引用本文: 姚雨佳, 李佳駿, 王蘇豫, 李柯然. G蛋白抑制性α亞單位1/3介導神經軸突導向因子-1信號轉導并調控血管生成. 中華眼底病雜志, 2024, 40(10): 781-789. doi: 10.3760/cma.j.cn511434-20240222-00078 復制
版權信息: ?四川大學華西醫院華西期刊社《中華眼底病雜志》版權所有,未經授權不得轉載、改編
糖尿病視網膜病變(DR)是糖尿病的重要并發癥,尤其在工作年齡段人群中是視力損害的主要原因[1]。高血糖引發的神經炎癥和微血管損傷破壞血視網膜屏障,促進病理性血管生成,是DR發展的關鍵環節[2]。軸突導向因子-1(NTN1)是一種在神經發育中促進軸突生長的蛋白,也被發現參與內皮細胞活化和血管生成,具有濃度依賴性的雙相效應。低濃度的NTN1可激活內皮細胞,促進血管生成,但具體機制尚不完全清楚[3-5]。G蛋白抑制性α亞單位(Gαi1/Gαi3)在多種生長因子信號轉導中發揮作用,通過活化蛋白激酶B(Akt)-哺乳動物雷帕霉素靶蛋白(mTOR)和細胞外信號調節激酶(Erk)-絲裂原活化蛋白激酶(MAPK)等通路,影響細胞增殖和遷移[6-11]。Gαi1/Gαi3介導的血管內皮生長因子(VEGF)信號通路對視網膜血管新生至關重要,提示其在病理性血管生成中作為潛在治療靶點的重要性[11-12]。本研究旨在探索Gαi1/Gαi3在NTN1信號轉導和血管生成中的作用及其分子機制,通過建立糖尿病小鼠模型,深入了解血管生成的分子基礎,以期為DR的早期干預和治療策略提供新的理論依據和潛在靶標。
1 材料和方法
1.1 實驗動物及主要材料、儀器
雄性C57BL/6J小鼠55只,其中6~8周齡20只,體重20~25 g;2周齡35只,體重約10 g;均為無特定病原體級,由南京君科生物工程有限公司提供。所有小鼠飼養于標準(12 h明/暗循環)清潔級環境中。飼養環境及實驗操作均符合國家科學技術委員會《實驗動物管理條例》規定,并獲得南京醫科大學實驗動物倫理委員會許可[許可證號:SYXK(蘇)2018-0020]。
人臍靜脈內皮細胞(HUVEC)細胞株由本實驗室自行保存。攜綠色熒光蛋白(GFP)Gαi1/Gαi3 shRNA重組腺相關病毒(AAV5)載體(AAV5-TIE1-Gαi1/Gαi3 shRNA)、僅攜帶GFP的空白AAV5載體、Gαi1/Gαi3 shRNA重組慢病毒、僅攜帶GFP的空白慢病毒(上海吉凱基因醫學科技股份有限公司)。無水葡萄糖(德國Biofroxx公司);內皮細胞培養基(ECM培養基)(美國Sciencell公司);4',6-二脒基-2-苯基吲哚(DAPI)、嘌呤霉素、辣根過氧化物酶(HRP)二抗(上海碧云天生物技術有限公司);鏈脲佐菌素(STZ)、磷酸鹽緩沖液(PBS)(北京蘭杰柯科技有限公司);20倍洗膜緩沖液(TBST)(北京索萊寶科技有限公司);核糖體蛋白S6激酶(S6K)抗體、磷酸化S6K(p-S6K)抗體(美國Cell Signaling Technology公司);Erk1/2抗體、磷酸化Erk1/2(p-Erk1/2)抗體、Akt抗體、磷酸化Akt(p-Akt)抗體(杭州華安生物技術有限公司);NTN1(美國Enzo公司);β-肌動蛋白(actin)抗體(成都正能生物技術有限責任公司)。EdU-555細胞增殖檢測試劑盒(上海碧云天生物技術有限公司);Transwell共培養板、高蛋白濃度基質膠(美國Corning公司);同工凝集素B4(IB4)(美國Sigma公司);細胞/組織總RNA提取試劑盒、逆轉錄預混液、通用型高靈敏度染料法定量聚合酶鏈反應(qPCR)檢測試劑盒(南京諾唯贊生物科技有限公司);PIKOReal 96實時熒光qPCR(qRT-PCR)儀(美國Thermo公司);倒置熒光顯微鏡(日本Olympus公司)。
1.2 實驗動物分組和糖尿病模型建立
采用隨機數字表法將20只6~8周齡小鼠分為正常對照組、糖尿病組,每組各10只。糖尿病組小鼠空腹饑餓12 h(期間可自由進水)后,按10 mg/kg劑量予以小鼠腹腔注射STZ。1周后測空腹血糖≥16.7 mmol/L為造模成功,繼續高糖、高脂飼養,期間定期監測血糖,使血糖維持在成模范圍內直至實驗結束。
將35只2周齡小鼠隨機分為正常對照組、玻璃體腔注射NTN1組(NTN1組)、視網膜內皮細胞Gαi1/Gαi3特異性敲低+玻璃體腔注射NTN1組(Gαi1/Gαi3 eKD+NTN1組),其中正常對照組、NTN1組每組各15只,Gαi1/Gαi3 eKD+NTN1組5只。NTN1組于小鼠2周齡時玻璃體腔注射50 ng/ml NTN1 2 μl。Gαi1/Gαi3 eKD+NTN1組于小鼠2周齡時玻璃體腔注射AAV5-TIE1-Gαi1/Gαi3 shRNA 2 μl,1周后玻璃體腔注射50 ng/ml NTN1 2 μl。玻璃體腔注射方式:腹腔注射氯胺酮和甲苯噻嗪麻醉小鼠,散瞳后微量注射器抽取AAV5-TIE1-Gαi1/Gαi3 shRNA 2 μl于小鼠眼鼻上方角膜緣后約1 mm處垂直穿刺進入玻璃體腔,向眼球中心方向進針,將藥液緩慢注入玻璃體腔,眼內停留約30 s后拔出針頭,涂左氧氟沙星眼膏。
1.3 細胞培養、轉染分組
細胞培養。將HUVEC細胞株置于含5%胎牛血清和1%雙抗的ECM培養基中常規培養。取對數生長期細胞用于實驗。
將HUVEC分為陰性對照慢病毒組(shC組)、陰性對照慢病毒+NTN1處理組(shC+NTN1組)、Gαi1/Gαi3敲低組(shGαi1/Gαi3組)、Gαi1/Gαi3敲低+NTN1處理組(shGαi1/Gαi3+NTN1組)。shC組細胞加入空白慢病毒;shGαi1/Gαi3組細胞加入Gαi1/Gαi3 shRNA重組慢病毒誘導。轉染20 h后換為正常完全培養基,然后正常傳代,穩定細胞株通過含有嘌呤霉素的完全培養基篩選1周。shC+NTN1組細胞及shGαi1/Gαi3+NTN1組細胞在嘌呤霉素篩選后使用NTN1(50 ng/ml)預處理24 h后進行后續實驗。
1.4 IB4染色法檢測Gαi1/Gαi3敲低對NTN1誘導的視網膜新生血管的影響
正常對照組、NTN1組、Gαi1/Gαi3 eKD+NTN1組小鼠4周齡時腹腔注射氯胺酮和甲苯噻嗪麻醉,將彎鑷沿顳側眶壁進入呈先垂直再平行向鼻側走形,至眼球下方夾住視神經,摘取完整眼球,置于4%多聚甲醛中固定40 min,轉移至含PBS的10 cm細胞培養皿中。用眼科剪和眼科鑷去除眼球周圍組織,沿角膜緣剪開眼球,去除眼前節,移除晶狀體和玻璃體,分離視網膜,切成4瓣。固定、通透封閉,IB4染料(1∶50)4℃孵育過夜,PBS洗滌3次,10 min/次。在每個視網膜瓣的中間區域隨機選取視野,于熒光顯微鏡下按序拍攝。
1.5 EdU實驗觀察細胞增殖能力
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞以8×104個/孔的密度接種于24孔板中,每孔加入10 μmol/L EdU工作液,于37℃孵育箱中培養2 h,固定及洗滌后,DAPI避光染色10 min,PBS洗滌3次,5 min/次。熒光顯微鏡觀察并計數。細胞核呈藍色熒光。實驗重復3次。
1.6 Transwell實驗觀察細胞的遷移能力
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞以8×104個/孔的密度接種于Transwell小室上室內,無血清培養基培養;下室加入含5%胎牛血清的ECM培養基。細胞培養板置于孵箱中培育24 h,多聚甲醛固定下室表面細胞15 min,棉簽擦去上室未向下室遷移的細胞,常規結晶紫染色3 min。光學顯微鏡下觀察拍照。每組隨機選取6~8個視野以計數小室底膜上、下室側附著的細胞,即為發生遷移的細胞數。每組各設3個復孔,重復3次。
1.7 Matrigel實驗檢測細胞管腔形成能力
將4℃高蛋白濃度基質膠40 μl緩慢加入24孔板中,于37℃孵育箱中培養30 min。shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞以8×104個/孔的密度接種于24孔板中孵育,6 h后觀察管腔形成情況。計數相同視野面積下的管腔形成數量。實驗重復3次。
1.8 qPCR檢測小鼠視網膜及HUVEC中Ntn1、Gαi1、Gαi3、甘油醛-3-磷酸脫氫酶(Gapdh)基因的mRNA相對表達量
過量麻醉處死小鼠,摘除眼球剝離視網膜,RNA提取試劑盒提取總RNA;HUVEC傳代后鋪于6孔板,細胞匯合度為90%~100%時RNA提取試劑盒提取總RNA。反轉錄試劑逆轉錄成cDNA,按相應基因配置SYBR Green qPCR擴增反應體系進行qPCR檢測。應用Express 3.0軟件設計引物序列(表1)。置于qRT-PCR儀中進行擴增并輸出循環閾值(Ct值),以GAPDH為內參照,依照公式2-ΔΔCt計算mRNA相對表達量。
1.9 蛋白質免疫印跡法(Western blot)檢測小鼠視網膜組織及HUVEC中NTN1、Gαi1、Gαi3、Akt、p-Akt、S6K、p-S6K、Erk1/2、p-Erk1/2蛋白表達
收集各組細胞總蛋白,調整蛋白濃度。每個樣孔加入10 μl待測樣品,十二烷基硫酸鈉聚丙烯酰胺凝膠電泳中上樣電泳;140 V恒壓50 min,400 mA轉膜40 min。5%脫脂牛奶封閉2 h,1∶1 000一抗4℃孵育過夜(β-actin作為陽性對照),TBST洗膜10 min,重復3次;加入辣根過氧化物偶聯的二抗,室溫孵育1 h,TBST洗膜10 min,重復3次;加入化學發光劑,暗室曝光。Image J軟件分析蛋白條帶的灰度值,目的蛋白相對表達量=目的蛋白條帶灰度值/內參照β-actin蛋白條帶灰度值,磷酸化蛋白相對總蛋白相對表達量=磷酸化蛋白條帶灰度值/總蛋白條帶灰度值。
1.10 統計學方法
采用SPSS20.0軟件進行統計學分析。計量數據以均數±標準差(x±s)表示。兩組間比較采用t檢驗;三組間比較采用單因素方差分析;多因素比較采用事后Dunnett檢驗。采用Graph-prism軟件對所獲得數據進行圖表整理。P<0.05為差異有統計學意義。
2 結果
2.1 DR模型小鼠視網膜組織中Ntn1、Gαi1、Gαi3 mRNA和蛋白表達升高
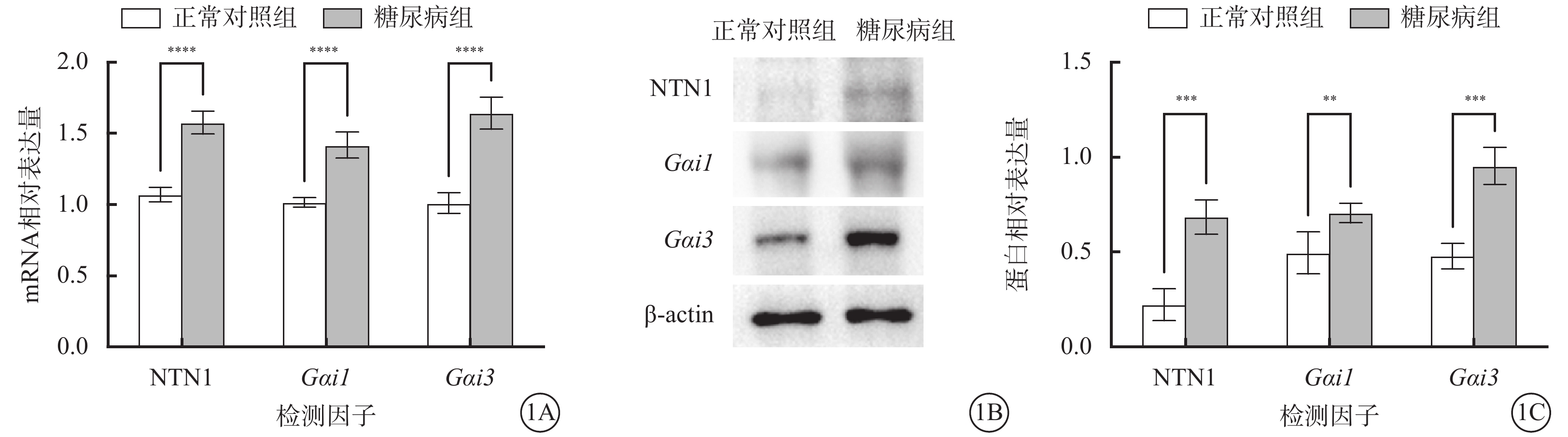
與正常對照組比較,糖尿病組小鼠視網膜內Ntn1、Gαi1、Gαi3 mRNA和蛋白相對表達量顯著增加,差異有統計學意義(t=11.800、9.298、10.620、7.503、3.432、8.037,P<0.000 1)(圖1)。
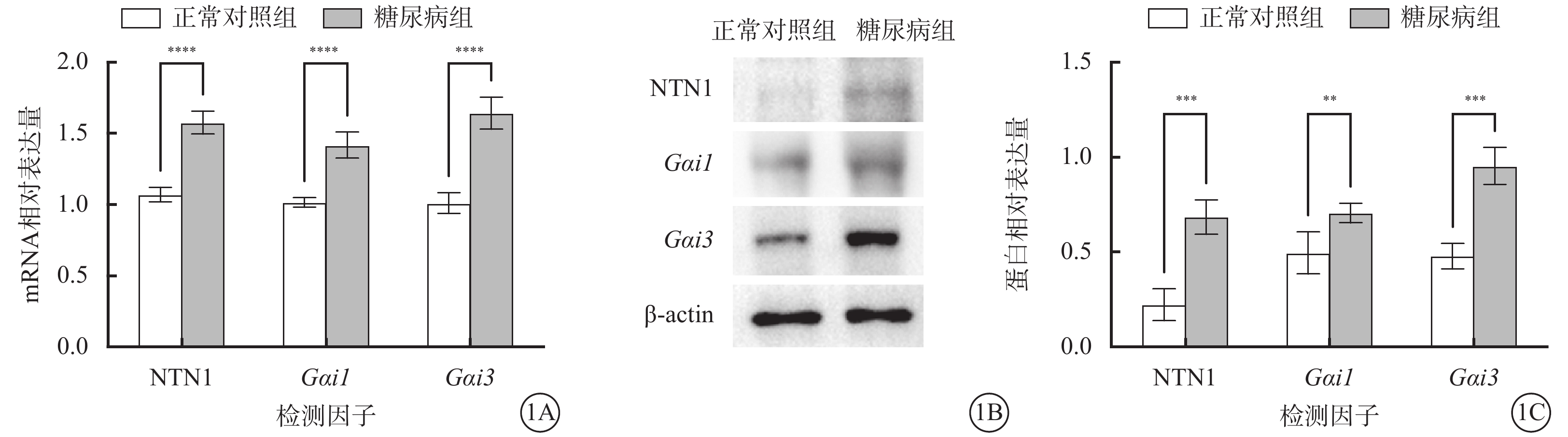 圖1
正常對照組、糖尿病組小鼠視網膜中Ntn1、Gαi1、Gαi3 mRNA及蛋白相對表達量比較(n=3) 1A示Ntn1、Gαi1、Gαi3 mRNA相對表達量,****P<0.000 1;1B示NTN1、Gαi1、Gαi3蛋白表達電泳圖;1C示NTN1、Gαi1、Gαi3蛋白相對表達量比較,**P<0.01,***P<0.001 NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;正常對照組:不做任何處理;糖尿病組:腹腔注射鏈脲佐菌素10 mg/kg
圖1
正常對照組、糖尿病組小鼠視網膜中Ntn1、Gαi1、Gαi3 mRNA及蛋白相對表達量比較(n=3) 1A示Ntn1、Gαi1、Gαi3 mRNA相對表達量,****P<0.000 1;1B示NTN1、Gαi1、Gαi3蛋白表達電泳圖;1C示NTN1、Gαi1、Gαi3蛋白相對表達量比較,**P<0.01,***P<0.001 NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;正常對照組:不做任何處理;糖尿病組:腹腔注射鏈脲佐菌素10 mg/kg
2.2 Gαi1/Gαi3 shRNA重組慢病毒誘導后HUVEC中 Gαi1、Gαi3 mRNA和蛋白相對表達量受到抑制
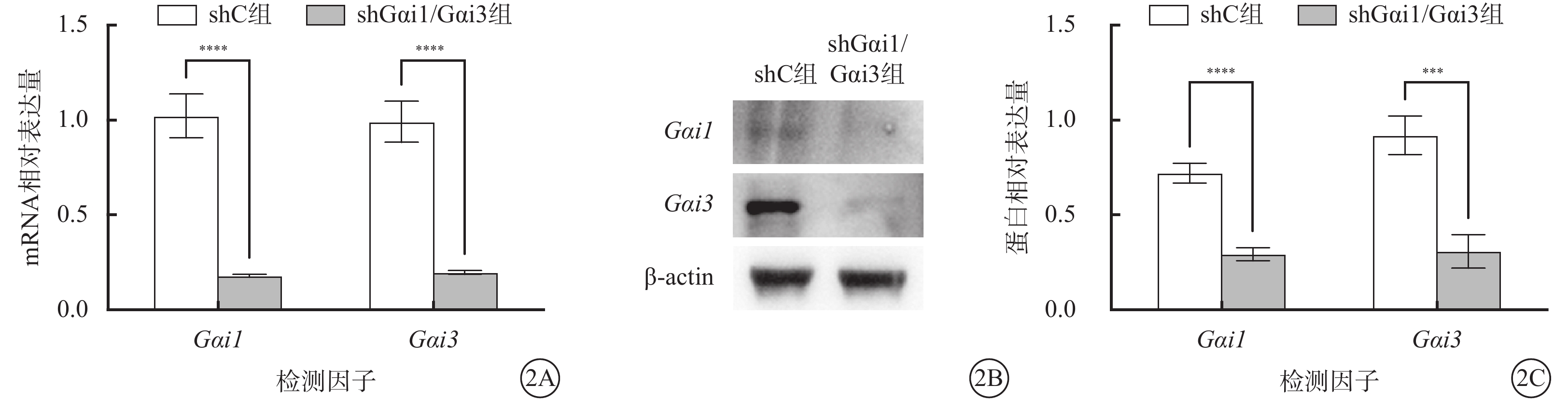
與shC組比較,shGαi1/Gαi3組HUVEC中Gαi1、Gαi3 mRNA和蛋白相對表達量顯著降低,差異有統計學意義(t=16.310、16.300、13.600、9.068,P<0.000 1)(圖2)。
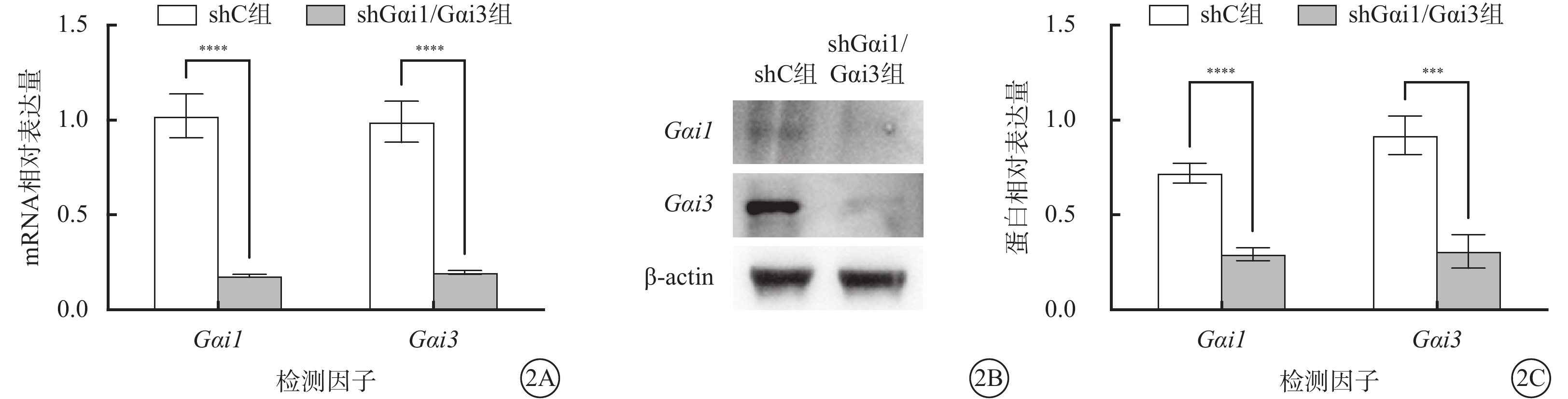 圖2
shC組、shGαi1/Gαi3組HUVEC中Gαi1、Gαi3 mRNA及蛋白相對表達量比較(n=3) 2A示Gαi1、Gαi3 mRNA相對表達量結果,****P<0.000 1;2B示Gαi1、Gαi3蛋白表達電泳圖;2C示中Gαi1、Gαi3蛋白相對表達量,***P<0.001, ****P<0.000 1 HUVEC:人臍靜脈內皮細胞;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;actin:肌動蛋白;shC組:空白慢病毒誘導;shGαi1/Gαi3組:加入Gαi1/Gαi3 shRNA重組慢病毒誘導
圖2
shC組、shGαi1/Gαi3組HUVEC中Gαi1、Gαi3 mRNA及蛋白相對表達量比較(n=3) 2A示Gαi1、Gαi3 mRNA相對表達量結果,****P<0.000 1;2B示Gαi1、Gαi3蛋白表達電泳圖;2C示中Gαi1、Gαi3蛋白相對表達量,***P<0.001, ****P<0.000 1 HUVEC:人臍靜脈內皮細胞;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;actin:肌動蛋白;shC組:空白慢病毒誘導;shGαi1/Gαi3組:加入Gαi1/Gαi3 shRNA重組慢病毒誘導
2.3 敲低Gαi1/Gαi3基因抑制NTN1誘導的HUVEC增殖、遷移與管腔形成能力
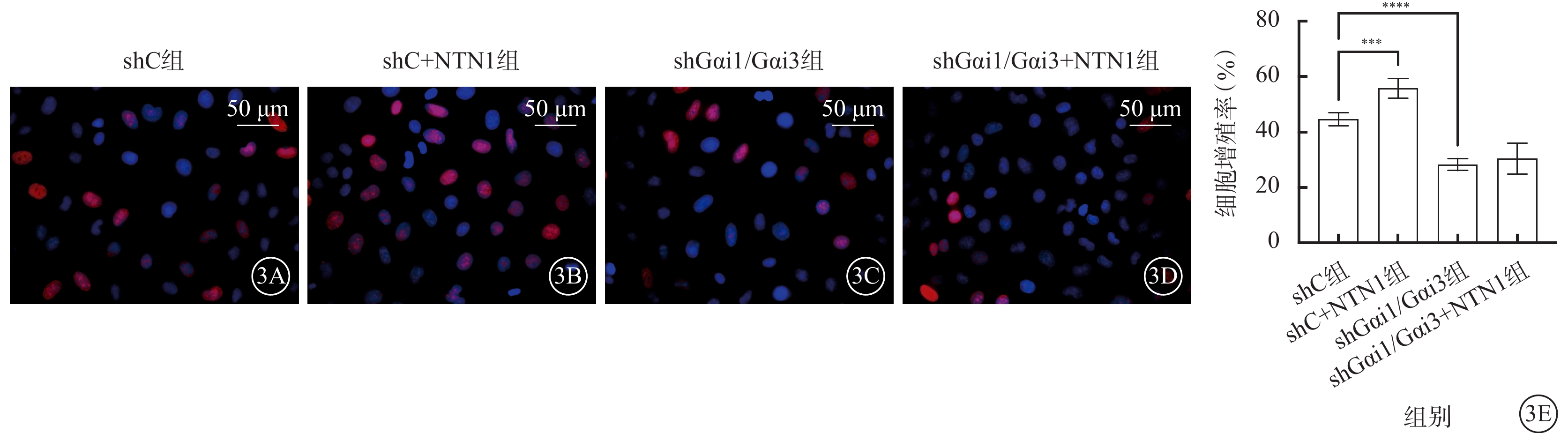
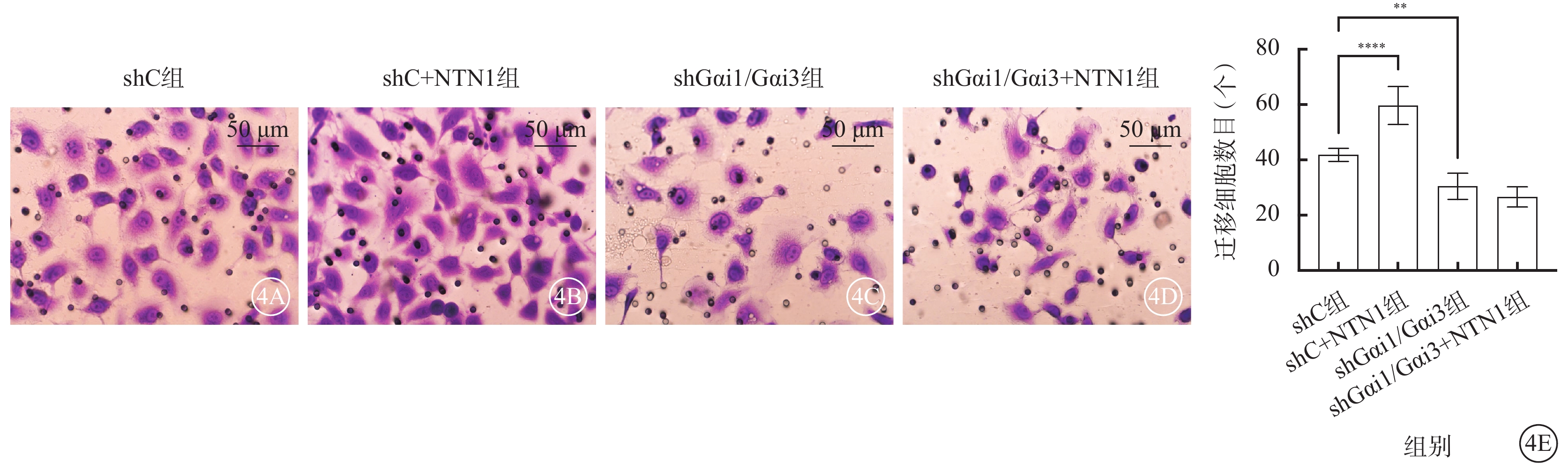
與shC組比較,shC+NTN1組HUVEC增殖率、遷移數量、管腔形成數量均顯著增加,shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組HUVEC增殖率、遷移數量、管腔形成數量均顯著減少,差異均有統計學意義(F=62.750、49.830、54.900,P<0.000 1)(圖3~5)。
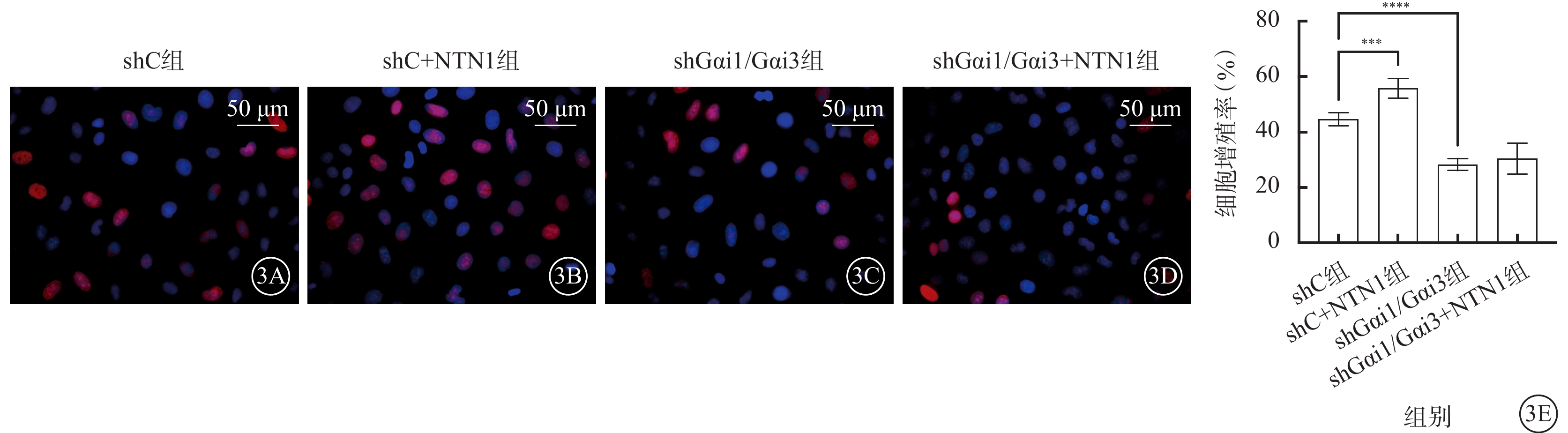 圖3
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組HUVEC增殖率比較(n=3) 3A~3D分別示shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組熒光顯微鏡像(DAPI染色,標尺:50 μm),與shC組比較,shC+NTN1組細胞增殖率顯著增加,shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞增殖率無明顯變化;3E示四組細胞增殖率比較,***P<0.001,****P<0.000 1 NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;shC組:加入空白慢病毒誘導;shC+NTN1組:加入空白慢病毒,用50 ng/ml NTN1誘導;shGαi1/Gαi3組:加入Gαi1/Gαi3 shRNA重組慢病毒誘導;shGαi1/Gαi3+NTN1組:加入Gαi1/Gαi3 shRNA重組慢病毒,用50 ng/ml NTN1誘導;DAPI:4',6-二脒基-2-苯基吲哚
圖3
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組HUVEC增殖率比較(n=3) 3A~3D分別示shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組熒光顯微鏡像(DAPI染色,標尺:50 μm),與shC組比較,shC+NTN1組細胞增殖率顯著增加,shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞增殖率無明顯變化;3E示四組細胞增殖率比較,***P<0.001,****P<0.000 1 NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;shC組:加入空白慢病毒誘導;shC+NTN1組:加入空白慢病毒,用50 ng/ml NTN1誘導;shGαi1/Gαi3組:加入Gαi1/Gαi3 shRNA重組慢病毒誘導;shGαi1/Gαi3+NTN1組:加入Gαi1/Gαi3 shRNA重組慢病毒,用50 ng/ml NTN1誘導;DAPI:4',6-二脒基-2-苯基吲哚
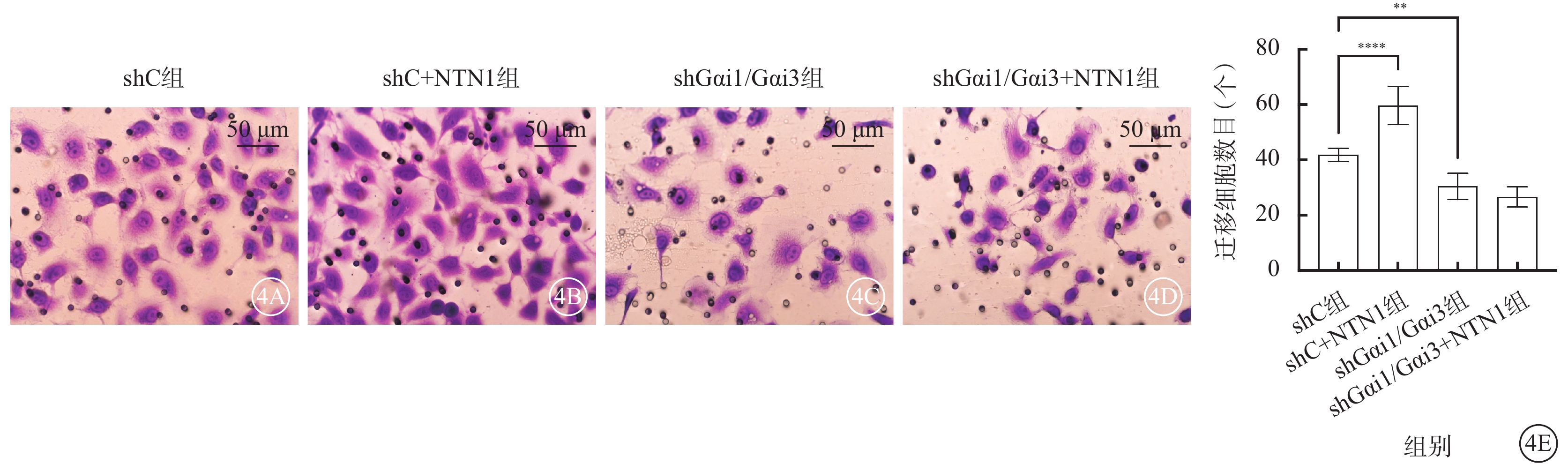 圖4
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組HUVEC遷移數量比較(n=3) 4A~4D分別示shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組光學顯微鏡像(結晶紫染色,標尺:50 μm),與shC組比較,shC+NTN1組細胞遷移數量顯著增加,shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞遷移數量無明顯變化;4E示四組細胞遷移數量比較,**P<0.01,****P<0.000 1 NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;shC組:加入空白慢病毒誘導;shC+NTN1組:加入空白慢病毒,用50 ng/ml NTN1誘導;shGαi1/Gαi3組:加入Gαi1/Gαi3 shRNA重組慢病毒誘導;shGαi1/Gαi3+NTN1組:加入Gαi1/Gαi3 shRNA重組慢病毒,用50 ng/ml NTN1誘導
圖4
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組HUVEC遷移數量比較(n=3) 4A~4D分別示shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組光學顯微鏡像(結晶紫染色,標尺:50 μm),與shC組比較,shC+NTN1組細胞遷移數量顯著增加,shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞遷移數量無明顯變化;4E示四組細胞遷移數量比較,**P<0.01,****P<0.000 1 NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;shC組:加入空白慢病毒誘導;shC+NTN1組:加入空白慢病毒,用50 ng/ml NTN1誘導;shGαi1/Gαi3組:加入Gαi1/Gαi3 shRNA重組慢病毒誘導;shGαi1/Gαi3+NTN1組:加入Gαi1/Gαi3 shRNA重組慢病毒,用50 ng/ml NTN1誘導
 圖5
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組HUVEC管腔形成情況比較(n=3) 5A~5D分別示shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞倒置相差顯微鏡像(標尺:50 μm),與shC組比較,shC+NTN1組細胞管腔形成數量顯著增加,shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞管腔形成數量顯著減少;5E示四組細胞管腔形成數目比較,*P<0.05 ,****P<0.000 1 NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;shC組:加入空白慢病毒誘導;shC+NTN1組:加入空白慢病毒,用50 ng/ml NTN1誘導;shGαi1/Gαi3組:加入Gαi1/Gαi3 shRNA重組慢病毒誘導;shGαi1/Gαi3+NTN1組:加入Gαi1/Gαi3 shRNA重組慢病毒,用50 ng/ml NTN1誘導
圖5
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組HUVEC管腔形成情況比較(n=3) 5A~5D分別示shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞倒置相差顯微鏡像(標尺:50 μm),與shC組比較,shC+NTN1組細胞管腔形成數量顯著增加,shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞管腔形成數量顯著減少;5E示四組細胞管腔形成數目比較,*P<0.05 ,****P<0.000 1 NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;shC組:加入空白慢病毒誘導;shC+NTN1組:加入空白慢病毒,用50 ng/ml NTN1誘導;shGαi1/Gαi3組:加入Gαi1/Gαi3 shRNA重組慢病毒誘導;shGαi1/Gαi3+NTN1組:加入Gαi1/Gαi3 shRNA重組慢病毒,用50 ng/ml NTN1誘導
2.4 敲低Gαi1/Gαi3基因檢測結果抑制NTN1誘導的Gαi1、Gαi3 mRNA和蛋白表達
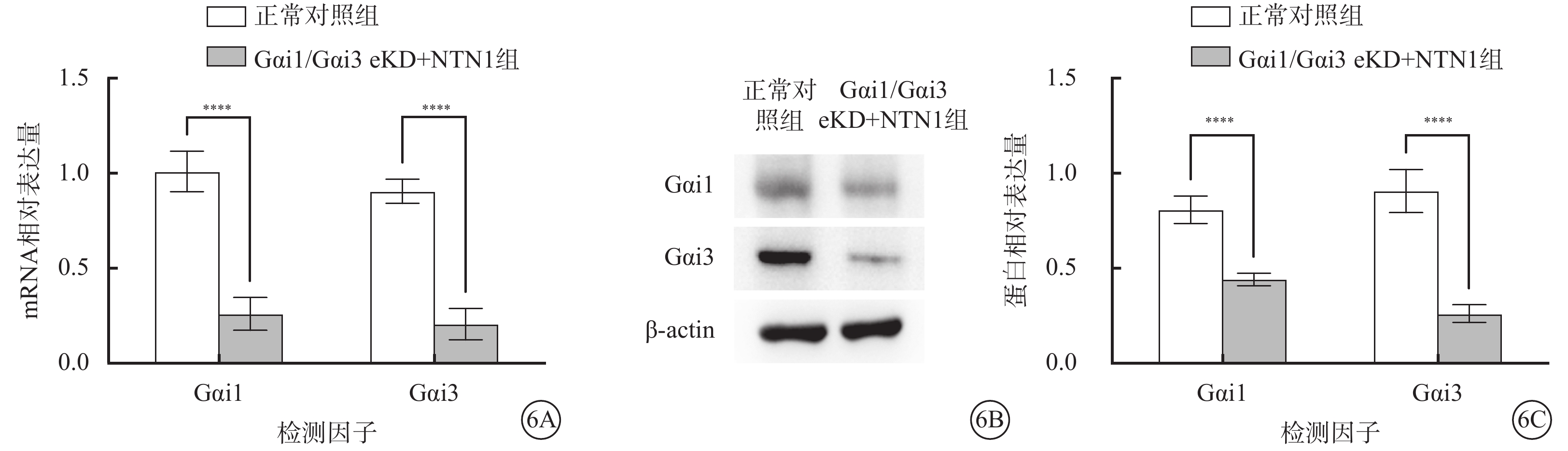
qPCR、Western blot檢測結果顯示,與正常對照組比較,Gαi1/Gαi3 eKD+NTN1組小鼠視網膜組織中Gαi1、Gαi3 mRNA和蛋白相對表達量顯著下降,差異有統計學意義(t=10.920、13.460、9.219、10.500,P<0.000 1)(圖6)。
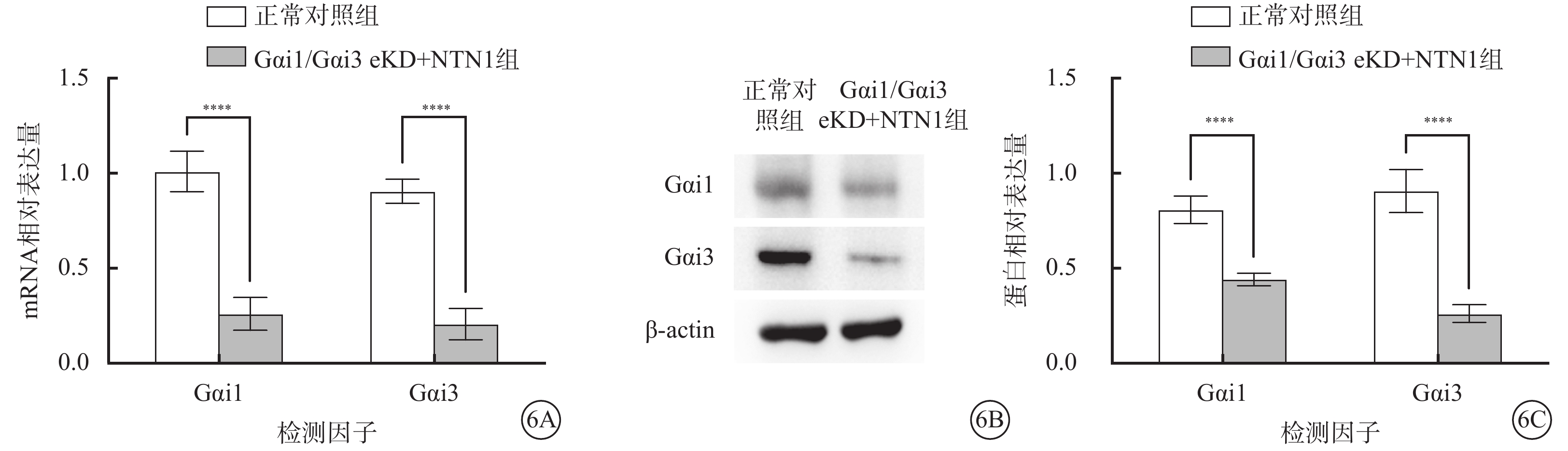 圖6
正常對照組、Gαi1/Gαi3 eKD組小鼠視網膜組織中Gαi1、Gαi3 mRNA及蛋白相對表達量比較(n=3) 6A示Gαi1、Gαi3 mRNA相對表達量;6B示Gαi1、Gαi3蛋白表達電泳圖;6C示Gαi1、Gαi3蛋白相對表達量,****P<0.000 1 Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;AAV:腺相關病毒;TIE1:人酪氨酸蛋白激酶受體tie-1;正常對照組:不做任何處理;Gαi1/Gαi3 eKD+NTN1組:玻璃體腔注射AAV5-TIE1-Gαi1/Gαi3,1周后玻璃體腔注射 50 ng/ml NTN1 2 μl
圖6
正常對照組、Gαi1/Gαi3 eKD組小鼠視網膜組織中Gαi1、Gαi3 mRNA及蛋白相對表達量比較(n=3) 6A示Gαi1、Gαi3 mRNA相對表達量;6B示Gαi1、Gαi3蛋白表達電泳圖;6C示Gαi1、Gαi3蛋白相對表達量,****P<0.000 1 Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;AAV:腺相關病毒;TIE1:人酪氨酸蛋白激酶受體tie-1;正常對照組:不做任何處理;Gαi1/Gαi3 eKD+NTN1組:玻璃體腔注射AAV5-TIE1-Gαi1/Gαi3,1周后玻璃體腔注射 50 ng/ml NTN1 2 μl
2.5 敲低Gαi1/Gαi3基因抑制NTN1誘導的視網膜新生血管形成
免疫熒光染色結果顯示,與正常對照組相比,NTN1組視網膜新生血管形成面積顯著增加,Gαi1/Gαi3 eKD+NTN1組視網膜新生血管形成面積顯著減小,差異有統計學意義(F=24.010,P<0.000 1)(圖7)。
 圖7
正常對照組、NTN1組、Gαi1/Gαi3 eKD+NTN1組視網膜新生血管面積比較(n=3) 7A~7C分別示正常對照組、NTN1組、Gαi1/Gαi3 eKD+NTN1組視網膜熒光顯微鏡像(IB4染色,標尺:200 μm),下圖為上圖黃框區域放大像,與正常對照組相比,NTN1組視網膜新生血管形成面積顯著增加,Gαi1/Gαi3 eKD+NTN1組視網膜新生血管形成面積顯著減小;7D示三組視網膜新生血管面積比較,*P<0.05,**P<0.01 NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;AAV:腺相關病毒;正常對照組:不做任何處理;NTN1組:玻璃體腔注射2 μl 50 ng/ml NTN1;Gαi1/Gαi3 eKD組+NTN1組:玻璃體腔注射AAV5-TIE1-Gαi1/Gαi3 shRNA 2 μl,1周后玻璃體腔注射 50 ng/ml NTN1 2 μl;IB4:同工凝集素B4
圖7
正常對照組、NTN1組、Gαi1/Gαi3 eKD+NTN1組視網膜新生血管面積比較(n=3) 7A~7C分別示正常對照組、NTN1組、Gαi1/Gαi3 eKD+NTN1組視網膜熒光顯微鏡像(IB4染色,標尺:200 μm),下圖為上圖黃框區域放大像,與正常對照組相比,NTN1組視網膜新生血管形成面積顯著增加,Gαi1/Gαi3 eKD+NTN1組視網膜新生血管形成面積顯著減小;7D示三組視網膜新生血管面積比較,*P<0.05,**P<0.01 NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;AAV:腺相關病毒;正常對照組:不做任何處理;NTN1組:玻璃體腔注射2 μl 50 ng/ml NTN1;Gαi1/Gαi3 eKD組+NTN1組:玻璃體腔注射AAV5-TIE1-Gαi1/Gαi3 shRNA 2 μl,1周后玻璃體腔注射 50 ng/ml NTN1 2 μl;IB4:同工凝集素B4
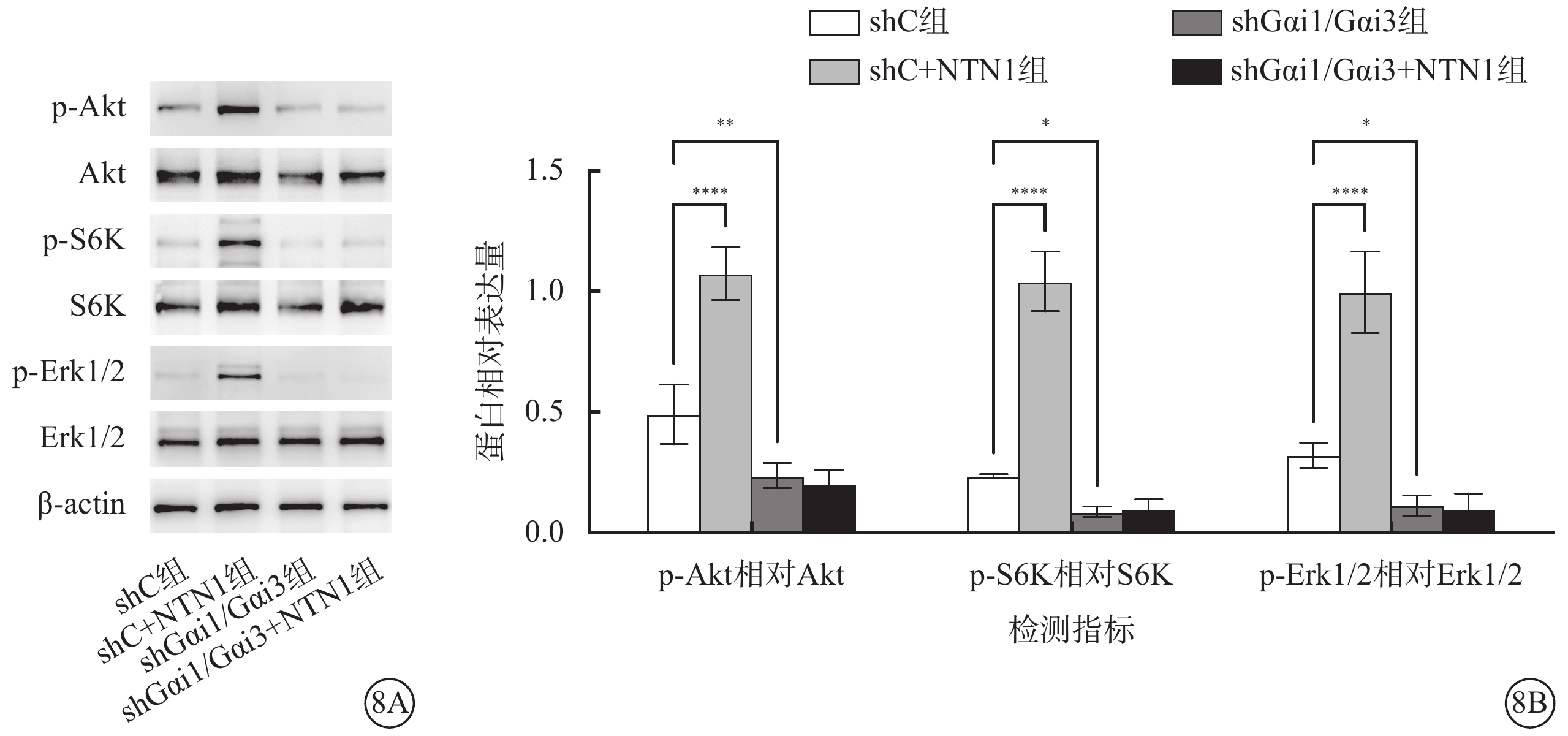
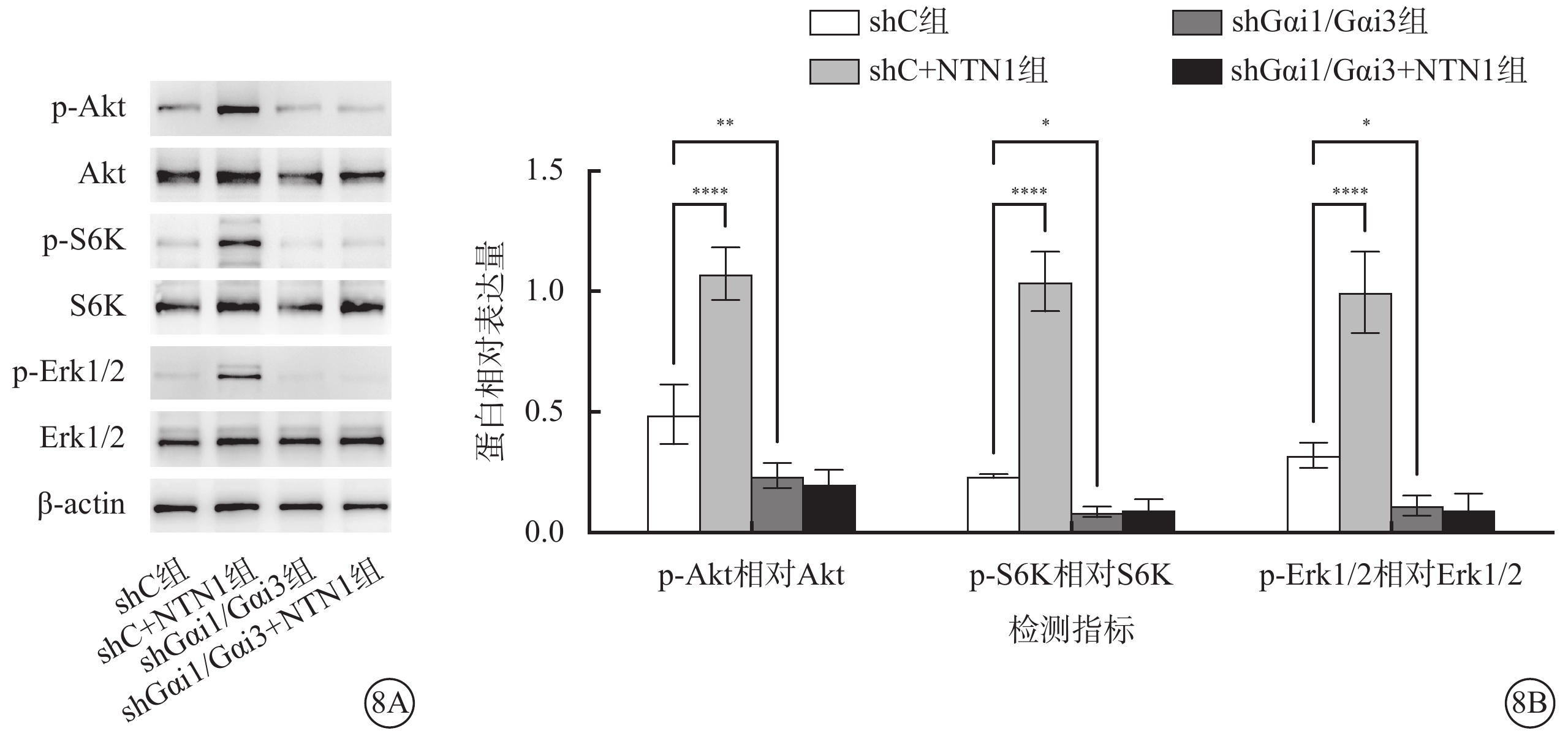 圖8
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組HUVEC中p-Akt、Akt、p-S6K、S6K、p-Erk1/2、Erk1/2蛋白表達情況(n=3) 8A示電泳圖;8B示各組HUVEC中p-Akt相對Akt、p-S6K相對S6K、p-Erk1/2相對Erk1/2蛋白相對表達量比較,*P<0.05,**P<0.01,****P<0.000 1 Akt:蛋白激酶B;p-Akt:磷酸化Akt;S6K:核糖體蛋白S6激酶;p-S6K:磷酸化S6K;Erk1/2:細胞外調節蛋白激酶;磷酸化Erk1/2;NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;shC組:加入空白慢病毒誘導;shC+NTN1組:加入空白慢病毒,用50 ng/ml NTN1誘導;shGαi1/Gαi3組:加入Gαi1/Gαi3 shRNA重組慢病毒誘導;shGαi1/Gαi3+NTN1組:加入Gαi1/Gαi3 shRNA重組慢病毒,用50 ng/ml NTN1誘導
圖8
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組HUVEC中p-Akt、Akt、p-S6K、S6K、p-Erk1/2、Erk1/2蛋白表達情況(n=3) 8A示電泳圖;8B示各組HUVEC中p-Akt相對Akt、p-S6K相對S6K、p-Erk1/2相對Erk1/2蛋白相對表達量比較,*P<0.05,**P<0.01,****P<0.000 1 Akt:蛋白激酶B;p-Akt:磷酸化Akt;S6K:核糖體蛋白S6激酶;p-S6K:磷酸化S6K;Erk1/2:細胞外調節蛋白激酶;磷酸化Erk1/2;NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;shC組:加入空白慢病毒誘導;shC+NTN1組:加入空白慢病毒,用50 ng/ml NTN1誘導;shGαi1/Gαi3組:加入Gαi1/Gαi3 shRNA重組慢病毒誘導;shGαi1/Gαi3+NTN1組:加入Gαi1/Gαi3 shRNA重組慢病毒,用50 ng/ml NTN1誘導
2.6 敲低Gαi1/Gαi3基因抑制NTN1誘導的HUVEC中Akt-mTOR和Erk的信號通路激活
Western blot檢測結果顯示,與shC組相比,shC+NTN1組p-Akt相對Akt、p-S6K相對S6K、p-Erk1/2相對Erk1/2蛋白相對表達量顯著增加,shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組p-Akt相對Akt、p-S6K相對S6K、p-Erk1/2相對Erk1/2蛋白相對表達量顯著減少,差異有統計學意義(F=78.610、144.400、77.010,P<0.000 1)(圖8)。
3 討論
DR作為一種典型的微血管并發癥,其發病機制涉及多種生長因子和信號通路的復雜交互。Gαi1/Gαi3介導的VEGF信號通路的活化是VEGF促視網膜血管新生的核心機制,為病理性血管新生提供了新的治療靶點。通過敲低Gαi1和Gαi3基因可顯著抑制堿燒傷誘導的小鼠角膜新生血管形成,并減少了氧誘導視網膜病變(OIR)小鼠的視網膜新生血管形成[11-12]。磷酸烯醇丙酮酸羧激酶1作為催化糖異生的關鍵限速酶,通過促進Gαi3蛋白表達增加和Akt-mTOR信號通路激活,在體內外發揮促新生血管作用[13]。此外,Gαi1和Gαi3還是R-脊椎蛋白3誘導的Akt-mTOR激活和新生血管生成所需的關鍵信號蛋白[14]。
研究表明,NTN1具有雙向調節血管活性的作用。在大鼠角膜堿燒傷模型中,角膜上皮中Ntn1 mRNA的表達水平降低,抑制角膜新生血管形成[15]。NTN1也可以作為促血管生成因子,正向調控內皮細胞遷移,促進DR等血管新生相關疾病的進程[16-17]。研究報道,在OIR小鼠模型中,視網膜組織Ntn1基因表達水平顯著升高,抑制Ntn1基因表達后視網膜新生血管減少[18]。在大鼠DR模型中,靜脈注射100 ng/ml NTN1加速視網膜新生血管的形成[6]。研究報道,大鼠DR模型的視網膜組織中Ntn1 mRNA及蛋白表達均明顯增加,這提示其可能促進內皮細胞的增殖和分化,進而誘導視網膜新生血管形成[19]。NTN1在不同模型及微環境下的差異性表達和作用,反映其可能在血管生成調控中具有復雜性和多功能性。
本研究發現,12周糖尿病模型小鼠視網膜中Ntn1、Gαi1和Gαi3基因的mRNA和蛋白表達均顯著增加,這提示DR過程中持續異常分泌的NTN1蛋白在血管生成過程中的重要作用。外源性添加低濃度NTN1(50 ng/ml)蛋白刺激后,HUVEC增殖、遷移與細胞成管能力增強,小鼠視網膜新生血管增多,這與以往研究結果一致[15-16]。為了進一步明確Ntn1基因促進新生血管的具體機制,我們在HUVEC中轉染shRNA Gαi1和shRNA Gαi3沉默Gαi1/Gαi3,隨后發現NTN1誘導的促血管生成作用被抑制,這表明Gαi1和Gαi3介導了NTN1誘導的體內外促血管生成作用。既往研究報道,NTN1誘導下游Akt-mTOR信號通路和Erk激活[20-22]。本研究證實,NTN1可以在HUVEC中誘導下游Akt-mTOR信號通路和Erk激活,而這種激活作用在Gαi1和Gαi3基因敲除后幾乎被完全抑制。據此我們推測,Gαi1和Gαi3蛋白是介導NTN1誘導的下游Akt-mTOR與Erk-MAPK信號通路激活和血管生成的信號蛋白。這進一步表明,NTN1-Gαi1/Gαi3信號轉導增加可能與DR異常血管生成有關。
在DR的進程中,NTN1可能作為一種促血管生成因子,誘導新生血管的形成。而這種作用可能與NTN1與細胞膜上特定受體,如結腸癌缺失蛋白、不協調5同系物、黑色素瘤細胞黏附分子等的結合有關,從而激活下游的信號通路[23-24]。Gαi1和Gαi3是包括VEGF受體及白細胞介素-4受體內吞和內化的關鍵蛋白,而接頭蛋白Grb2關聯結合蛋白1招募往往在Gαi1和Gαi3蛋白誘導的下游信號轉導中發揮重要作用[11, 14, 25]。這說明在DR中,Gαi1和Gαi3基因可能通過類似的機制參與NTN1誘導的血管生成過程。
DR被認為是一種微血管疾病,以選擇性視網膜周細胞丟失、基底膜增厚、毛細血管閉塞等毛細血管內皮損害、血視網膜屏障功能受損、微血管滲漏、廣泛的視網膜缺血缺氧促發VEGF等生長因子表達增加,最終導致視網膜新生血管形成。目前臨床使用的VEGF單克隆抗體在一定程度上能夠阻止血管滲漏和新生血管的形成,但由于DR病理性血管生成過程涉及其他生長因子和各種炎癥細胞因子的參與,部分患者對VEGF單克隆抗體敏感性有限且治療后易復發[26]。因此確定控制DR進展的新的治療靶點是極其重要的。本研究結果表明,NTN1-Gαi1/Gαi3信號轉導可能是DR異常血管生成的重要原因之一,這種級聯反應提示DR分子治療的新靶點。然而目前研究存在一定局限性。首先,目前結果側重于體外細胞實驗,尚無法全方位立體模擬錯綜復雜的體內環境。其次,在研究方法上,本研究僅通過敲低Gαi1和Gαi3基因以觀察其對血管生成的影響,雖揭示Gαi1和Gαi3基因在介導NTN1促血管生成效應中的作用,但對于這一信號通路更深入的分子機制、調控細節及其與其他信號通路的交互作用,并未進行深入探究。為實現以干預NTN1-Gαi1/Gαi3信號為靶點的治療方法,未來需進一步探索體內外實驗,通過基因敲低、敲除、過表達、顯性陰性突變、挽救Ntn1、Gαi1、Gαi3基因等多種方法,正負向調節NTN1、Gαi1、Gαi3蛋白表達或活化水平,探索影響其血管生成的具體機制。結合蛋白和轉錄組學技術,全面分析NTN1刺激下細胞內的基因轉錄和蛋白質表達的變化。在未來需重點關注NTN1-Gαi1/Gαi3參與血管新生調控的具體機制,為視網膜新生血管性疾病的治療提供新的思路和策略。
糖尿病視網膜病變(DR)是糖尿病的重要并發癥,尤其在工作年齡段人群中是視力損害的主要原因[1]。高血糖引發的神經炎癥和微血管損傷破壞血視網膜屏障,促進病理性血管生成,是DR發展的關鍵環節[2]。軸突導向因子-1(NTN1)是一種在神經發育中促進軸突生長的蛋白,也被發現參與內皮細胞活化和血管生成,具有濃度依賴性的雙相效應。低濃度的NTN1可激活內皮細胞,促進血管生成,但具體機制尚不完全清楚[3-5]。G蛋白抑制性α亞單位(Gαi1/Gαi3)在多種生長因子信號轉導中發揮作用,通過活化蛋白激酶B(Akt)-哺乳動物雷帕霉素靶蛋白(mTOR)和細胞外信號調節激酶(Erk)-絲裂原活化蛋白激酶(MAPK)等通路,影響細胞增殖和遷移[6-11]。Gαi1/Gαi3介導的血管內皮生長因子(VEGF)信號通路對視網膜血管新生至關重要,提示其在病理性血管生成中作為潛在治療靶點的重要性[11-12]。本研究旨在探索Gαi1/Gαi3在NTN1信號轉導和血管生成中的作用及其分子機制,通過建立糖尿病小鼠模型,深入了解血管生成的分子基礎,以期為DR的早期干預和治療策略提供新的理論依據和潛在靶標。
1 材料和方法
1.1 實驗動物及主要材料、儀器
雄性C57BL/6J小鼠55只,其中6~8周齡20只,體重20~25 g;2周齡35只,體重約10 g;均為無特定病原體級,由南京君科生物工程有限公司提供。所有小鼠飼養于標準(12 h明/暗循環)清潔級環境中。飼養環境及實驗操作均符合國家科學技術委員會《實驗動物管理條例》規定,并獲得南京醫科大學實驗動物倫理委員會許可[許可證號:SYXK(蘇)2018-0020]。
人臍靜脈內皮細胞(HUVEC)細胞株由本實驗室自行保存。攜綠色熒光蛋白(GFP)Gαi1/Gαi3 shRNA重組腺相關病毒(AAV5)載體(AAV5-TIE1-Gαi1/Gαi3 shRNA)、僅攜帶GFP的空白AAV5載體、Gαi1/Gαi3 shRNA重組慢病毒、僅攜帶GFP的空白慢病毒(上海吉凱基因醫學科技股份有限公司)。無水葡萄糖(德國Biofroxx公司);內皮細胞培養基(ECM培養基)(美國Sciencell公司);4',6-二脒基-2-苯基吲哚(DAPI)、嘌呤霉素、辣根過氧化物酶(HRP)二抗(上海碧云天生物技術有限公司);鏈脲佐菌素(STZ)、磷酸鹽緩沖液(PBS)(北京蘭杰柯科技有限公司);20倍洗膜緩沖液(TBST)(北京索萊寶科技有限公司);核糖體蛋白S6激酶(S6K)抗體、磷酸化S6K(p-S6K)抗體(美國Cell Signaling Technology公司);Erk1/2抗體、磷酸化Erk1/2(p-Erk1/2)抗體、Akt抗體、磷酸化Akt(p-Akt)抗體(杭州華安生物技術有限公司);NTN1(美國Enzo公司);β-肌動蛋白(actin)抗體(成都正能生物技術有限責任公司)。EdU-555細胞增殖檢測試劑盒(上海碧云天生物技術有限公司);Transwell共培養板、高蛋白濃度基質膠(美國Corning公司);同工凝集素B4(IB4)(美國Sigma公司);細胞/組織總RNA提取試劑盒、逆轉錄預混液、通用型高靈敏度染料法定量聚合酶鏈反應(qPCR)檢測試劑盒(南京諾唯贊生物科技有限公司);PIKOReal 96實時熒光qPCR(qRT-PCR)儀(美國Thermo公司);倒置熒光顯微鏡(日本Olympus公司)。
1.2 實驗動物分組和糖尿病模型建立
采用隨機數字表法將20只6~8周齡小鼠分為正常對照組、糖尿病組,每組各10只。糖尿病組小鼠空腹饑餓12 h(期間可自由進水)后,按10 mg/kg劑量予以小鼠腹腔注射STZ。1周后測空腹血糖≥16.7 mmol/L為造模成功,繼續高糖、高脂飼養,期間定期監測血糖,使血糖維持在成模范圍內直至實驗結束。
將35只2周齡小鼠隨機分為正常對照組、玻璃體腔注射NTN1組(NTN1組)、視網膜內皮細胞Gαi1/Gαi3特異性敲低+玻璃體腔注射NTN1組(Gαi1/Gαi3 eKD+NTN1組),其中正常對照組、NTN1組每組各15只,Gαi1/Gαi3 eKD+NTN1組5只。NTN1組于小鼠2周齡時玻璃體腔注射50 ng/ml NTN1 2 μl。Gαi1/Gαi3 eKD+NTN1組于小鼠2周齡時玻璃體腔注射AAV5-TIE1-Gαi1/Gαi3 shRNA 2 μl,1周后玻璃體腔注射50 ng/ml NTN1 2 μl。玻璃體腔注射方式:腹腔注射氯胺酮和甲苯噻嗪麻醉小鼠,散瞳后微量注射器抽取AAV5-TIE1-Gαi1/Gαi3 shRNA 2 μl于小鼠眼鼻上方角膜緣后約1 mm處垂直穿刺進入玻璃體腔,向眼球中心方向進針,將藥液緩慢注入玻璃體腔,眼內停留約30 s后拔出針頭,涂左氧氟沙星眼膏。
1.3 細胞培養、轉染分組
細胞培養。將HUVEC細胞株置于含5%胎牛血清和1%雙抗的ECM培養基中常規培養。取對數生長期細胞用于實驗。
將HUVEC分為陰性對照慢病毒組(shC組)、陰性對照慢病毒+NTN1處理組(shC+NTN1組)、Gαi1/Gαi3敲低組(shGαi1/Gαi3組)、Gαi1/Gαi3敲低+NTN1處理組(shGαi1/Gαi3+NTN1組)。shC組細胞加入空白慢病毒;shGαi1/Gαi3組細胞加入Gαi1/Gαi3 shRNA重組慢病毒誘導。轉染20 h后換為正常完全培養基,然后正常傳代,穩定細胞株通過含有嘌呤霉素的完全培養基篩選1周。shC+NTN1組細胞及shGαi1/Gαi3+NTN1組細胞在嘌呤霉素篩選后使用NTN1(50 ng/ml)預處理24 h后進行后續實驗。
1.4 IB4染色法檢測Gαi1/Gαi3敲低對NTN1誘導的視網膜新生血管的影響
正常對照組、NTN1組、Gαi1/Gαi3 eKD+NTN1組小鼠4周齡時腹腔注射氯胺酮和甲苯噻嗪麻醉,將彎鑷沿顳側眶壁進入呈先垂直再平行向鼻側走形,至眼球下方夾住視神經,摘取完整眼球,置于4%多聚甲醛中固定40 min,轉移至含PBS的10 cm細胞培養皿中。用眼科剪和眼科鑷去除眼球周圍組織,沿角膜緣剪開眼球,去除眼前節,移除晶狀體和玻璃體,分離視網膜,切成4瓣。固定、通透封閉,IB4染料(1∶50)4℃孵育過夜,PBS洗滌3次,10 min/次。在每個視網膜瓣的中間區域隨機選取視野,于熒光顯微鏡下按序拍攝。
1.5 EdU實驗觀察細胞增殖能力
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞以8×104個/孔的密度接種于24孔板中,每孔加入10 μmol/L EdU工作液,于37℃孵育箱中培養2 h,固定及洗滌后,DAPI避光染色10 min,PBS洗滌3次,5 min/次。熒光顯微鏡觀察并計數。細胞核呈藍色熒光。實驗重復3次。
1.6 Transwell實驗觀察細胞的遷移能力
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞以8×104個/孔的密度接種于Transwell小室上室內,無血清培養基培養;下室加入含5%胎牛血清的ECM培養基。細胞培養板置于孵箱中培育24 h,多聚甲醛固定下室表面細胞15 min,棉簽擦去上室未向下室遷移的細胞,常規結晶紫染色3 min。光學顯微鏡下觀察拍照。每組隨機選取6~8個視野以計數小室底膜上、下室側附著的細胞,即為發生遷移的細胞數。每組各設3個復孔,重復3次。
1.7 Matrigel實驗檢測細胞管腔形成能力
將4℃高蛋白濃度基質膠40 μl緩慢加入24孔板中,于37℃孵育箱中培養30 min。shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞以8×104個/孔的密度接種于24孔板中孵育,6 h后觀察管腔形成情況。計數相同視野面積下的管腔形成數量。實驗重復3次。
1.8 qPCR檢測小鼠視網膜及HUVEC中Ntn1、Gαi1、Gαi3、甘油醛-3-磷酸脫氫酶(Gapdh)基因的mRNA相對表達量
過量麻醉處死小鼠,摘除眼球剝離視網膜,RNA提取試劑盒提取總RNA;HUVEC傳代后鋪于6孔板,細胞匯合度為90%~100%時RNA提取試劑盒提取總RNA。反轉錄試劑逆轉錄成cDNA,按相應基因配置SYBR Green qPCR擴增反應體系進行qPCR檢測。應用Express 3.0軟件設計引物序列(表1)。置于qRT-PCR儀中進行擴增并輸出循環閾值(Ct值),以GAPDH為內參照,依照公式2-ΔΔCt計算mRNA相對表達量。
1.9 蛋白質免疫印跡法(Western blot)檢測小鼠視網膜組織及HUVEC中NTN1、Gαi1、Gαi3、Akt、p-Akt、S6K、p-S6K、Erk1/2、p-Erk1/2蛋白表達
收集各組細胞總蛋白,調整蛋白濃度。每個樣孔加入10 μl待測樣品,十二烷基硫酸鈉聚丙烯酰胺凝膠電泳中上樣電泳;140 V恒壓50 min,400 mA轉膜40 min。5%脫脂牛奶封閉2 h,1∶1 000一抗4℃孵育過夜(β-actin作為陽性對照),TBST洗膜10 min,重復3次;加入辣根過氧化物偶聯的二抗,室溫孵育1 h,TBST洗膜10 min,重復3次;加入化學發光劑,暗室曝光。Image J軟件分析蛋白條帶的灰度值,目的蛋白相對表達量=目的蛋白條帶灰度值/內參照β-actin蛋白條帶灰度值,磷酸化蛋白相對總蛋白相對表達量=磷酸化蛋白條帶灰度值/總蛋白條帶灰度值。
1.10 統計學方法
采用SPSS20.0軟件進行統計學分析。計量數據以均數±標準差(x±s)表示。兩組間比較采用t檢驗;三組間比較采用單因素方差分析;多因素比較采用事后Dunnett檢驗。采用Graph-prism軟件對所獲得數據進行圖表整理。P<0.05為差異有統計學意義。
2 結果
2.1 DR模型小鼠視網膜組織中Ntn1、Gαi1、Gαi3 mRNA和蛋白表達升高
與正常對照組比較,糖尿病組小鼠視網膜內Ntn1、Gαi1、Gαi3 mRNA和蛋白相對表達量顯著增加,差異有統計學意義(t=11.800、9.298、10.620、7.503、3.432、8.037,P<0.000 1)(圖1)。
圖1
正常對照組、糖尿病組小鼠視網膜中Ntn1、Gαi1、Gαi3 mRNA及蛋白相對表達量比較(n=3) 1A示Ntn1、Gαi1、Gαi3 mRNA相對表達量,****P<0.000 1;1B示NTN1、Gαi1、Gαi3蛋白表達電泳圖;1C示NTN1、Gαi1、Gαi3蛋白相對表達量比較,**P<0.01,***P<0.001 NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;正常對照組:不做任何處理;糖尿病組:腹腔注射鏈脲佐菌素10 mg/kg
2.2 Gαi1/Gαi3 shRNA重組慢病毒誘導后HUVEC中 Gαi1、Gαi3 mRNA和蛋白相對表達量受到抑制
與shC組比較,shGαi1/Gαi3組HUVEC中Gαi1、Gαi3 mRNA和蛋白相對表達量顯著降低,差異有統計學意義(t=16.310、16.300、13.600、9.068,P<0.000 1)(圖2)。
圖2
shC組、shGαi1/Gαi3組HUVEC中Gαi1、Gαi3 mRNA及蛋白相對表達量比較(n=3) 2A示Gαi1、Gαi3 mRNA相對表達量結果,****P<0.000 1;2B示Gαi1、Gαi3蛋白表達電泳圖;2C示中Gαi1、Gαi3蛋白相對表達量,***P<0.001, ****P<0.000 1 HUVEC:人臍靜脈內皮細胞;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;actin:肌動蛋白;shC組:空白慢病毒誘導;shGαi1/Gαi3組:加入Gαi1/Gαi3 shRNA重組慢病毒誘導
2.3 敲低Gαi1/Gαi3基因抑制NTN1誘導的HUVEC增殖、遷移與管腔形成能力
與shC組比較,shC+NTN1組HUVEC增殖率、遷移數量、管腔形成數量均顯著增加,shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組HUVEC增殖率、遷移數量、管腔形成數量均顯著減少,差異均有統計學意義(F=62.750、49.830、54.900,P<0.000 1)(圖3~5)。
圖3
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組HUVEC增殖率比較(n=3) 3A~3D分別示shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組熒光顯微鏡像(DAPI染色,標尺:50 μm),與shC組比較,shC+NTN1組細胞增殖率顯著增加,shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞增殖率無明顯變化;3E示四組細胞增殖率比較,***P<0.001,****P<0.000 1 NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;shC組:加入空白慢病毒誘導;shC+NTN1組:加入空白慢病毒,用50 ng/ml NTN1誘導;shGαi1/Gαi3組:加入Gαi1/Gαi3 shRNA重組慢病毒誘導;shGαi1/Gαi3+NTN1組:加入Gαi1/Gαi3 shRNA重組慢病毒,用50 ng/ml NTN1誘導;DAPI:4',6-二脒基-2-苯基吲哚
圖4
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組HUVEC遷移數量比較(n=3) 4A~4D分別示shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組光學顯微鏡像(結晶紫染色,標尺:50 μm),與shC組比較,shC+NTN1組細胞遷移數量顯著增加,shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞遷移數量無明顯變化;4E示四組細胞遷移數量比較,**P<0.01,****P<0.000 1 NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;shC組:加入空白慢病毒誘導;shC+NTN1組:加入空白慢病毒,用50 ng/ml NTN1誘導;shGαi1/Gαi3組:加入Gαi1/Gαi3 shRNA重組慢病毒誘導;shGαi1/Gαi3+NTN1組:加入Gαi1/Gαi3 shRNA重組慢病毒,用50 ng/ml NTN1誘導
圖5
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組HUVEC管腔形成情況比較(n=3) 5A~5D分別示shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞倒置相差顯微鏡像(標尺:50 μm),與shC組比較,shC+NTN1組細胞管腔形成數量顯著增加,shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組細胞管腔形成數量顯著減少;5E示四組細胞管腔形成數目比較,*P<0.05 ,****P<0.000 1 NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;shC組:加入空白慢病毒誘導;shC+NTN1組:加入空白慢病毒,用50 ng/ml NTN1誘導;shGαi1/Gαi3組:加入Gαi1/Gαi3 shRNA重組慢病毒誘導;shGαi1/Gαi3+NTN1組:加入Gαi1/Gαi3 shRNA重組慢病毒,用50 ng/ml NTN1誘導
2.4 敲低Gαi1/Gαi3基因檢測結果抑制NTN1誘導的Gαi1、Gαi3 mRNA和蛋白表達
qPCR、Western blot檢測結果顯示,與正常對照組比較,Gαi1/Gαi3 eKD+NTN1組小鼠視網膜組織中Gαi1、Gαi3 mRNA和蛋白相對表達量顯著下降,差異有統計學意義(t=10.920、13.460、9.219、10.500,P<0.000 1)(圖6)。
圖6
正常對照組、Gαi1/Gαi3 eKD組小鼠視網膜組織中Gαi1、Gαi3 mRNA及蛋白相對表達量比較(n=3) 6A示Gαi1、Gαi3 mRNA相對表達量;6B示Gαi1、Gαi3蛋白表達電泳圖;6C示Gαi1、Gαi3蛋白相對表達量,****P<0.000 1 Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;AAV:腺相關病毒;TIE1:人酪氨酸蛋白激酶受體tie-1;正常對照組:不做任何處理;Gαi1/Gαi3 eKD+NTN1組:玻璃體腔注射AAV5-TIE1-Gαi1/Gαi3,1周后玻璃體腔注射 50 ng/ml NTN1 2 μl
2.5 敲低Gαi1/Gαi3基因抑制NTN1誘導的視網膜新生血管形成
免疫熒光染色結果顯示,與正常對照組相比,NTN1組視網膜新生血管形成面積顯著增加,Gαi1/Gαi3 eKD+NTN1組視網膜新生血管形成面積顯著減小,差異有統計學意義(F=24.010,P<0.000 1)(圖7)。
圖7
正常對照組、NTN1組、Gαi1/Gαi3 eKD+NTN1組視網膜新生血管面積比較(n=3) 7A~7C分別示正常對照組、NTN1組、Gαi1/Gαi3 eKD+NTN1組視網膜熒光顯微鏡像(IB4染色,標尺:200 μm),下圖為上圖黃框區域放大像,與正常對照組相比,NTN1組視網膜新生血管形成面積顯著增加,Gαi1/Gαi3 eKD+NTN1組視網膜新生血管形成面積顯著減小;7D示三組視網膜新生血管面積比較,*P<0.05,**P<0.01 NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;AAV:腺相關病毒;正常對照組:不做任何處理;NTN1組:玻璃體腔注射2 μl 50 ng/ml NTN1;Gαi1/Gαi3 eKD組+NTN1組:玻璃體腔注射AAV5-TIE1-Gαi1/Gαi3 shRNA 2 μl,1周后玻璃體腔注射 50 ng/ml NTN1 2 μl;IB4:同工凝集素B4
圖8
shC組、shC+NTN1組、shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組HUVEC中p-Akt、Akt、p-S6K、S6K、p-Erk1/2、Erk1/2蛋白表達情況(n=3) 8A示電泳圖;8B示各組HUVEC中p-Akt相對Akt、p-S6K相對S6K、p-Erk1/2相對Erk1/2蛋白相對表達量比較,*P<0.05,**P<0.01,****P<0.000 1 Akt:蛋白激酶B;p-Akt:磷酸化Akt;S6K:核糖體蛋白S6激酶;p-S6K:磷酸化S6K;Erk1/2:細胞外調節蛋白激酶;磷酸化Erk1/2;NTN1:軸突導向因子1;Gαi1:G蛋白抑制性α亞單位1;Gαi3:G蛋白抑制性α亞單位3;shC組:加入空白慢病毒誘導;shC+NTN1組:加入空白慢病毒,用50 ng/ml NTN1誘導;shGαi1/Gαi3組:加入Gαi1/Gαi3 shRNA重組慢病毒誘導;shGαi1/Gαi3+NTN1組:加入Gαi1/Gαi3 shRNA重組慢病毒,用50 ng/ml NTN1誘導
2.6 敲低Gαi1/Gαi3基因抑制NTN1誘導的HUVEC中Akt-mTOR和Erk的信號通路激活
Western blot檢測結果顯示,與shC組相比,shC+NTN1組p-Akt相對Akt、p-S6K相對S6K、p-Erk1/2相對Erk1/2蛋白相對表達量顯著增加,shGαi1/Gαi3組、shGαi1/Gαi3+NTN1組p-Akt相對Akt、p-S6K相對S6K、p-Erk1/2相對Erk1/2蛋白相對表達量顯著減少,差異有統計學意義(F=78.610、144.400、77.010,P<0.000 1)(圖8)。
3 討論
DR作為一種典型的微血管并發癥,其發病機制涉及多種生長因子和信號通路的復雜交互。Gαi1/Gαi3介導的VEGF信號通路的活化是VEGF促視網膜血管新生的核心機制,為病理性血管新生提供了新的治療靶點。通過敲低Gαi1和Gαi3基因可顯著抑制堿燒傷誘導的小鼠角膜新生血管形成,并減少了氧誘導視網膜病變(OIR)小鼠的視網膜新生血管形成[11-12]。磷酸烯醇丙酮酸羧激酶1作為催化糖異生的關鍵限速酶,通過促進Gαi3蛋白表達增加和Akt-mTOR信號通路激活,在體內外發揮促新生血管作用[13]。此外,Gαi1和Gαi3還是R-脊椎蛋白3誘導的Akt-mTOR激活和新生血管生成所需的關鍵信號蛋白[14]。
研究表明,NTN1具有雙向調節血管活性的作用。在大鼠角膜堿燒傷模型中,角膜上皮中Ntn1 mRNA的表達水平降低,抑制角膜新生血管形成[15]。NTN1也可以作為促血管生成因子,正向調控內皮細胞遷移,促進DR等血管新生相關疾病的進程[16-17]。研究報道,在OIR小鼠模型中,視網膜組織Ntn1基因表達水平顯著升高,抑制Ntn1基因表達后視網膜新生血管減少[18]。在大鼠DR模型中,靜脈注射100 ng/ml NTN1加速視網膜新生血管的形成[6]。研究報道,大鼠DR模型的視網膜組織中Ntn1 mRNA及蛋白表達均明顯增加,這提示其可能促進內皮細胞的增殖和分化,進而誘導視網膜新生血管形成[19]。NTN1在不同模型及微環境下的差異性表達和作用,反映其可能在血管生成調控中具有復雜性和多功能性。
本研究發現,12周糖尿病模型小鼠視網膜中Ntn1、Gαi1和Gαi3基因的mRNA和蛋白表達均顯著增加,這提示DR過程中持續異常分泌的NTN1蛋白在血管生成過程中的重要作用。外源性添加低濃度NTN1(50 ng/ml)蛋白刺激后,HUVEC增殖、遷移與細胞成管能力增強,小鼠視網膜新生血管增多,這與以往研究結果一致[15-16]。為了進一步明確Ntn1基因促進新生血管的具體機制,我們在HUVEC中轉染shRNA Gαi1和shRNA Gαi3沉默Gαi1/Gαi3,隨后發現NTN1誘導的促血管生成作用被抑制,這表明Gαi1和Gαi3介導了NTN1誘導的體內外促血管生成作用。既往研究報道,NTN1誘導下游Akt-mTOR信號通路和Erk激活[20-22]。本研究證實,NTN1可以在HUVEC中誘導下游Akt-mTOR信號通路和Erk激活,而這種激活作用在Gαi1和Gαi3基因敲除后幾乎被完全抑制。據此我們推測,Gαi1和Gαi3蛋白是介導NTN1誘導的下游Akt-mTOR與Erk-MAPK信號通路激活和血管生成的信號蛋白。這進一步表明,NTN1-Gαi1/Gαi3信號轉導增加可能與DR異常血管生成有關。
在DR的進程中,NTN1可能作為一種促血管生成因子,誘導新生血管的形成。而這種作用可能與NTN1與細胞膜上特定受體,如結腸癌缺失蛋白、不協調5同系物、黑色素瘤細胞黏附分子等的結合有關,從而激活下游的信號通路[23-24]。Gαi1和Gαi3是包括VEGF受體及白細胞介素-4受體內吞和內化的關鍵蛋白,而接頭蛋白Grb2關聯結合蛋白1招募往往在Gαi1和Gαi3蛋白誘導的下游信號轉導中發揮重要作用[11, 14, 25]。這說明在DR中,Gαi1和Gαi3基因可能通過類似的機制參與NTN1誘導的血管生成過程。
DR被認為是一種微血管疾病,以選擇性視網膜周細胞丟失、基底膜增厚、毛細血管閉塞等毛細血管內皮損害、血視網膜屏障功能受損、微血管滲漏、廣泛的視網膜缺血缺氧促發VEGF等生長因子表達增加,最終導致視網膜新生血管形成。目前臨床使用的VEGF單克隆抗體在一定程度上能夠阻止血管滲漏和新生血管的形成,但由于DR病理性血管生成過程涉及其他生長因子和各種炎癥細胞因子的參與,部分患者對VEGF單克隆抗體敏感性有限且治療后易復發[26]。因此確定控制DR進展的新的治療靶點是極其重要的。本研究結果表明,NTN1-Gαi1/Gαi3信號轉導可能是DR異常血管生成的重要原因之一,這種級聯反應提示DR分子治療的新靶點。然而目前研究存在一定局限性。首先,目前結果側重于體外細胞實驗,尚無法全方位立體模擬錯綜復雜的體內環境。其次,在研究方法上,本研究僅通過敲低Gαi1和Gαi3基因以觀察其對血管生成的影響,雖揭示Gαi1和Gαi3基因在介導NTN1促血管生成效應中的作用,但對于這一信號通路更深入的分子機制、調控細節及其與其他信號通路的交互作用,并未進行深入探究。為實現以干預NTN1-Gαi1/Gαi3信號為靶點的治療方法,未來需進一步探索體內外實驗,通過基因敲低、敲除、過表達、顯性陰性突變、挽救Ntn1、Gαi1、Gαi3基因等多種方法,正負向調節NTN1、Gαi1、Gαi3蛋白表達或活化水平,探索影響其血管生成的具體機制。結合蛋白和轉錄組學技術,全面分析NTN1刺激下細胞內的基因轉錄和蛋白質表達的變化。在未來需重點關注NTN1-Gαi1/Gαi3參與血管新生調控的具體機制,為視網膜新生血管性疾病的治療提供新的思路和策略。