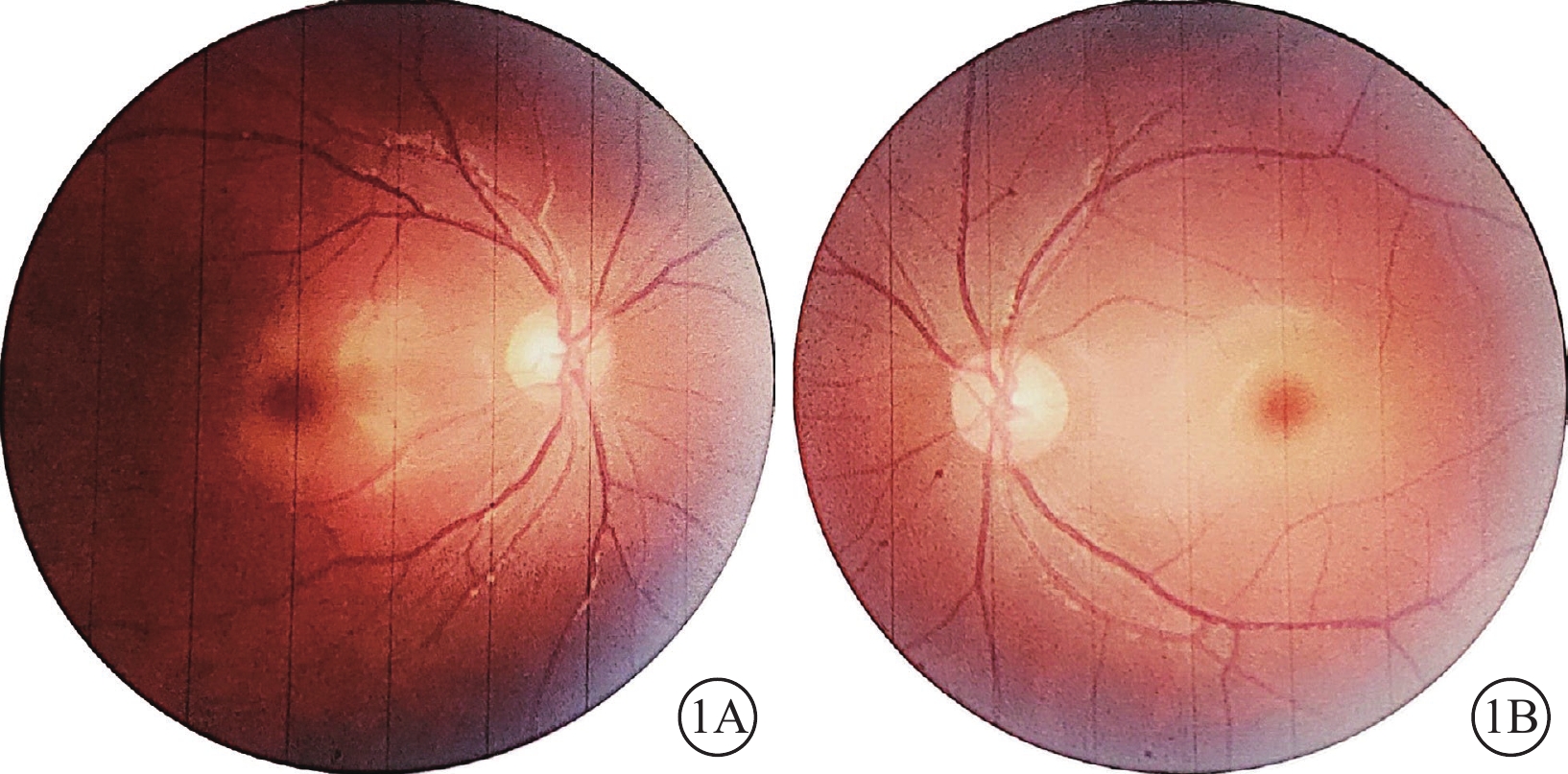
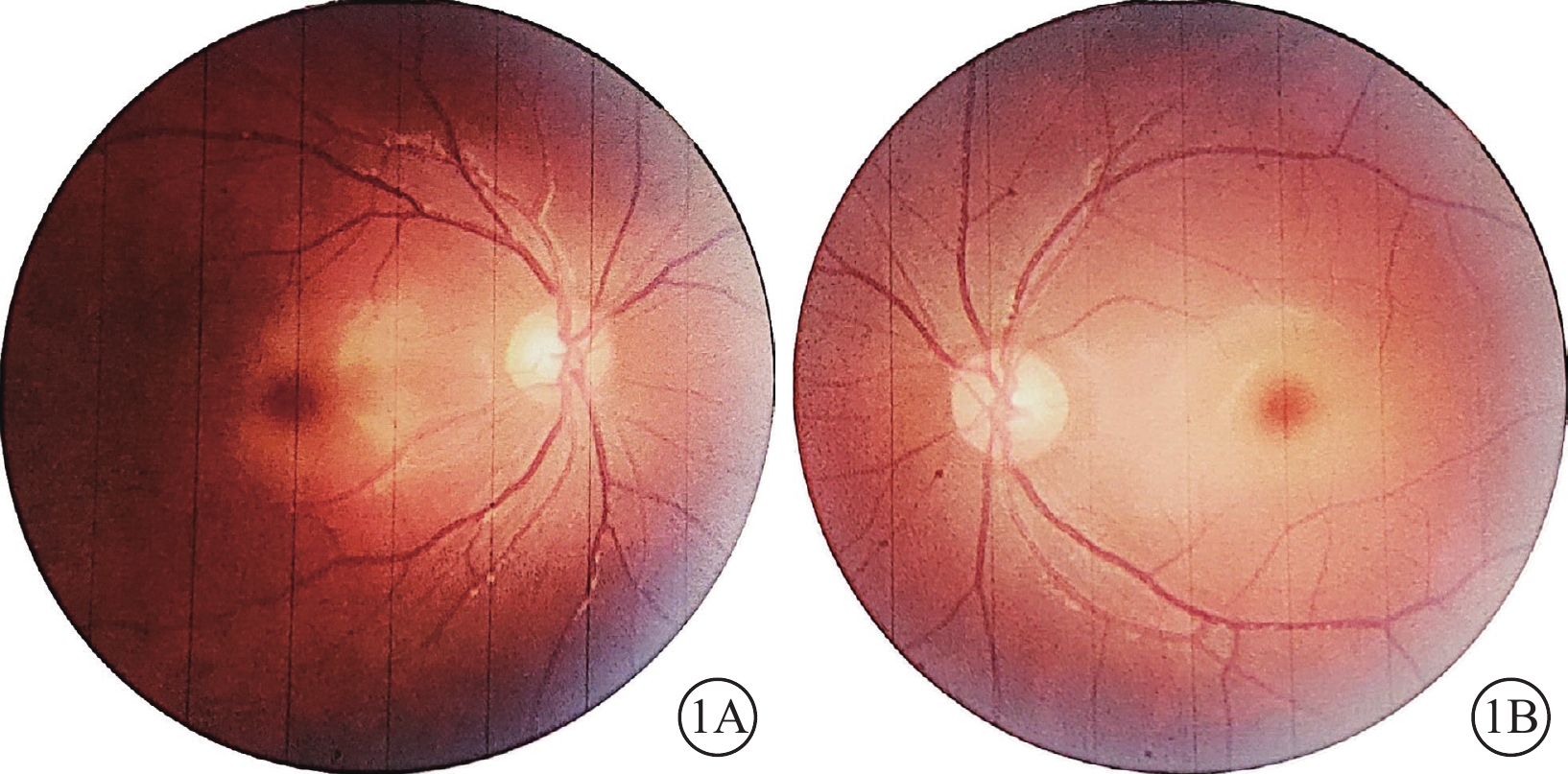 圖1
Ⅰ型唾液酸沉積癥并發黃斑部“櫻桃紅斑”患者雙眼彩色眼底像
圖1
Ⅰ型唾液酸沉積癥并發黃斑部“櫻桃紅斑”患者雙眼彩色眼底像
1A示右眼;1B示左眼。雙眼視盤顏色淡、邊界清晰,黃斑部“櫻桃紅斑”改變
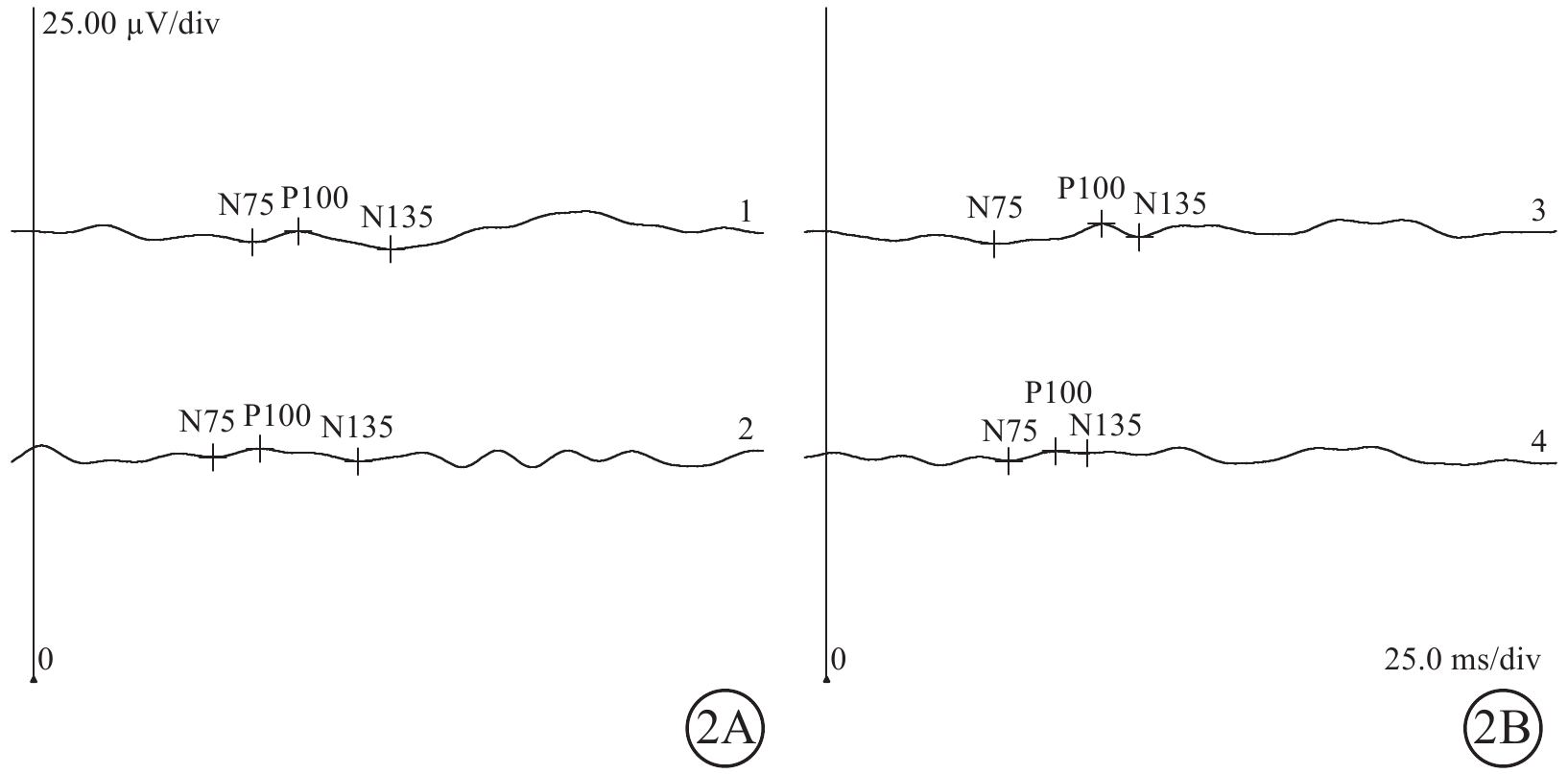
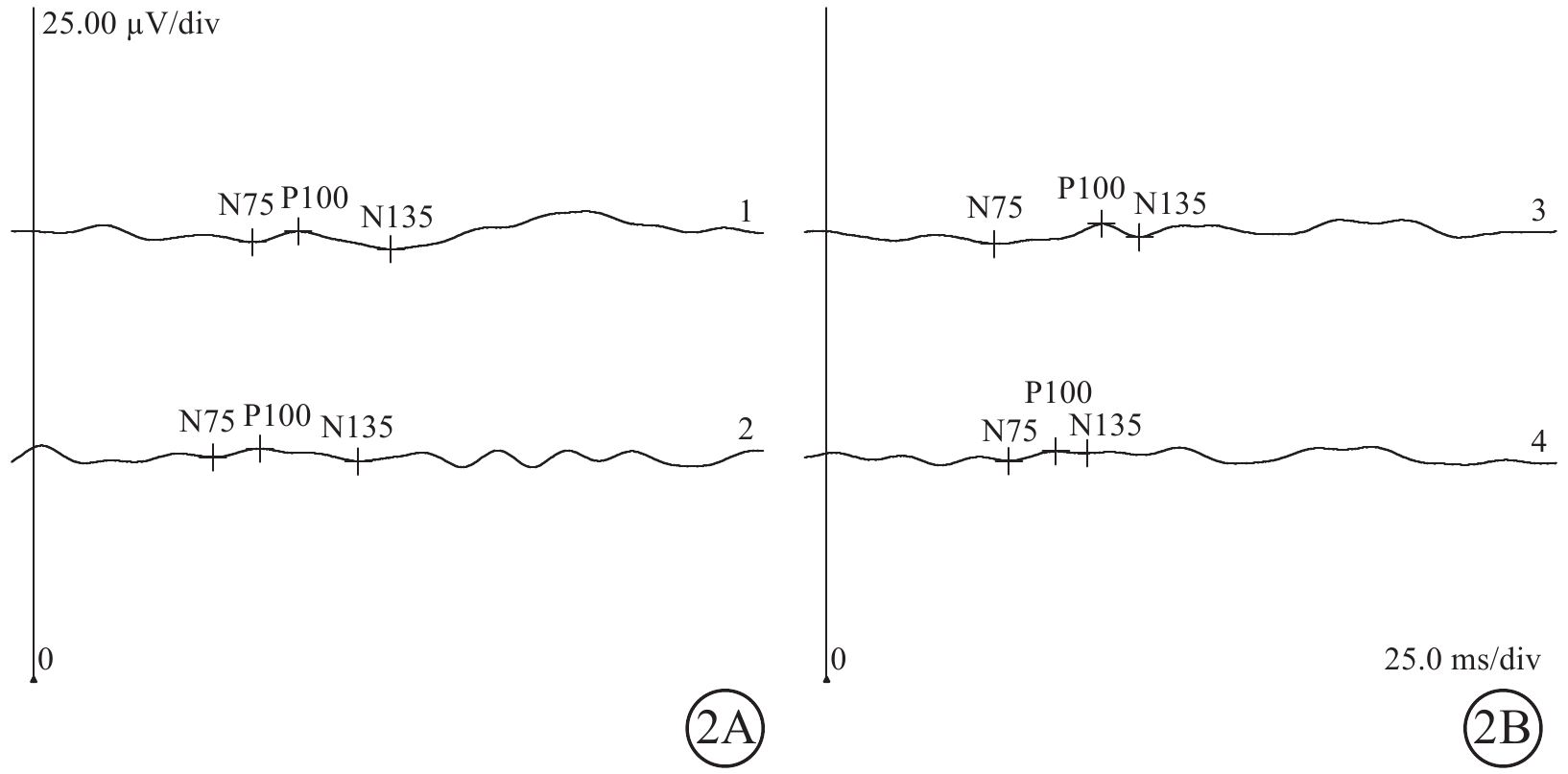 圖2
Ⅰ型唾液酸沉積癥并發黃斑部“櫻桃紅斑”患者雙眼視覺誘發電位檢查像
圖2
Ⅰ型唾液酸沉積癥并發黃斑部“櫻桃紅斑”患者雙眼視覺誘發電位檢查像
2A示右眼;2B示左眼。可見P100波潛時大致正常,振幅明顯降低
取得患者及其監護人知情同意后,取患者及其父母外周血行基因檢測。結果顯示,患者存在NEU1基因chr6:31829889 exon2 c.239C>T(p.P80L)雜合突變(來源于父親)、NEU1基因chr6:31827858 exon5 c.982G>A(p.G328S)雜合突變(來源于母親)(圖4)。根據臨床表現、黃斑部“櫻桃紅斑”改變及基因檢測結果,最終診斷:Ⅰ型唾液酸沉積癥(SD)。
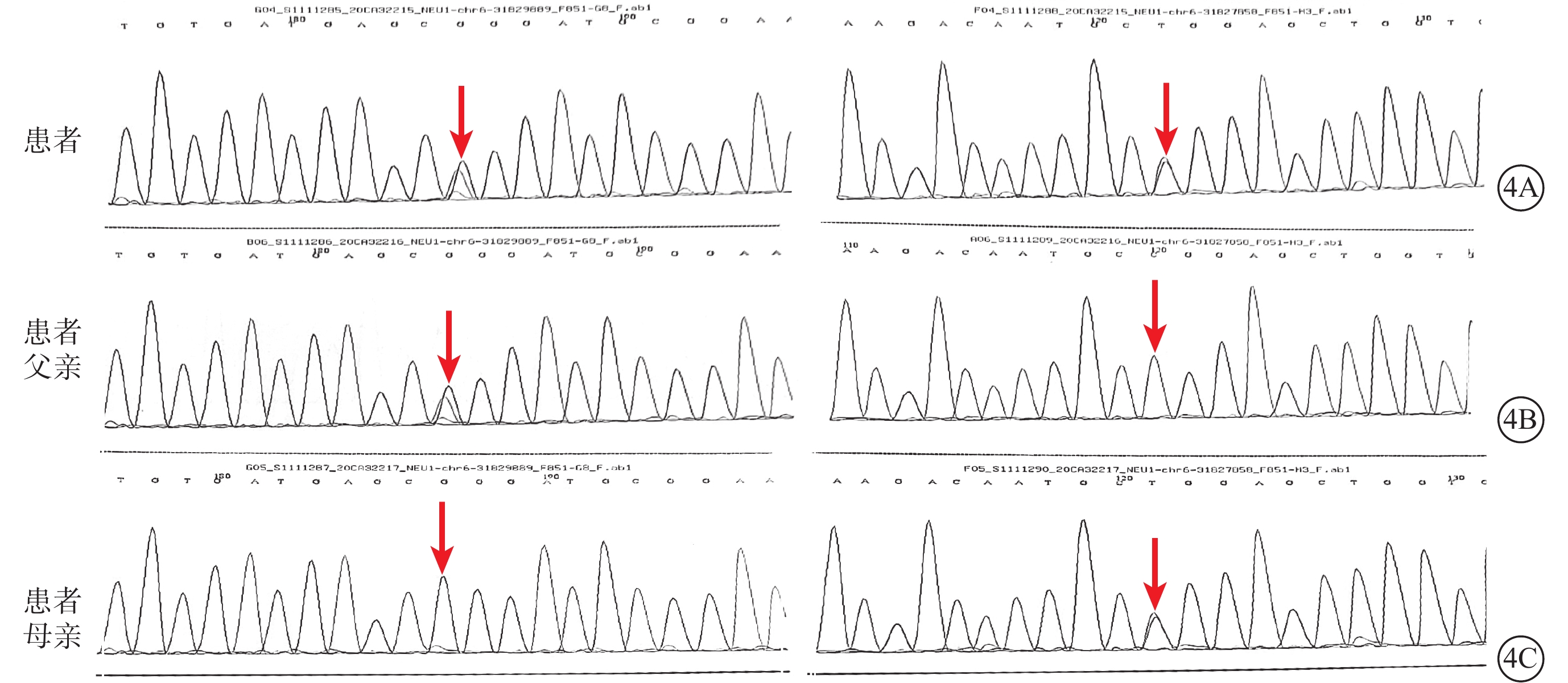
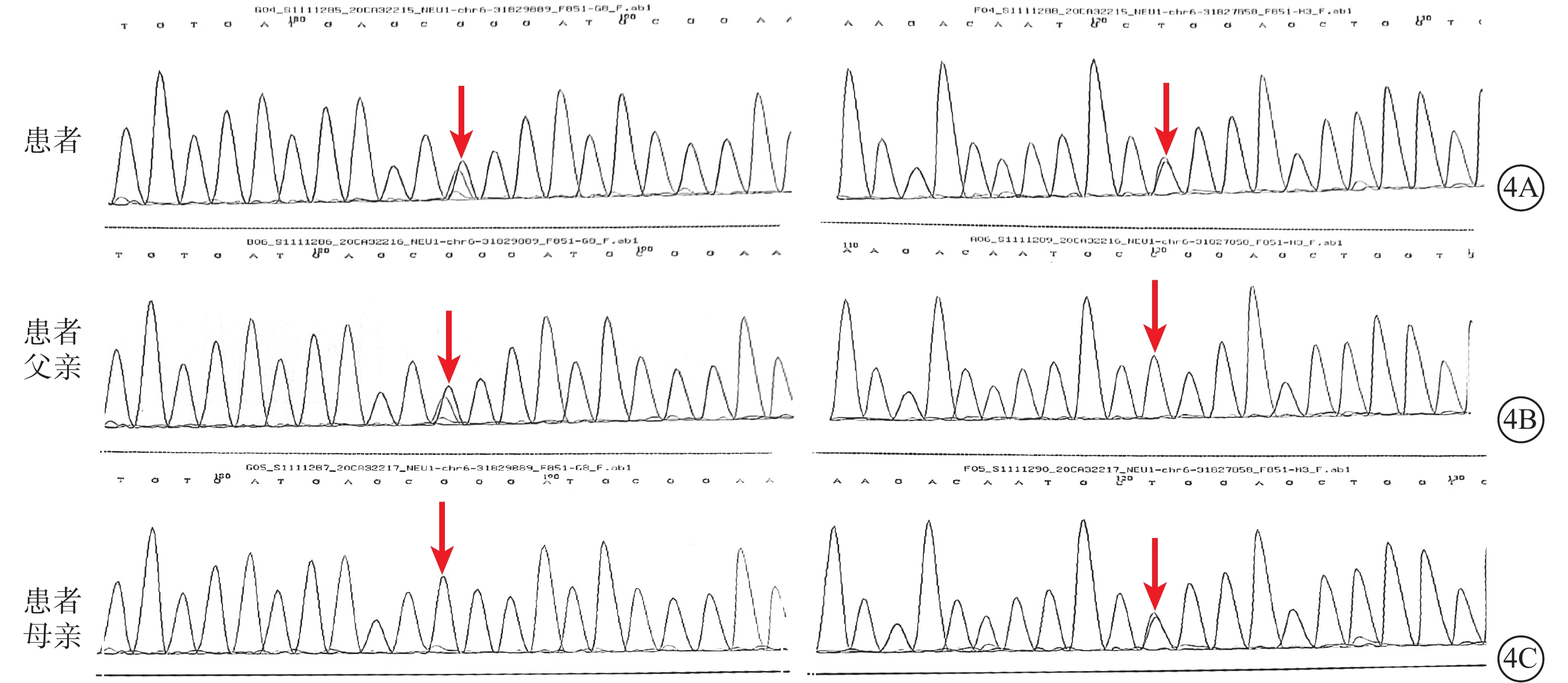 圖4
Ⅰ型唾液酸沉積癥并發黃斑部“櫻桃紅斑”患者及其父母基因測序圖
圖4
Ⅰ型唾液酸沉積癥并發黃斑部“櫻桃紅斑”患者及其父母基因測序圖
4A示患者存在
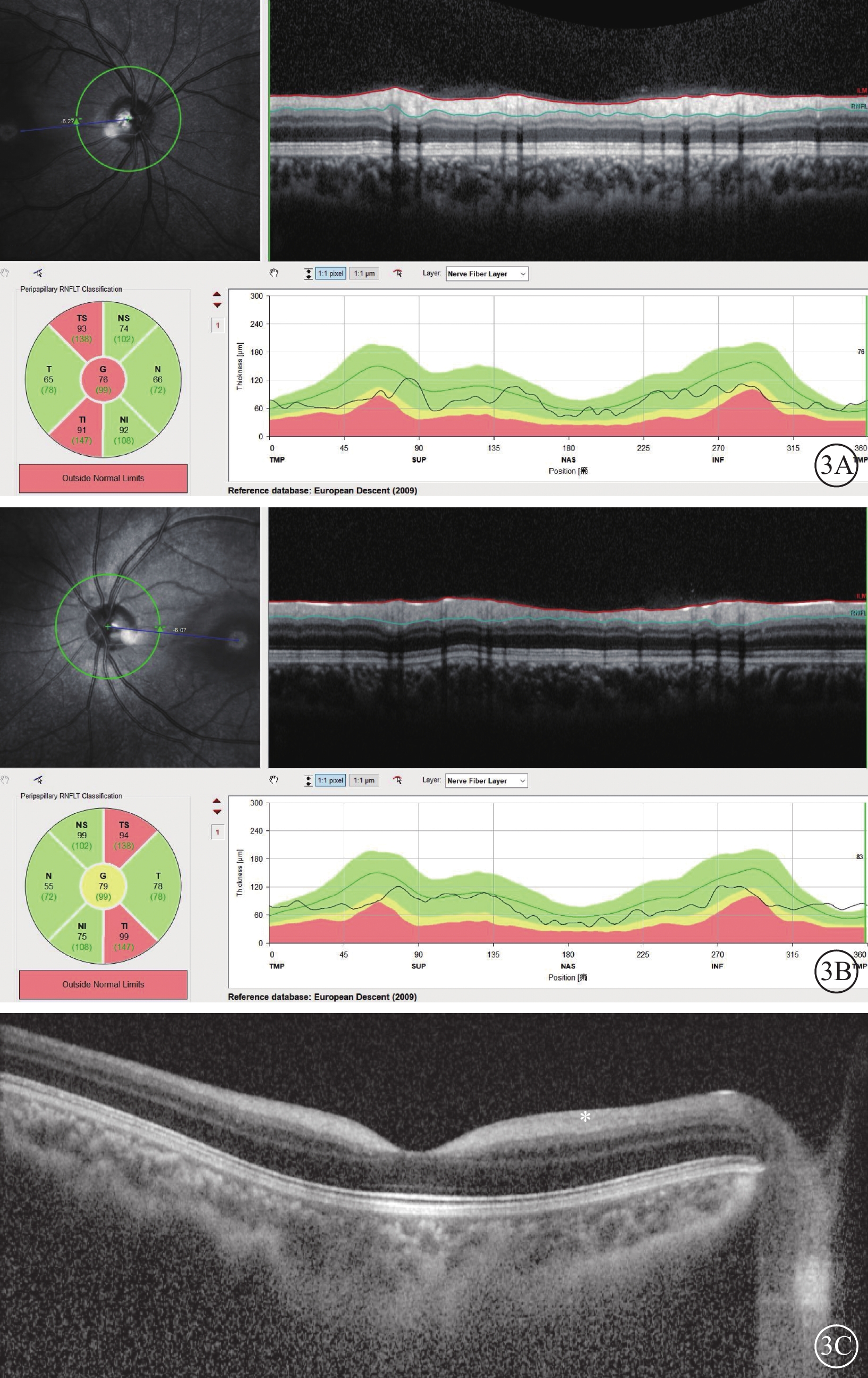
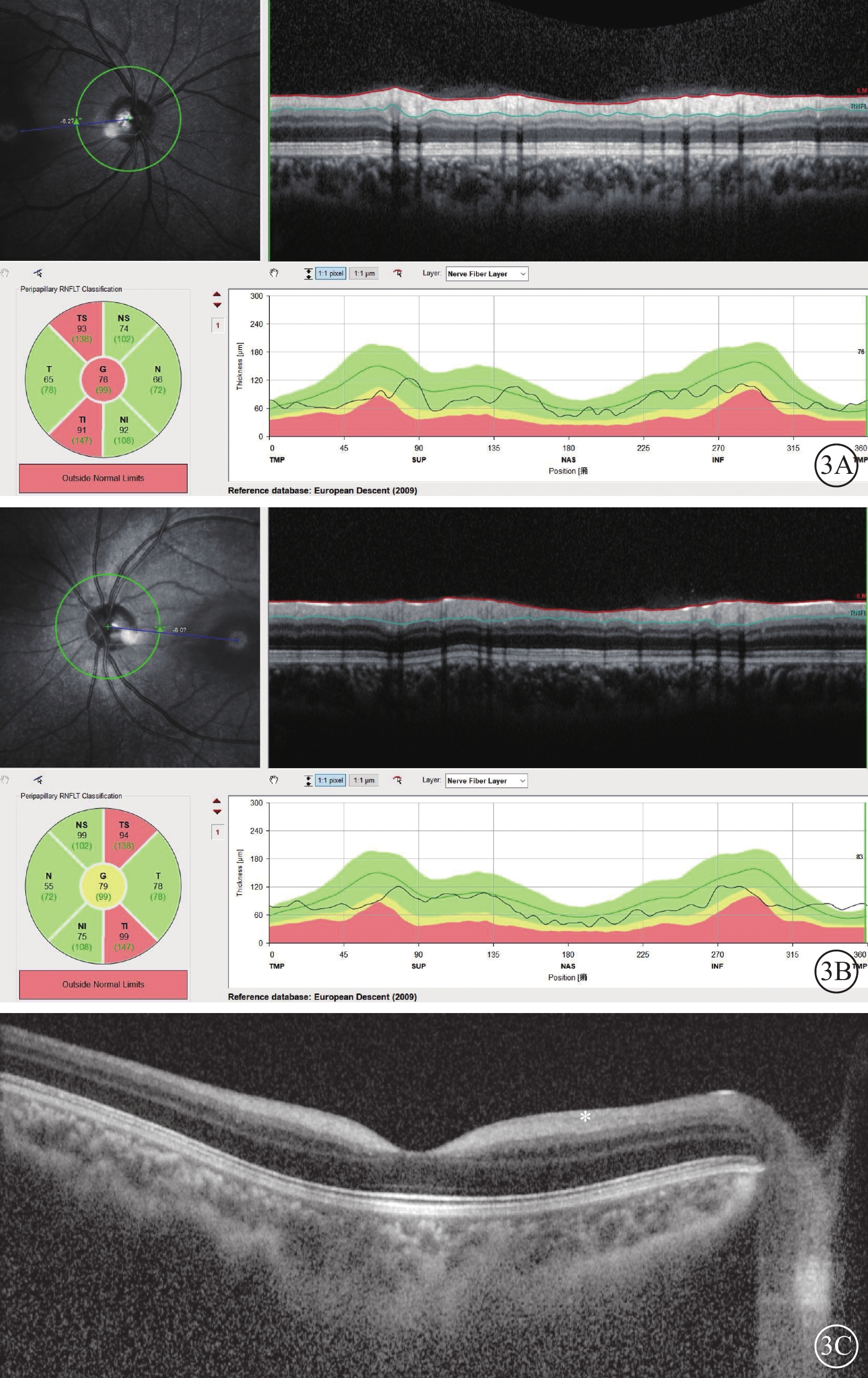 圖3
Ⅰ型唾液酸沉積癥并發黃斑部“櫻桃紅斑”患者雙眼光相干斷層掃描像
圖3
Ⅰ型唾液酸沉積癥并發黃斑部“櫻桃紅斑”患者雙眼光相干斷層掃描像
3A、3B分別示右眼、左眼視盤掃描,左圖為掃描方向和部位,右圖為檢查結果。雙眼視盤周圍神經纖維層厚度丟失,主要發生在雙眼顳上和顳下象限。3C示示右眼黃斑掃描,神經節細胞層由于代謝產物沉積反光增強至與神經纖維層相同,界限不清(白星)
討論 SD是一種極為罕見的常染色體隱性遺傳的神經代謝類疾病。它系NEU1基因突變導致α-N-乙酰神經氨酸酶(即唾液酸酶)異常,糖蛋白和糖脂的正常代謝分解受阻,導致唾液酸寡糖在細胞內異常蓄積所致。根據發病年齡和臨床癥狀SD分為兩型:Ⅰ型SD發病率為1/250 000~2 000 000成活嬰兒,發病年齡多在10歲之后[1]。初期癥狀較輕,隨著病情發展出現中樞神經系統表現,以肌陣攣最常見,另可伴有共濟失調(88.3%)和癲癇(72.5%)。顱腦MRI多表現為正常[2]。臨床上因其缺乏特異性表現常常造成誤診或漏診。眼部在早期即可出現視力下降,發生率高達68.4%[2]。黃斑“櫻桃紅斑”具有明顯的特異性,為診斷此病提供了重要線索。Ⅱ型SD于患兒出生即發病,表現為面部粗糙、肝膽脾腫大、骨骼異常、脊柱畸形及嚴重發育遲緩,常常在1歲內死亡[3-4]。本例患者最初癥狀表現為走路不穩,逐步進行性加重的肌陣攣和癲癇發作,與SD特征性肌陣攣、癲癇發作、小腦共濟失調表現相一致,因此此類患者常首診于神經內科。因患者出現視力進行性下降,經眼科檢查發現眼底特征性黃斑“櫻桃紅斑”表現,特別注意的是我們發現其OCT顯示唾液酸化代謝產物沉積在黃斑周圍的神經節細胞層,導致其反射增強與神經纖維層界限不清。VEP檢查提示雙眼傳導通路異常。基因檢測結果發現,本例患者攜帶NEU1基因c.239C>T和c.982G>A復合雜合突變,分別來自表型正常的父母。結合以上信息,該患兒符合Ⅰ型SD診斷。
目前國內有關兒童SD鮮見報道。呂瑞娟等[5]納入77例全球范圍內發表的Ⅰ型SD患者數據進行分析,發現中國內地Ⅰ型SD患者的發病年齡明顯偏小,為(10.8±2.7)歲;視力損害的發生率較其他亞洲地區更高,約81.8%~100.0%;39/72(54.2%)的患者存在黃斑“櫻桃紅斑”。“櫻桃紅斑”在臨床上具有辨識度和特異性,既往研究報道Ⅰ型SD患者的“櫻桃紅斑”發生率在50%以上[6-7]。Ahn等[8]認為,發病早的患者較發病晚的患者,更容易出現“櫻桃紅斑”。SD患者出現“櫻桃紅斑”的可能機制是,唾液酸化代謝產物沉積在黃斑周圍的神經節細胞層中,導致黃斑中心凹周圍神經節細胞層透明度下降而呈現白色外觀,中心凹缺乏神經節細胞而透出脈絡膜紅色,在白色背景的映襯下呈“櫻桃紅斑”的改變[9]。黃斑部神經節細胞最多,呈3~4層排列,隨著大量的唾液酸寡糖或糖肽的代謝產物沉積,在OCT圖像上表現為反射增強至與神經纖維層相同,兩層間界線模糊、融合為一層;同時造成視神經逐步萎縮,導致視力損傷[10]。本例患者黃斑部神經節細胞層呈乳白色、在OCT上表現為反射增強與神經纖維層相同,給唾液酸代謝物沉積所致的發病機制提供了臨床支持。
黃斑“櫻桃紅斑”還可見于其他溶酶體沉積癥疾病,如神經節苷脂沉積癥、神經元蠟樣脂褐質沉積癥、戈謝病、黏多糖病等。但這些疾病都有各自特點,臨床上較容易鑒別[11]。如嬰幼兒神經節苷脂沉積癥發病較早,多在出生后6~7個月出現發育倒退,伴有表情淡漠、對聲音刺激的“驚跳”現象、抽搐以及肌力改變等[12]。神經元蠟樣脂褐質沉積癥以進行性癡呆、發育倒退、癲癇、視力喪失、神經元內蠟樣脂褐素貯積為表現。戈謝病患兒多表現為明顯生長發育落后、骨骼受累、脾臟腫大、脾功能亢進血小板減少、貧血及眼球運動異常等。黏多糖病以面容粗陋、肝脾腫大和多發性骨發育不良為特征表現,伴或不伴進行性中樞神經系統疾病異常。因此,“櫻桃紅斑”對臨床上診斷SD具有較高的特異性,是確診SD的重要線索。
本例SD患者雙眼VEP檢查顯示P100波振幅明顯降低。Fan等[2]發現,幾乎所有的Ⅰ型SD患者均可見潛時延長和巨大的體感誘發電位(SSEP),即使在沒有視覺癥狀的SD患者中,VEP和SSEP也可能是Ⅰ型SD患者視力受損敏感的神經電生理標記物。Lai等[13]報道的17例Ⅰ型SD患者中,僅4例(23.5%)有視覺損害的臨床癥狀,而16例(94.1%)發現伴有VEP異常表現,因此VEP檢查對該病的視力損害有早期診斷價值,尤其適用于無法配合視力、OCT或視野檢查的低齡兒童。
本例患者同時伴有癲癇、肌陣攣、共濟失調等神經系統癥狀,盡管就診時顱腦MRI檢查及ERG早期無明顯異常。出現癲癇的SD患者腦電圖常顯多棘波和多棘-慢波,隨著病情進展,顱腦MRI檢查顯示大腦皮質萎縮或小腦萎縮[11, 14-15]。Lu等[15]通過Ⅰ型SD患者顱腦MRI檢查發現,顱腦后部皮質受損最嚴重位于枕葉和顳葉,而患者出現大腦微結構損傷最初發生在后部視覺區域,隨病情加重向更廣泛的大腦功能區域擴展。病理學研究發現,在Ⅰ型SD患者的大腦中樞神經元內積累的唾液酸寡糖[16],癲癇發作可能提示病變范圍增加或病情加重。
患者男,15歲。因走路易跌倒3年至北京兒童醫院神經內科就診,因雙眼進行性視力下降轉診于眼科。患兒1個月前無發熱性抽搐2次,表現為雙眼上翻,口唇發紺,牙關緊閉,約1~2 min緩解。無全身病史及家族遺傳病史。眼科檢查:右眼、左眼最佳矯正視力均為20/100;屈光狀態:右眼-4.50 DS/-2.75 DC×180°、 左眼-2.00 DS/-2.75 DC×5°。眼壓:右眼、左眼分別為19、16 mm Hg(1 mm Hg=0.133 kPa)。雙眼眼球運動正常;瞳孔圓,直徑2.5 mm,對光反射存在;晶狀體可見少量白色點狀混濁。眼底檢查,雙眼視盤顏色淡、邊界清楚,杯盤比約0.3~0.4,黃斑呈“櫻桃紅斑”改變(圖1)。視覺誘發電位(VEP)檢查,雙眼在低、中、高空間頻率刺激下P100波振幅明顯降低(圖2)。視網膜電圖(ERG)檢查,雙眼分別在明、暗適應刺激下a、b波振幅大致正常。光相干斷層掃描(OCT)檢查,雙眼視盤周圍神經纖維層厚度降低,右眼、左眼分別為76、79 μm;雙眼黃斑中心凹形態大致正常,神經節細胞層因唾液酸沉積反射增強,與神經纖維層反射強度相同,界限不清,融合為一層,兩層厚度正常(圖3)。神經系統檢查,四肢肌力及肌張力正常,腱反射正常,雙側巴賓斯基征(+),踝陣攣(-)。共濟運動失調檢測,指鼻試驗不準,走直線不穩。顱腦核磁共振成像(MRI)檢查及腦電圖檢查未見異常。眼科初步診斷:雙眼視神經病變、雙眼黃斑病變。神經內科建議進一步完善外周血基因檢查明確診斷。
圖1
Ⅰ型唾液酸沉積癥并發黃斑部“櫻桃紅斑”患者雙眼彩色眼底像
1A示右眼;1B示左眼。雙眼視盤顏色淡、邊界清晰,黃斑部“櫻桃紅斑”改變
圖2
Ⅰ型唾液酸沉積癥并發黃斑部“櫻桃紅斑”患者雙眼視覺誘發電位檢查像
2A示右眼;2B示左眼。可見P100波潛時大致正常,振幅明顯降低
取得患者及其監護人知情同意后,取患者及其父母外周血行基因檢測。結果顯示,患者存在NEU1基因chr6:31829889 exon2 c.239C>T(p.P80L)雜合突變(來源于父親)、NEU1基因chr6:31827858 exon5 c.982G>A(p.G328S)雜合突變(來源于母親)(圖4)。根據臨床表現、黃斑部“櫻桃紅斑”改變及基因檢測結果,最終診斷:Ⅰ型唾液酸沉積癥(SD)。
圖4
Ⅰ型唾液酸沉積癥并發黃斑部“櫻桃紅斑”患者及其父母基因測序圖
4A示患者存在
圖3
Ⅰ型唾液酸沉積癥并發黃斑部“櫻桃紅斑”患者雙眼光相干斷層掃描像
3A、3B分別示右眼、左眼視盤掃描,左圖為掃描方向和部位,右圖為檢查結果。雙眼視盤周圍神經纖維層厚度丟失,主要發生在雙眼顳上和顳下象限。3C示示右眼黃斑掃描,神經節細胞層由于代謝產物沉積反光增強至與神經纖維層相同,界限不清(白星)
討論 SD是一種極為罕見的常染色體隱性遺傳的神經代謝類疾病。它系NEU1基因突變導致α-N-乙酰神經氨酸酶(即唾液酸酶)異常,糖蛋白和糖脂的正常代謝分解受阻,導致唾液酸寡糖在細胞內異常蓄積所致。根據發病年齡和臨床癥狀SD分為兩型:Ⅰ型SD發病率為1/250 000~2 000 000成活嬰兒,發病年齡多在10歲之后[1]。初期癥狀較輕,隨著病情發展出現中樞神經系統表現,以肌陣攣最常見,另可伴有共濟失調(88.3%)和癲癇(72.5%)。顱腦MRI多表現為正常[2]。臨床上因其缺乏特異性表現常常造成誤診或漏診。眼部在早期即可出現視力下降,發生率高達68.4%[2]。黃斑“櫻桃紅斑”具有明顯的特異性,為診斷此病提供了重要線索。Ⅱ型SD于患兒出生即發病,表現為面部粗糙、肝膽脾腫大、骨骼異常、脊柱畸形及嚴重發育遲緩,常常在1歲內死亡[3-4]。本例患者最初癥狀表現為走路不穩,逐步進行性加重的肌陣攣和癲癇發作,與SD特征性肌陣攣、癲癇發作、小腦共濟失調表現相一致,因此此類患者常首診于神經內科。因患者出現視力進行性下降,經眼科檢查發現眼底特征性黃斑“櫻桃紅斑”表現,特別注意的是我們發現其OCT顯示唾液酸化代謝產物沉積在黃斑周圍的神經節細胞層,導致其反射增強與神經纖維層界限不清。VEP檢查提示雙眼傳導通路異常。基因檢測結果發現,本例患者攜帶NEU1基因c.239C>T和c.982G>A復合雜合突變,分別來自表型正常的父母。結合以上信息,該患兒符合Ⅰ型SD診斷。
目前國內有關兒童SD鮮見報道。呂瑞娟等[5]納入77例全球范圍內發表的Ⅰ型SD患者數據進行分析,發現中國內地Ⅰ型SD患者的發病年齡明顯偏小,為(10.8±2.7)歲;視力損害的發生率較其他亞洲地區更高,約81.8%~100.0%;39/72(54.2%)的患者存在黃斑“櫻桃紅斑”。“櫻桃紅斑”在臨床上具有辨識度和特異性,既往研究報道Ⅰ型SD患者的“櫻桃紅斑”發生率在50%以上[6-7]。Ahn等[8]認為,發病早的患者較發病晚的患者,更容易出現“櫻桃紅斑”。SD患者出現“櫻桃紅斑”的可能機制是,唾液酸化代謝產物沉積在黃斑周圍的神經節細胞層中,導致黃斑中心凹周圍神經節細胞層透明度下降而呈現白色外觀,中心凹缺乏神經節細胞而透出脈絡膜紅色,在白色背景的映襯下呈“櫻桃紅斑”的改變[9]。黃斑部神經節細胞最多,呈3~4層排列,隨著大量的唾液酸寡糖或糖肽的代謝產物沉積,在OCT圖像上表現為反射增強至與神經纖維層相同,兩層間界線模糊、融合為一層;同時造成視神經逐步萎縮,導致視力損傷[10]。本例患者黃斑部神經節細胞層呈乳白色、在OCT上表現為反射增強與神經纖維層相同,給唾液酸代謝物沉積所致的發病機制提供了臨床支持。
黃斑“櫻桃紅斑”還可見于其他溶酶體沉積癥疾病,如神經節苷脂沉積癥、神經元蠟樣脂褐質沉積癥、戈謝病、黏多糖病等。但這些疾病都有各自特點,臨床上較容易鑒別[11]。如嬰幼兒神經節苷脂沉積癥發病較早,多在出生后6~7個月出現發育倒退,伴有表情淡漠、對聲音刺激的“驚跳”現象、抽搐以及肌力改變等[12]。神經元蠟樣脂褐質沉積癥以進行性癡呆、發育倒退、癲癇、視力喪失、神經元內蠟樣脂褐素貯積為表現。戈謝病患兒多表現為明顯生長發育落后、骨骼受累、脾臟腫大、脾功能亢進血小板減少、貧血及眼球運動異常等。黏多糖病以面容粗陋、肝脾腫大和多發性骨發育不良為特征表現,伴或不伴進行性中樞神經系統疾病異常。因此,“櫻桃紅斑”對臨床上診斷SD具有較高的特異性,是確診SD的重要線索。
本例SD患者雙眼VEP檢查顯示P100波振幅明顯降低。Fan等[2]發現,幾乎所有的Ⅰ型SD患者均可見潛時延長和巨大的體感誘發電位(SSEP),即使在沒有視覺癥狀的SD患者中,VEP和SSEP也可能是Ⅰ型SD患者視力受損敏感的神經電生理標記物。Lai等[13]報道的17例Ⅰ型SD患者中,僅4例(23.5%)有視覺損害的臨床癥狀,而16例(94.1%)發現伴有VEP異常表現,因此VEP檢查對該病的視力損害有早期診斷價值,尤其適用于無法配合視力、OCT或視野檢查的低齡兒童。
本例患者同時伴有癲癇、肌陣攣、共濟失調等神經系統癥狀,盡管就診時顱腦MRI檢查及ERG早期無明顯異常。出現癲癇的SD患者腦電圖常顯多棘波和多棘-慢波,隨著病情進展,顱腦MRI檢查顯示大腦皮質萎縮或小腦萎縮[11, 14-15]。Lu等[15]通過Ⅰ型SD患者顱腦MRI檢查發現,顱腦后部皮質受損最嚴重位于枕葉和顳葉,而患者出現大腦微結構損傷最初發生在后部視覺區域,隨病情加重向更廣泛的大腦功能區域擴展。病理學研究發現,在Ⅰ型SD患者的大腦中樞神經元內積累的唾液酸寡糖[16],癲癇發作可能提示病變范圍增加或病情加重。