引用本文: 李霽竹, 馬媛, 劉寶怡, 劉亞萍, 陳子葉, 李濤. 人誘導多能干細胞和胚胎干細胞分化為血管周細胞和內皮細胞過程中差異表達基因分析. 中華眼底病雜志, 2024, 40(11): 869-877. doi: 10.3760/cma.j.cn511434-20241011-00379 復制
版權信息: ?四川大學華西醫院華西期刊社《中華眼底病雜志》版權所有,未經授權不得轉載、改編
視網膜局部組織缺血缺氧形成的病理性新生血管是糖尿病視網膜病變、視網膜靜脈阻塞、早產兒視網膜病變等致盲性視網膜血管疾病的共同病理基礎[1]。新生血管常伴有血管周細胞和內皮細胞丟失、基底膜增厚等病理改變,易發生滲漏和出血,加重視網膜缺血缺氧狀態[2]。近年來研究表明,干細胞在視網膜血管疾病中的應用潛力巨大[3-4],體外培養的人誘導多能干細胞(hiPSC)與胚胎干細胞(hESC)在特定誘導條件下可模擬體內血管發育過程,分化形成周細胞與內皮細胞,通過體內移植可以整合到病變的血管上,發揮修復血管的作用[5];此外,也可以體外培養形成3D血管類器官,用于篩選糖尿病微血管病變的治療藥物[6]。目前誘導干細胞分化為周細胞與內皮細胞的經典方案為先向中胚層方向誘導,再向血管方向誘導的“兩步法”[7]。但該方法耗時長、效率較低,且具體機制尚不明確。本研究對hiPSC與hESC分化為周細胞和內皮細胞過程中的差異表達基因(DEG)進行分析,分析調控分化進程的關鍵分子與信號通路,旨在為優化干細胞分化方案、揭示血管發育機制提供依據。
1 材料和方法
1.1 材料
人尿源性hiPSC細胞系(中山大學中山眼科中心劉春巧教授惠贈);hESC H9細胞系(中國科學院細胞庫)。Matrigel基質膠(美國BioCoat公司);mTeSR培養基、dispase消化液(加拿大StemCell公司);內皮細胞生長培養基-2套裝(EGM-2 kit培養基,瑞士LONZA公司);重組人Activin A蛋白、抗人CD31藻藍蛋白偶聯抗體(CD31-APC)、多聚甲醛(PFA)組織固定液、TRIzol裂解液(美國Thermo Fisher公司);重組人骨形成蛋白(BMP4)、Anti-Caldesmon抗體(英國Abcam公司);血管內皮生長因子(VEGF)、SB431542、CHIR因子、抗人血小板源性生長因子受體β抗體(PDGFR-β)(美國R&D公司);Gelatin基質膠(德國Sigma公司);測序試劑盒(美國Illumina公司)。
1.2 方法
hiPSC、hESC擴增培養和誘導分化。hiPSC、hESC分別接種于Matrigel基質膠包被的6孔板中,加入mTeSR培養基,37 ℃、5%CO2培養箱中進行擴增培養,細胞生長至40%~50%融合時記錄為0 d。加入中胚層分化培養液(mTeSR+25 μg/ml Activin A蛋白+30 μg/ml BMP4蛋白+50 μg/ml VEGF+4 mmol/L CHIR因子)培養3 d,其后更換為血管分化培養液(mTeSR+50 μg/ml VEGF+20 mmol/L SB431542因子)培養7 d。倒置顯微鏡觀察細胞生長狀況與形態變化,拍照記錄。
分離鑒定內皮細胞和周細胞。分別取hiPSC、hESC分化10 d時的細胞制成細胞懸液,加入CD31-APC抗體避光孵育45 min,流式細胞儀分離鑒定內皮細胞,FlowJo軟件分析獲得數據。將含有周細胞的剩余細胞懸液轉移至Gelatin包被的6孔板中,EGM-2 kit培養基繼續培養至周細胞融合。4% PFA組織固定液固定細胞爬片10 min,滴加含周細胞特征性標志物Caldesmon與PDGFR-β的一抗,4 ℃孵育過夜,滴加相應二抗,室溫避光孵育1 h,洗滌封片。熒光顯微鏡下觀察并拍照記錄。
1.3 RNA提取、轉錄組測序
分別收集hiPSC、hESC分化0、4、7、10 d的細胞,TRIzol裂解液裂解細胞,收集上清液,加入0.5 ml異丙醇沉淀RNA,10 000×g離心10 min,75%乙醇洗滌RNA沉淀,10 000×g離心10 min,棄上清,室溫干燥。加入無RNase的溶液溶解RNA,-80 ℃保存。不同細胞系實驗獨立重復3次。
誘導培養0、4、7、10 d(分化記作分化前、早期、中期、晚期)d分別收集hiPSC、hESC進行轉錄組測序。采用帶有Oligo的磁珠富集真核生物mRNA,打斷形成短片段,反轉錄合成cDNA鏈,末端修復后連接測序的接頭,瓊脂糖凝膠電泳對不同大小片段進行選擇,聚合酶鏈反應(PCR)擴增富集cDNA,完成文庫制備。應用Illumina HiSeq2500進行雙端測序。不同細胞系分別獨立重復測序3次。
1.4 DEG篩選和生物信息學分析
測序數據過濾,去除接頭、污染和低質量序列,采用hisat2將高質量序列匹配到參考基因組上。利用FeatureCounts軟件進行基因水平的定量分析,計算每個基因在各樣本中的讀段計數(read count)。標準化后,根據模型進行假設檢驗概率(P值)計算,多重假設檢驗校正,得到調整后的P值。以|log2[差異表達倍數(FC)]|>1且P<0.05為條件篩選DEG[8]。
應用R軟件中pheatmap函數繪制聚類熱圖,分析DEG變化,紅色越深表示DEG表達水平越高,藍色越深表示DEG表達水平越低;VENNY系統(http://bioinfogp.cnb.csic.es/tools/venny/index.html)繪制韋恩圖;R軟件中的phyper函數對分化過程中持續存在差異的DEG進行基因本體(GO,http://www.geneontology.org/)和京都基因與基因組百科全書(KEGG,https://www.kegg.jp/)富集分析,富集顯著性標準為P<0.05。
2 結果
2.1 hiPSC、hESC分化為周細胞和內皮細胞過程中形態變化與標志物表達情況
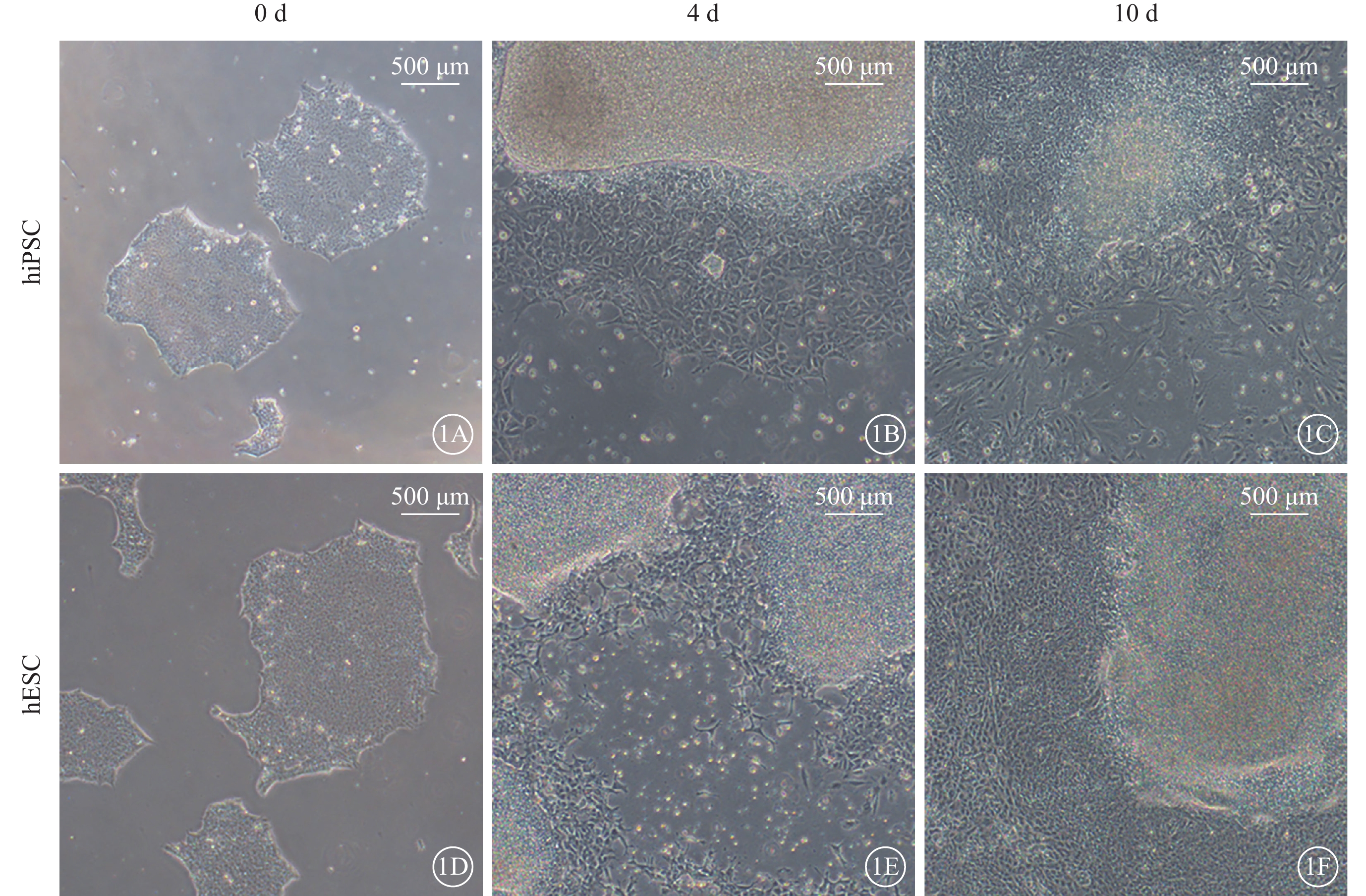
未分化hiPSC、hESC呈圓形克隆狀生長,邊界清晰,細胞排列緊密。誘導培養4 d時,可見分化的梭形細胞向外爬行生長;10 d時,梭形細胞數量明顯增多,部分區域呈融合生長,且hESC較hiPSC組分化細胞數量更多(圖1)。
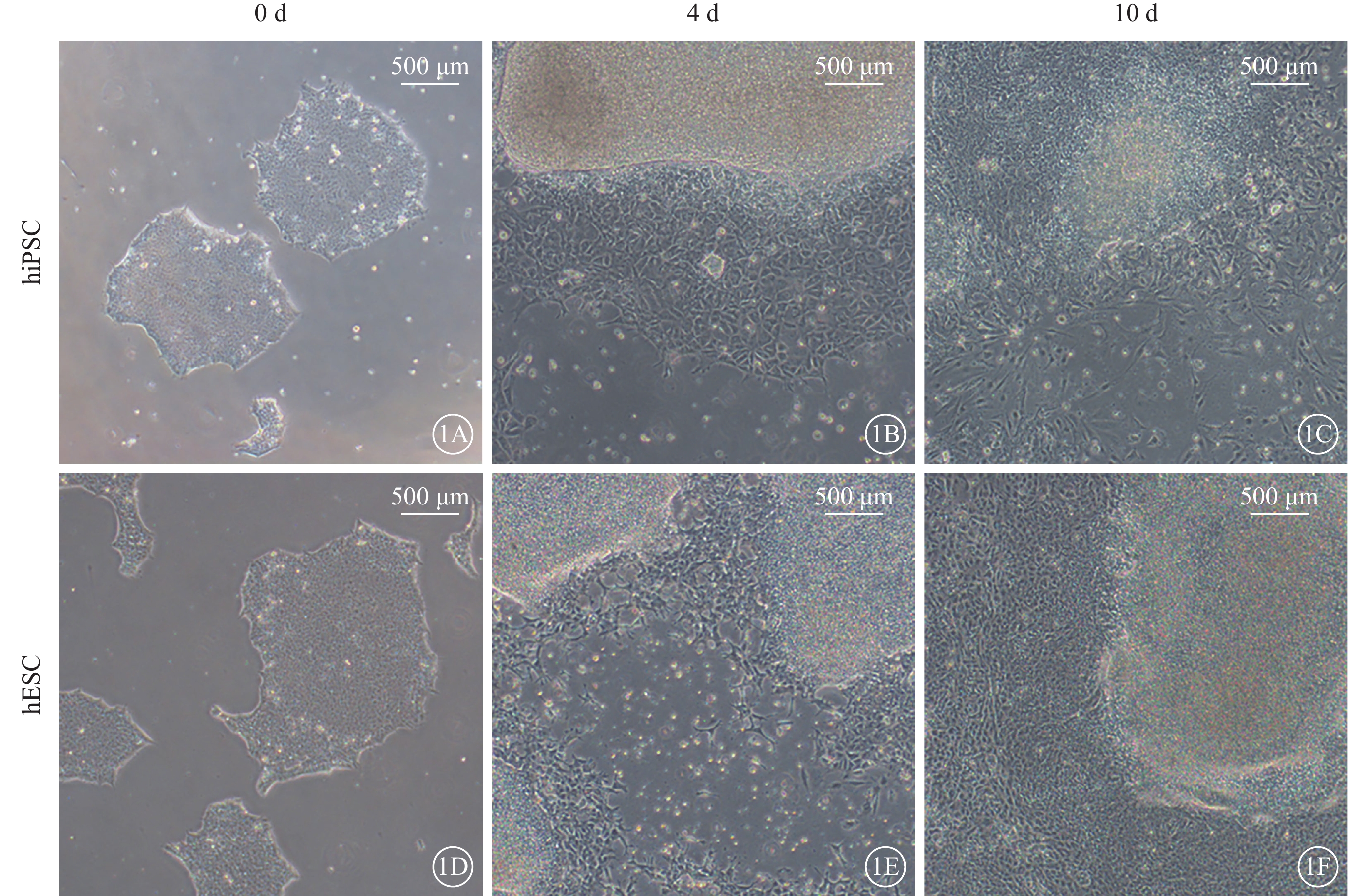 圖1
hiPSC、hESC分化過程中形態變化光學顯微鏡像(標尺:500 μm) 1A~1C分別示hiPSC誘導培養0、4、10 d,0 d時,hiPSC呈圓形克隆狀生長,邊界清晰;4 d時,分化細胞向外爬行生長呈梭形;10 d時,梭形細胞數量增多,局部呈融合生長。1D~1F分別示hESC誘導培養0、4、10 d,0、4 d時,形態變化與hiPSC一致;10 d時,分化細胞數量較hiPSC更多 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞
圖1
hiPSC、hESC分化過程中形態變化光學顯微鏡像(標尺:500 μm) 1A~1C分別示hiPSC誘導培養0、4、10 d,0 d時,hiPSC呈圓形克隆狀生長,邊界清晰;4 d時,分化細胞向外爬行生長呈梭形;10 d時,梭形細胞數量增多,局部呈融合生長。1D~1F分別示hESC誘導培養0、4、10 d,0、4 d時,形態變化與hiPSC一致;10 d時,分化細胞數量較hiPSC更多 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞
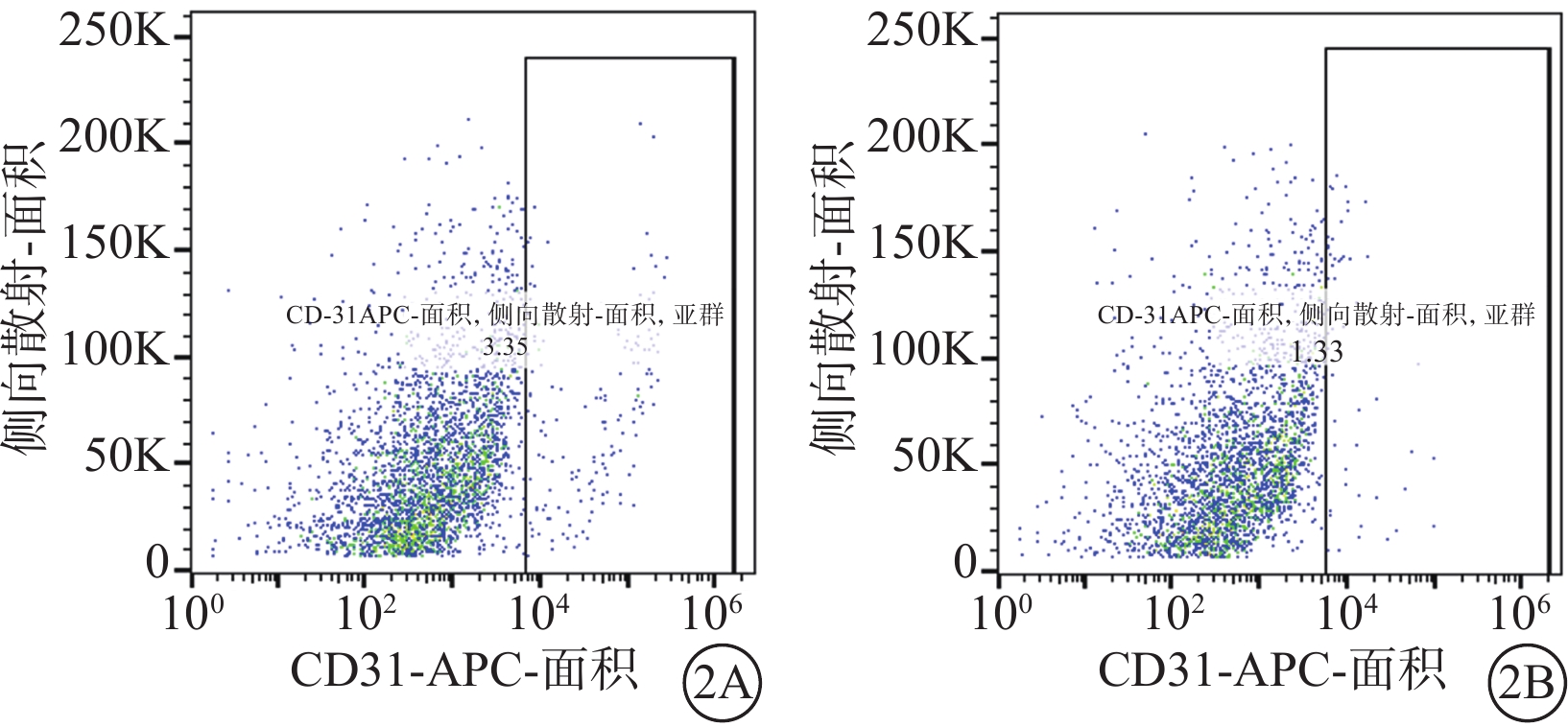
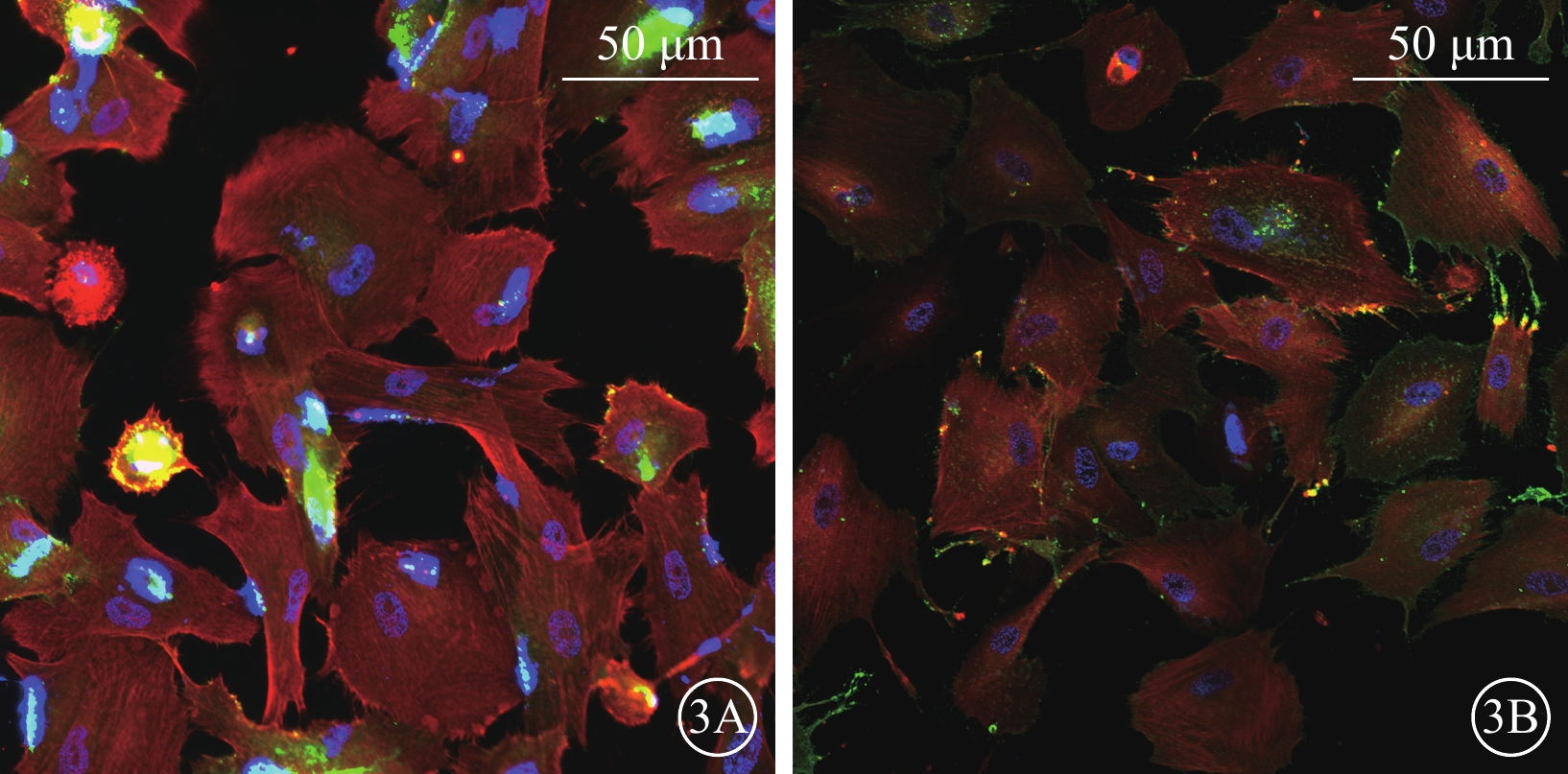
流式細胞儀檢測結果顯示,誘導培養10 d時,hiPSC、hESC中均存在CD31+的內皮細胞(圖2);免疫熒光染色結果顯示,hiPSC、hESC誘導培養的周細胞中均高表達特征性標志物Caldesmon、PDGFR-β(圖3)。
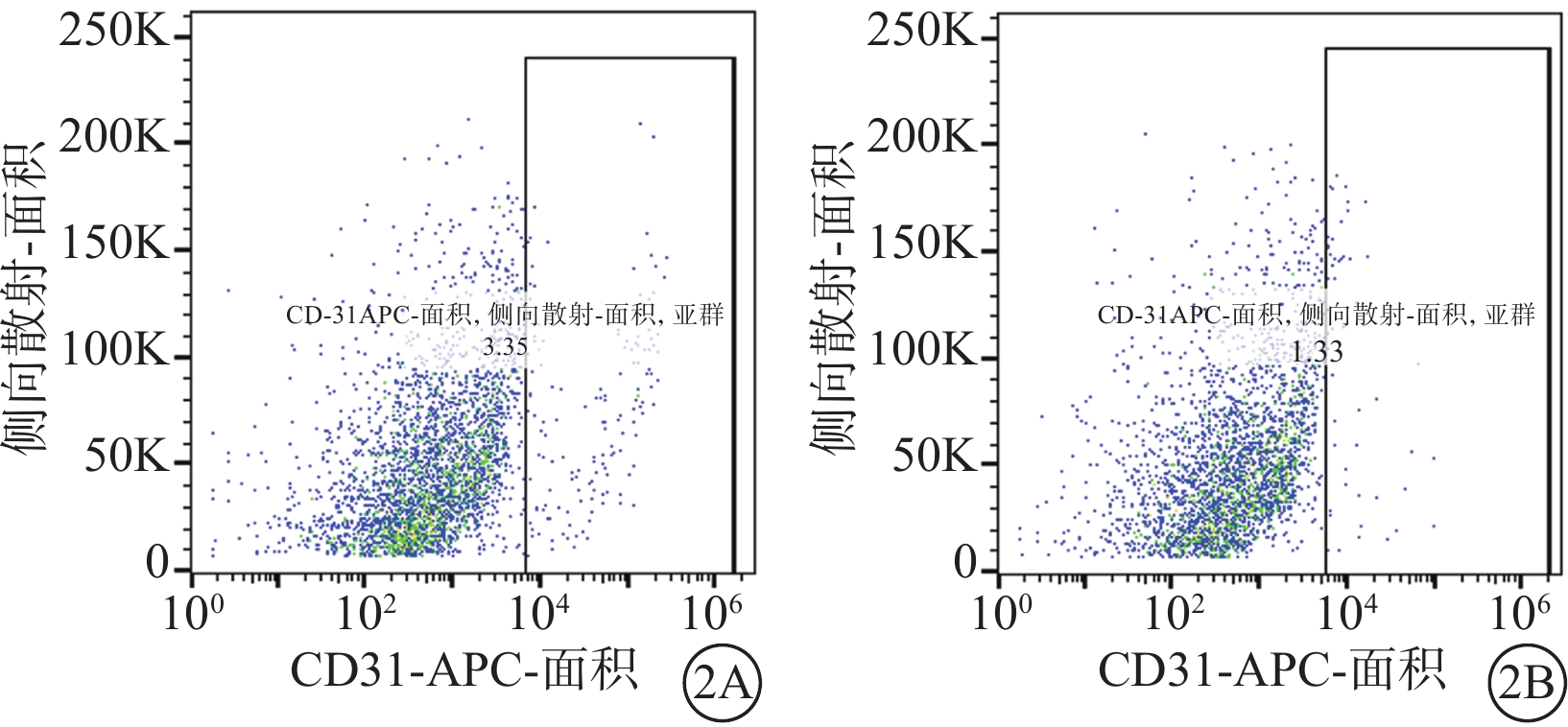 圖2
hiPSC、hESC誘導培養10 d時流式細胞儀檢測像 2A、2B分別示hiPSC、hESC,兩個細胞系均可見CD31+內皮細胞存在 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞;CD31-APC:抗人CD31藻藍蛋白偶聯抗體
圖2
hiPSC、hESC誘導培養10 d時流式細胞儀檢測像 2A、2B分別示hiPSC、hESC,兩個細胞系均可見CD31+內皮細胞存在 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞;CD31-APC:抗人CD31藻藍蛋白偶聯抗體
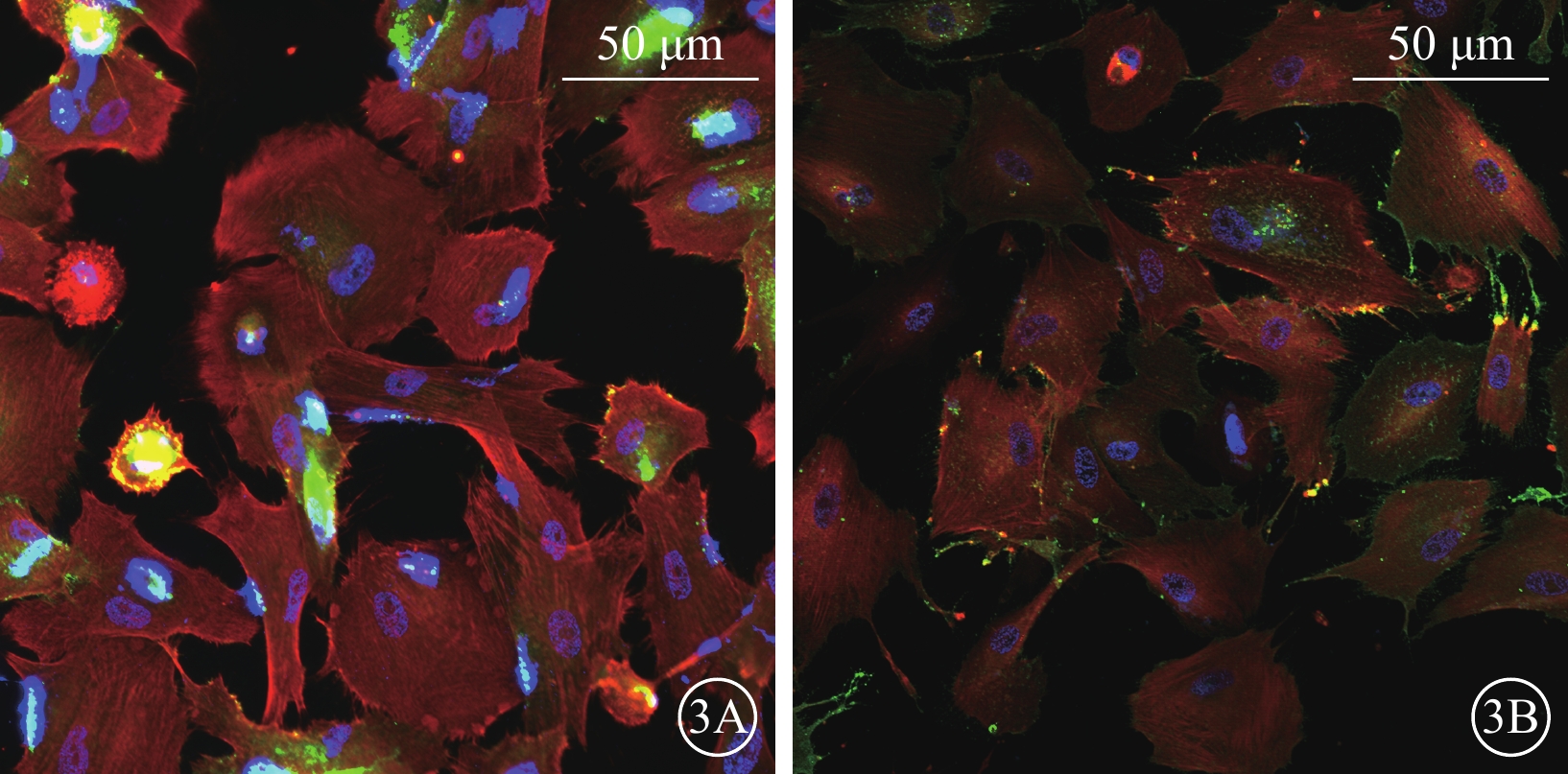 圖3
hiPSC、hESC誘導培養10 d時細胞熒光顯微鏡像(標尺:50 μm) 3A、3B分別示hiPSC、hESC,Caldesmon標記的周細胞呈紅色熒光,表達于細胞骨架;PDGFR-β標記的周細胞呈綠色熒光,表達于細胞膜;DAPI標記的細胞核呈藍色熒光 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞;PDGFR-β:血小板衍生生長因子受體-β:DAPI:4',6-二脒基-2-苯基吲哚
圖3
hiPSC、hESC誘導培養10 d時細胞熒光顯微鏡像(標尺:50 μm) 3A、3B分別示hiPSC、hESC,Caldesmon標記的周細胞呈紅色熒光,表達于細胞骨架;PDGFR-β標記的周細胞呈綠色熒光,表達于細胞膜;DAPI標記的細胞核呈藍色熒光 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞;PDGFR-β:血小板衍生生長因子受體-β:DAPI:4',6-二脒基-2-苯基吲哚
2.2 測序數據的分組與質量評估
測序結果顯示,hiPSC、hESC各有平均4.3 107的過濾后讀段計數,原始數據Q30比例為93.93%,匹配率均>95.9%(表1)。
107的過濾后讀段計數,原始數據Q30比例為93.93%,匹配率均>95.9%(表1)。
2.3 不同分化時間DEG表達譜分析
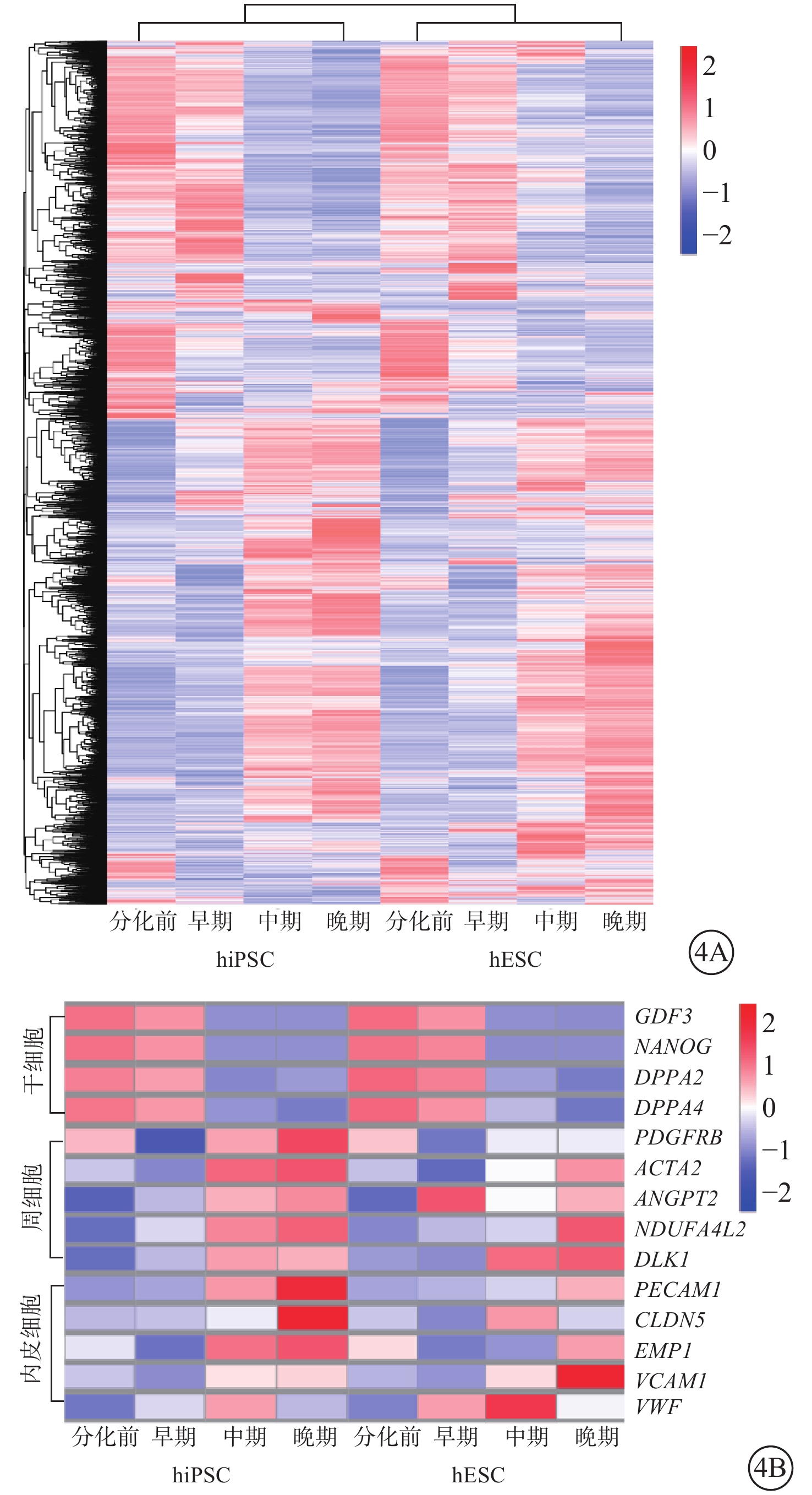
hiPSC、hESC隨分化時間延長,DEG顯著上調或下調且變化趨勢較為一致(圖4A)。分化中期,GDF3、NANOG、DPPA等干細胞標志性基因顯著下調;分化中期及晚期,周細胞標志基因PDGFR-β、ACTA2、ANGPT2、NDUFA4L2、DLK1,內皮細胞標志基因PECAM1、CLDN5、EMP1、VCAM1、VWF顯著上調(圖4B)。
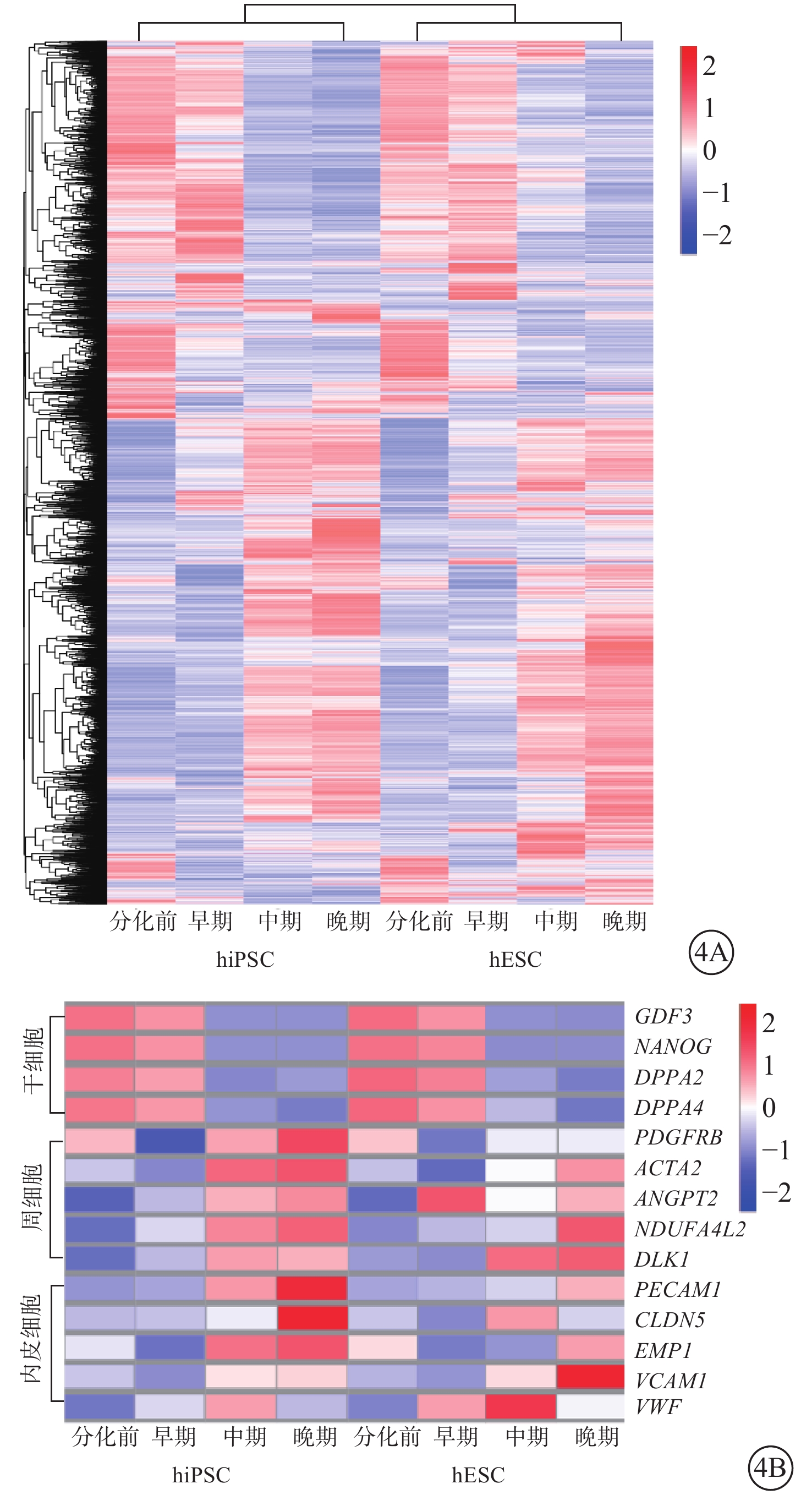 圖4
hiPSC、hESC不同分化時間DEG分析聚類熱圖(紅色越深表示FC越高,藍色越深表示FC越低) 4A示hiPSC、hESC中大量DEG隨分化時間顯著上調或下調,變化趨勢較為一致;4B示分化中期干細胞標志性基因顯著下調,分化中期及晚期周細胞與內皮細胞標志性基因顯著上調 DEG:差異表達基因;hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞;FC:差異表達倍數
圖4
hiPSC、hESC不同分化時間DEG分析聚類熱圖(紅色越深表示FC越高,藍色越深表示FC越低) 4A示hiPSC、hESC中大量DEG隨分化時間顯著上調或下調,變化趨勢較為一致;4B示分化中期干細胞標志性基因顯著下調,分化中期及晚期周細胞與內皮細胞標志性基因顯著上調 DEG:差異表達基因;hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞;FC:差異表達倍數
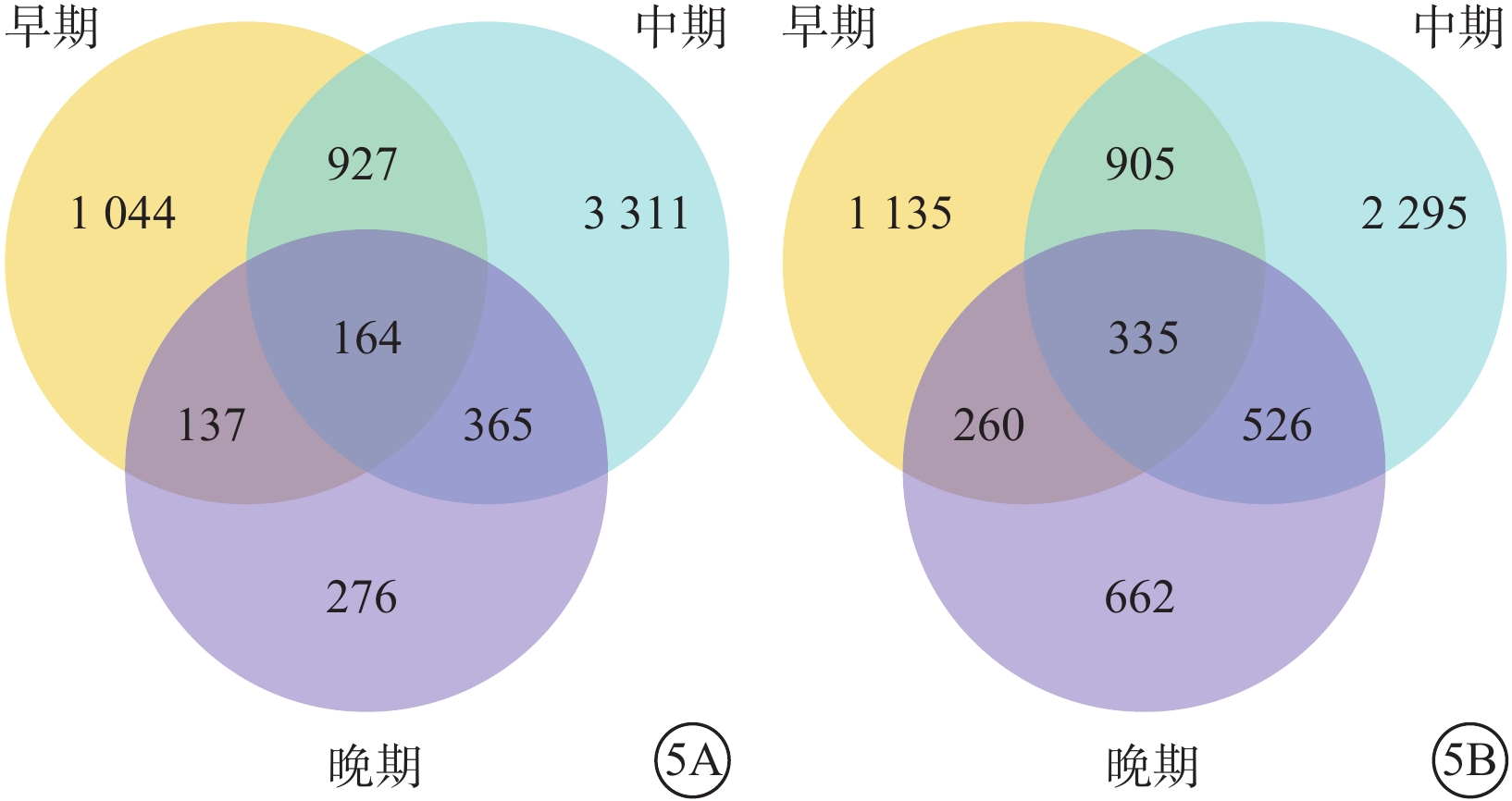
2.4 持續DEG的韋恩分析和GO富集分析
韋恩分析結果顯示,hiPSC與hESC在分化早期、中期、晚期共檢測到持續DEG 491個,其中hiPSC、hESC分別為164、335個(圖5)。hiPSC、hESC同時表達的基因為8個。其中,SLC30A3、LCK、TNFRSF8、PRDM14、GLB1L3持續表達下調;CLEC18C、CLEC18B、F2RL2持續表達上調(表2)。CLEC18C、CLEC18B為C型凝集素結構域家族;F2RL2為蛋白酶激活受體家族;SLC30A3為溶質載體家族;LCK為酪氨酸蛋白激酶家族;TNFRSF8為腫瘤壞死因子受體超家族;PRDM14為轉錄調節子家族;GLB1L3為編碼β-半乳糖樣蛋白。
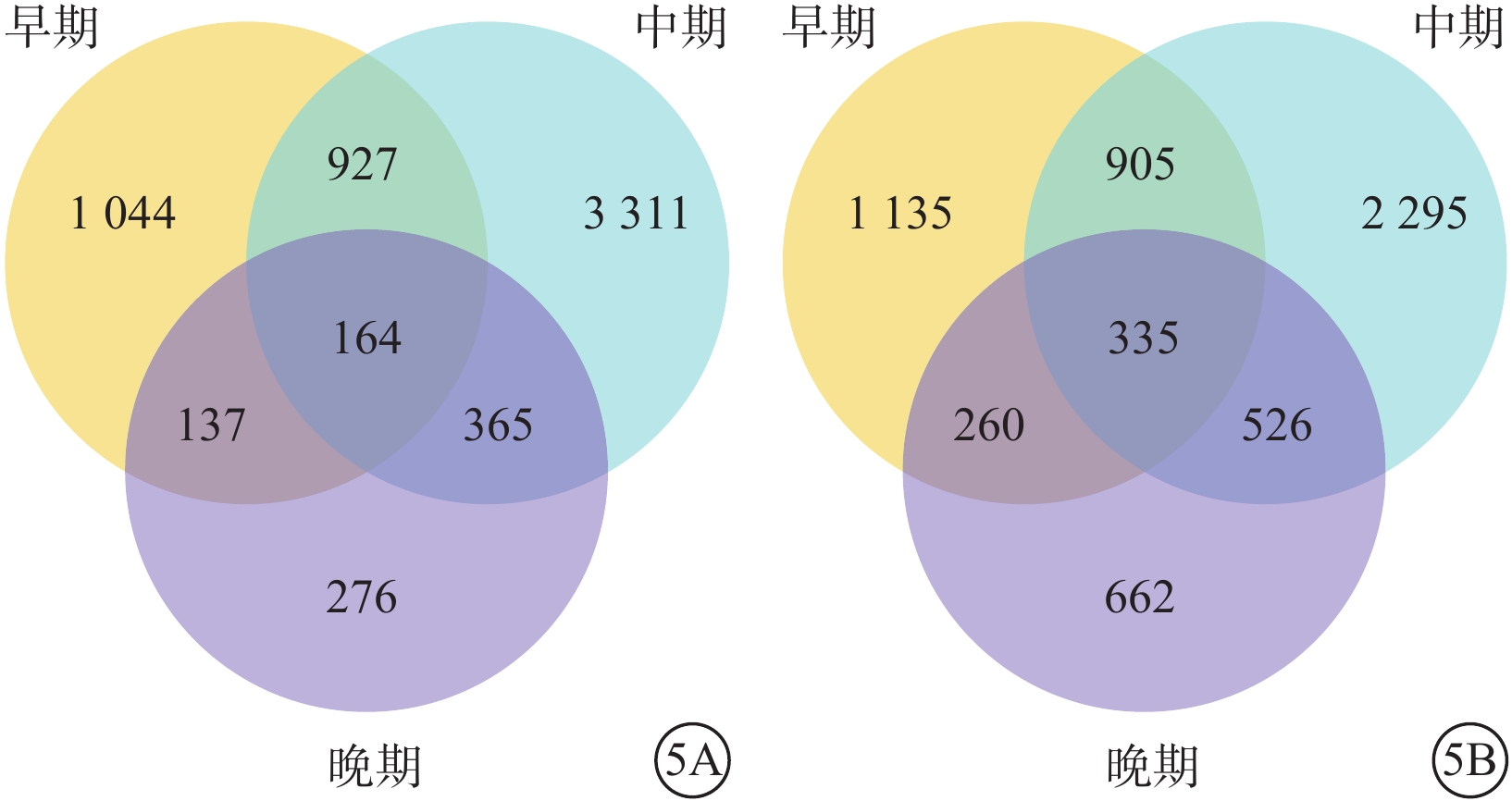 圖5
hiPSC、hESC不同分化時間的差異表達基因韋恩圖 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞
圖5
hiPSC、hESC不同分化時間的差異表達基因韋恩圖 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞
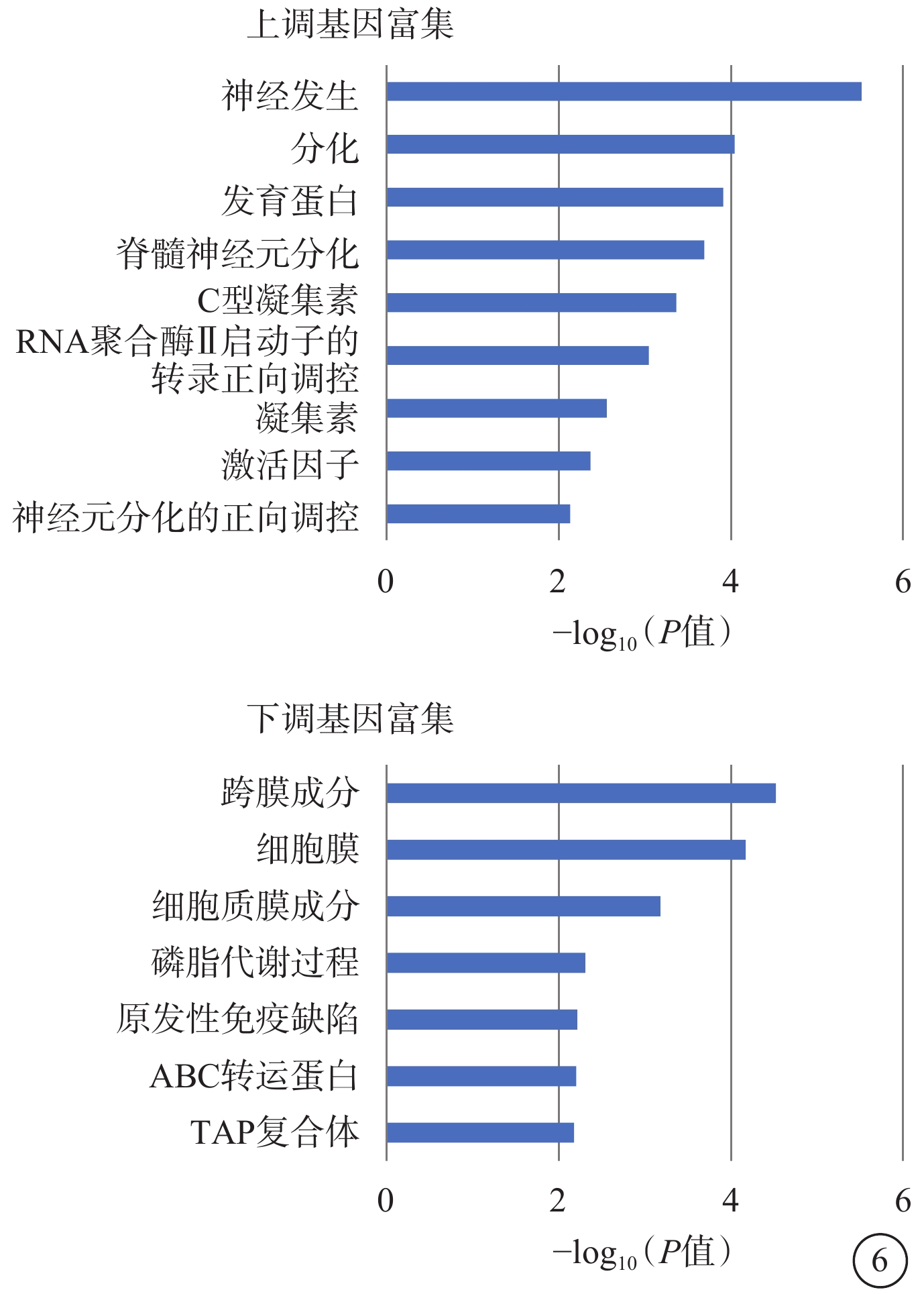
GO富集分析結果顯示,持續上調表達的DEG主要富集在神經發生、分化、發育蛋白等條目中;持續下調表達的DEG主要富集在膜結構、磷脂代謝過程等條目中(圖6)。
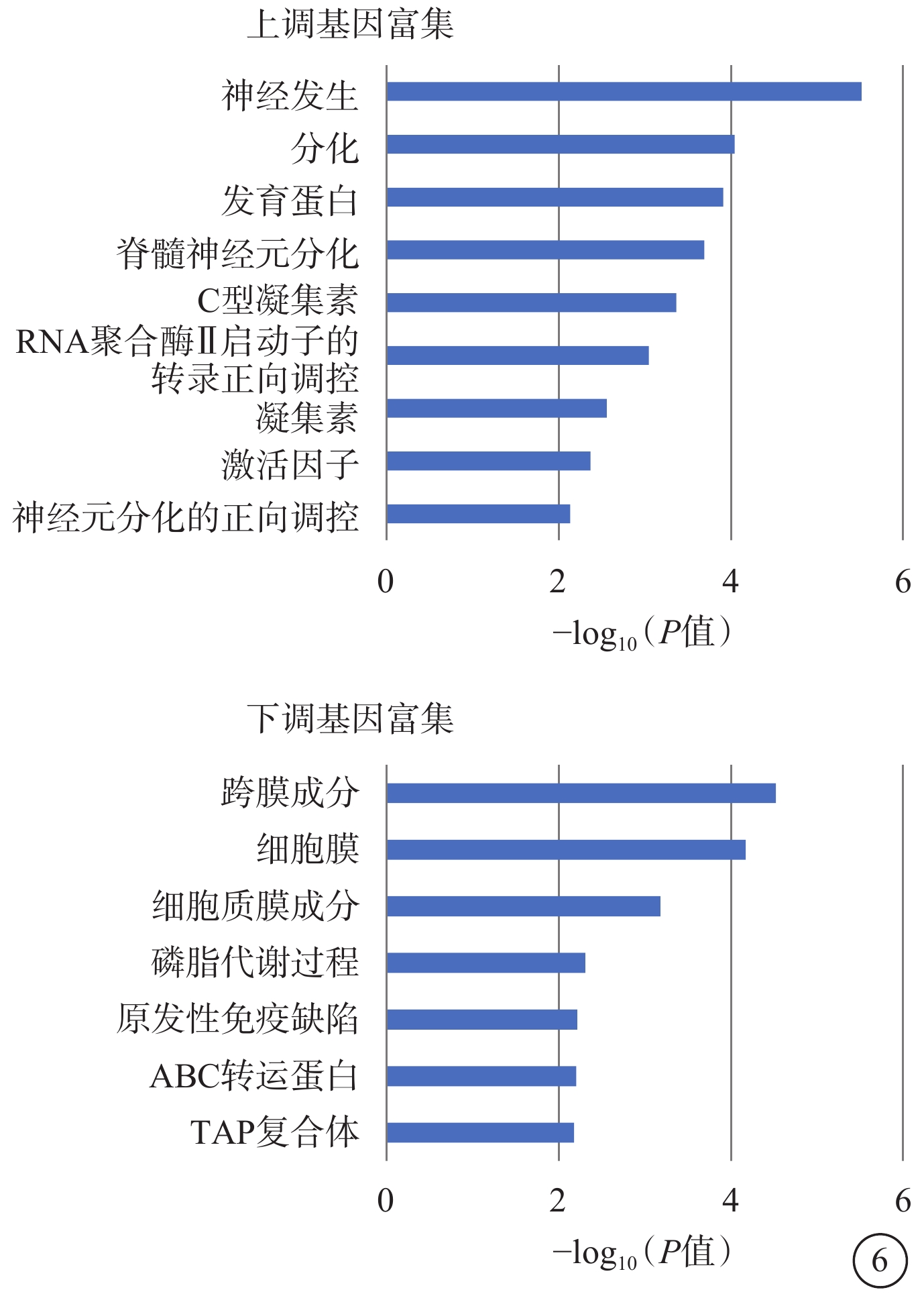 圖6
hiPSC、hESC持續DEG的GO富集分析圖 條柱代表富集GO條目的P值,柱線越高表示P值越小,富集越顯著 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞; DEG:差異表達基因;GO:基因本體
圖6
hiPSC、hESC持續DEG的GO富集分析圖 條柱代表富集GO條目的P值,柱線越高表示P值越小,富集越顯著 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞; DEG:差異表達基因;GO:基因本體
2.5 持續DEG的KEGG通路分析
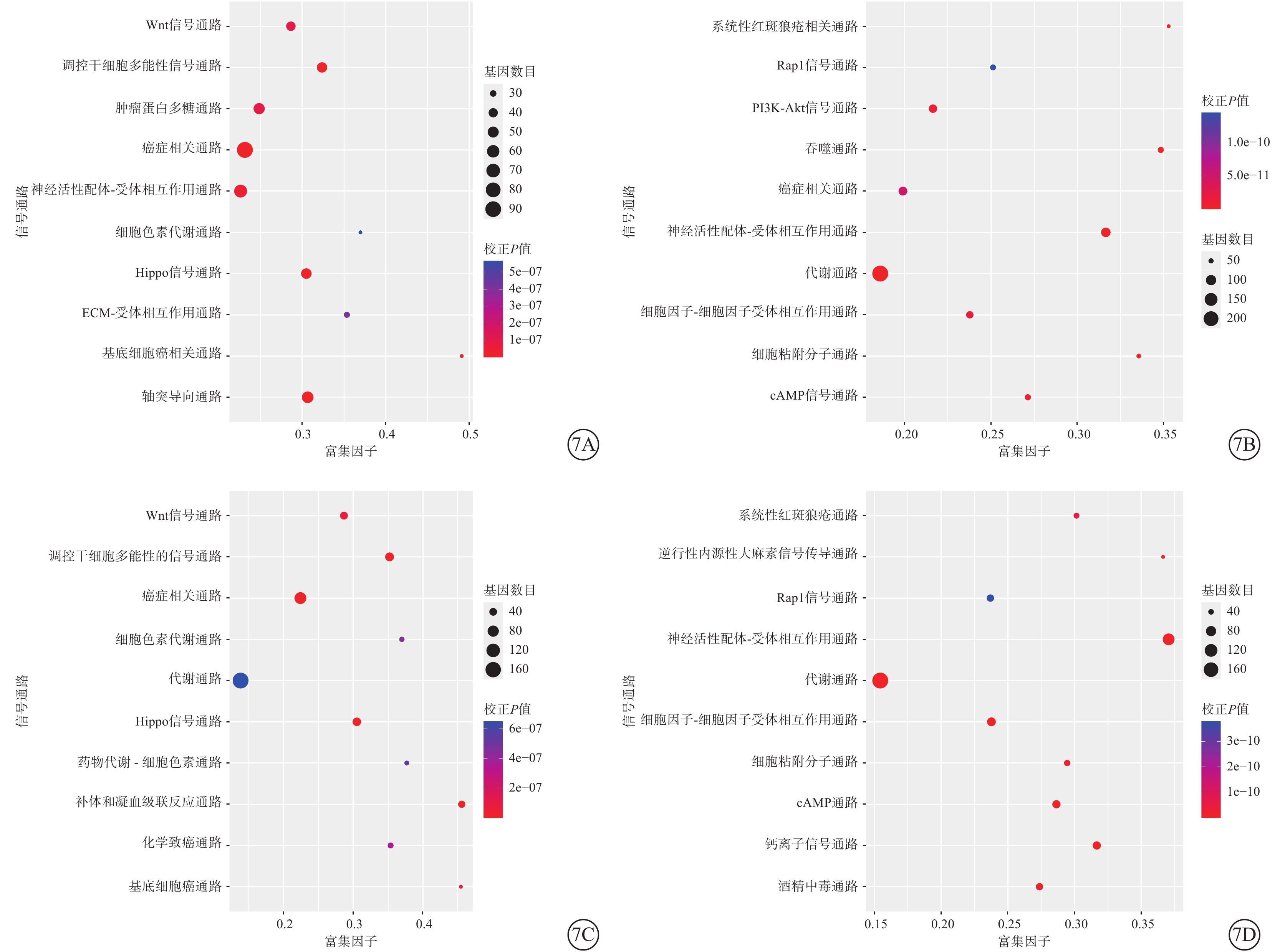
KEGG通路顯著性富集分析結果顯示,受分化進程影響,hiPSC、hESC中上調表達的基因主要富集于癌癥相關通路、干細胞多能性調節通路、Wnt信號通路、Hippo信號通路;下調表達的基因主要富集于代謝相關通路(圖7)。
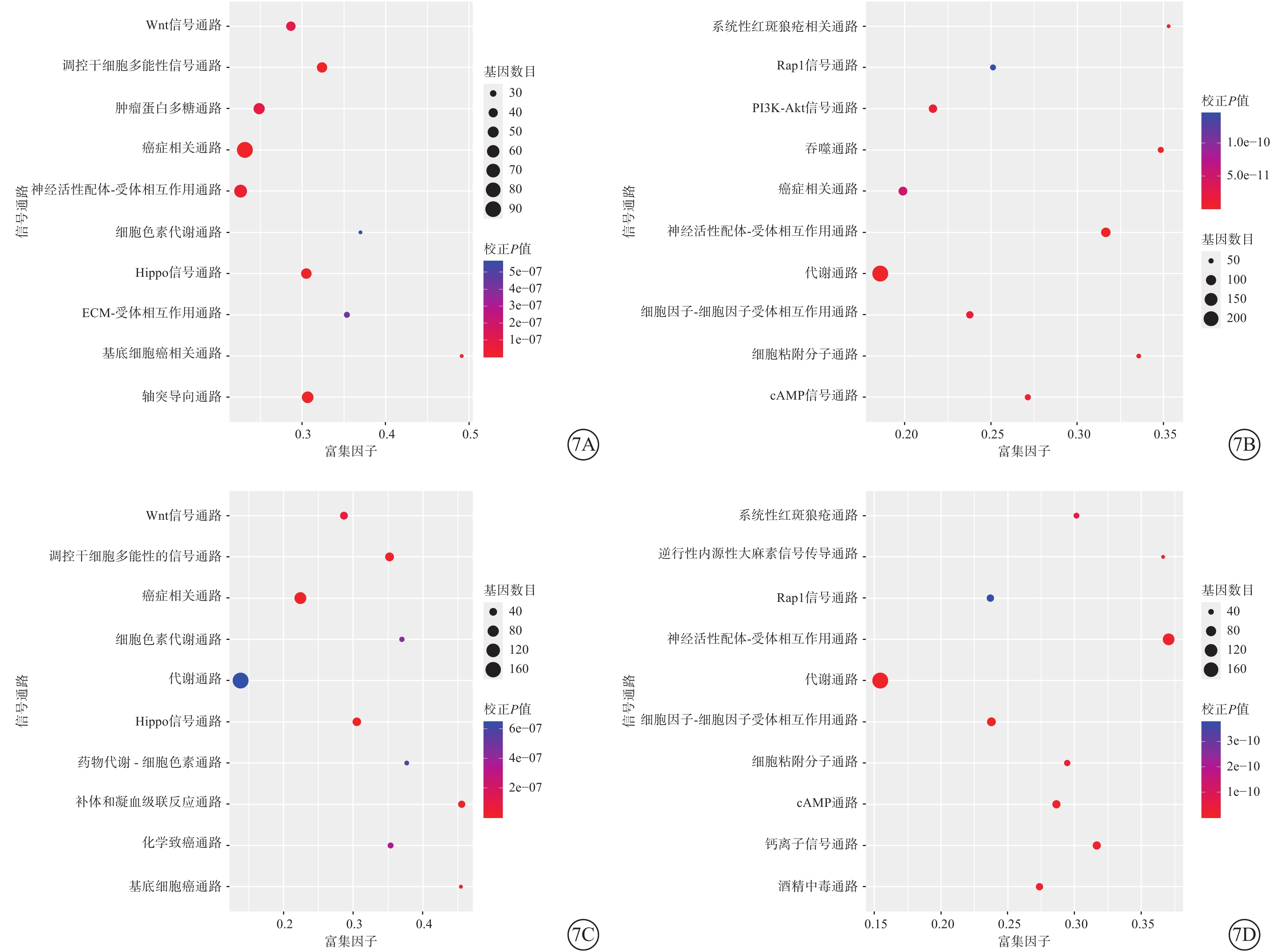 圖7
hiPSC、hESC持續DEG的KEGG富集排名前10的信號通路氣泡圖 7A、7B和7C、7D分別示hiPSC、hESC分化過程中排名前10的上調、下調信號通路 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞;DEG:差異表達基因;KEGG:京都基因與基因組百科全書;ECM:細胞外基質;PI3K-Akt:磷脂酰肌醇3激酶-絲裂原活化蛋白激酶;cAMP:環腺苷酸
圖7
hiPSC、hESC持續DEG的KEGG富集排名前10的信號通路氣泡圖 7A、7B和7C、7D分別示hiPSC、hESC分化過程中排名前10的上調、下調信號通路 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞;DEG:差異表達基因;KEGG:京都基因與基因組百科全書;ECM:細胞外基質;PI3K-Akt:磷脂酰肌醇3激酶-絲裂原活化蛋白激酶;cAMP:環腺苷酸
3 討論
本研究通過經典的“兩步法”成功誘導hiPSC和hESC分化為血管周細胞和內皮細胞,利用轉錄組測序技術分析識別了該分化過程中關鍵的DEG與相關通路,并分析參與調控分化的關鍵分子,為優化干細胞分化流程提供了科學依據。DEG聚類分析結果顯示,隨分化進程的進行,干細胞標志性基因(如NANOG、GDF3)的表達逐漸下調,表明多能性逐步喪失,而周細胞與內皮細胞標志性基因(如PDGFRB、PECAM1、VWF)則在中晚期顯著上調,支持hiPSC與hESC向特定血管相關類型分化的趨勢。在hiPSC與hESC的分化過程中,共檢測到8個具有持續特異性表達變化的DEG,包括表達上調的CLEC18C、CLEC18B、F2RL2,以及表達下調的SLC30A3、LCK、TNFRSF8、PRDM14、GLB1L3,這些DEG可能通過激活Wnt通路、Hippo通路,抑制代謝通路,解除對干細胞多能性的維持以及影響細胞周期、抑制細胞增殖等作用,促進周細胞與內皮細胞的分化。
CLEC18C、CLEC18B、F2RL2基因的持續上調表達增加了對細胞分化的支持作用。CLEC18C與CLEC18B屬于人C型凝集素家族,在多種細胞類型中參與調控細胞遷移和分化[9]。研究表明,CLEC18C在腫瘤和神經組織中高表達,通過激活Wnt信號通路促進細胞的增殖和遷移[10],提示其在分化過程中可能通過類似途徑支持干細胞向特定細胞類型的分化。F2RL2屬于蛋白酶激活受體家族,其高表達與膠質瘤、口腔鱗癌等腫瘤的預后不良有密切聯系[11],提示該基因在干細胞增殖和維持中也扮演類似關鍵角色。在本研究中,CLEC18C、CLEC18B和F2RL2的上調表達可能通過增強干細胞的增殖和遷移能力,為分化過程中細胞數量的增加提供支持,同時為特定細胞類型的分化奠定基礎。近年癌癥相關通路在干細胞研究中逐漸受到關注,因其在干細胞和癌細胞的自我更新和分化過程中可能共享一些調控機制,如Wnt和Notch通路既參與干細胞的自我更新,也在許多癌癥中被異常激活[12],癌癥相關信號通路的上調可幫助干細胞維持適當的增殖狀態,有助于維持細胞增殖和分化的動態平衡[13]。本研究KEGG通路富集性分析結果顯示,上調的DEG在癌癥通路存在顯著富集,可能反映了干細胞在分化過程中的增殖和存活需求,確保分化過程中有足夠的細胞數量,同時維持分化潛力。
Wnt與Hippo信號通路在調控hiPSC和hESC的分化中也發揮關鍵調控作用。在經典Wnt/β-連環蛋白(β-catenin)依賴型通路中,當Wnt配體與細胞膜上的Frizzled受體結合時,β-catenin得以穩定化并累積在細胞質中,隨后進入細胞核,激活與分化相關的目標基因[14]。這一過程不僅有助于干細胞向中胚層細胞的轉化,還通過上調PECAM1和VWF等標志性基因促進周細胞和內皮細胞的生成[15-17]。同時,Hippo信號通路通過調控YAP/TAZ轉錄共激活因子的活性,確保分化過程中細胞的增殖平衡,當Hippo通路被激活時,YAP/TAZ被磷酸化并保留在細胞質中,從而抑制細胞的增殖[18]。這種調控機制在確保分化過程中細胞數量適度增加的同時,避免了過度增殖帶來的異常增生風險。此外,Hippo通路與Wnt通路在內皮細胞的分化成熟階段具有協同作用,共同調控內皮細胞的生成和血管新生[19-21]。在本研究中,Wnt與Hippo信號通路在hiPSC和hESC分化過程中顯著上調,進一步提示其在血管周細胞和內皮細胞形成中的關鍵作用,我們推測Wnt信號通路的激活使干細胞逐步向血管相關細胞類型轉化,為周細胞和內皮細胞的形成奠定基礎,而Hippo通路的激活確保了細胞在分化過程中不會發生過度增殖,從而維持了分化進程的平衡與穩定。
持續特異性下調表達的LCK、PRDM14、GLB1L3基因在干細胞的多能性喪失中起到關鍵作用。LCK作為非受體酪氨酸激酶,在多種干細胞和造血細胞中高表達,與細胞增殖和存活密切相關[22-23]。LCK高表達于成年小鼠的造血細胞與hESC,且在胚胎干細胞分化為擬胚體過程中LCK表達水平顯著下降[24]。推測在干細胞分化過程中LCK的下調有助于降低細胞的增殖信號,促使細胞從增殖狀態進入分化狀態。PRDM14是維持胚胎干細胞多能性的特異性轉錄調節因子[25],在人原始生殖細胞中敲除該基因后會導致干細胞相關基因顯著下調,并促進分化相關基因上調[26]。其在分化過程中的下調,提示干細胞多能性基因逐漸關閉,為特異性分化路徑的開啟提供了條件。GLB1L3與胚胎干細胞的多能性密切相關[27],推測其下調表達可能通過解除干細胞多能性的維持,從而有利于分化的進行。
TNFRSF8、PRDM14、SLC30A3基因的持續特異性下調有助于抑制細胞周期,防止過度增殖。TNFRSF8編碼的蛋白CD30屬于腫瘤壞死因子受體家族,其下調表達可引起細胞周期蛋白激酶抑制因子CDKN2A、CDKN1A的表達上調,導致細胞阻滯于G1/S與G2/M周期,進而抑制細胞的過度增殖[28]。在293T細胞中,PRDM14通過抑制細胞周期G1/S期轉變,也可影響細胞增殖與遷移[29]。推測TNFRSF8、PRDM14的下調可能通過影響細胞周期,抑制干細胞的過度增殖達到促進分化的作用。SLC30A3編碼鋅轉運蛋白,參與調控細胞內鋅離子平衡,研究表明,鋅離子水平在細胞增殖和分化中具有重要調控作用,胚胎發育早期鋅水平的不足會抑制細胞周期與轉錄組水平,導致細胞增殖減少和分化受阻[30-31]。SLC30A3的下調可能通過減少細胞內鋅離子濃度,影響細胞周期和分裂速度,從而推動分化進程。
GO富集分析結果顯示,下調表達的DEG主要富集于膜結構、磷脂代謝相關生物過程。磷脂作為細胞膜的基本構成成分,參與細胞形態、遷移和增殖的調控[32]。分化過程中,磷脂代謝的下調可能通過調控細胞膜的流動性和穩定性,減少不必要的細胞遷移和增殖,從而增強分化過程的專一性和穩定性。此外,磷脂代謝的下調也可能通過減少細胞內能量的過度消耗,優化分化過程中對能量的需求。代謝通路在干細胞的多能性維持和分化中也扮演重要角色,代謝狀態的改變往往伴隨著干細胞狀態變化,研究表明,糖酵解和氧化磷酸化的平衡以及脂質代謝水平對干細胞分化具有深遠影響[33]。在本研究中,代謝相關通路的顯著下調可能與細胞代謝活動的降低及能量利用效率的提升有關,以滿足分化過程中對代謝的不同需求。
本研究存在的局限性包括:首先,研究結果主要依賴于轉錄組學數據分析,關鍵基因的功能未能通過基因敲除或過表達實驗進行驗證;其次,盡管Wnt通路、Hippo通路和代謝通路在分析結果中表現出重要性,但缺乏相關研究驗證其在分化過程中的實際分子機制;此外,本研究采用的干細胞系類型較為單一,樣本的多樣性較為局限。未來相關研究可進一步驗證關鍵基因和信號通路的功能,并通過多樣化的分析方法和更廣泛的樣本類型,深入探討干細胞分化的機制,揭示其在臨床應用中的潛力。
本研究使用經典的“兩步法”成功誘導hiPSC和hESC分化為血管周細胞和內皮細胞,通過轉錄組測序技術分析識別了該分化過程中關鍵的DEG與相關通路,為進一步優化干細胞分化流程、提高分化效率提供了科學依據,為血管性疾病的治療提供了新的研究方向和潛在的策略。
視網膜局部組織缺血缺氧形成的病理性新生血管是糖尿病視網膜病變、視網膜靜脈阻塞、早產兒視網膜病變等致盲性視網膜血管疾病的共同病理基礎[1]。新生血管常伴有血管周細胞和內皮細胞丟失、基底膜增厚等病理改變,易發生滲漏和出血,加重視網膜缺血缺氧狀態[2]。近年來研究表明,干細胞在視網膜血管疾病中的應用潛力巨大[3-4],體外培養的人誘導多能干細胞(hiPSC)與胚胎干細胞(hESC)在特定誘導條件下可模擬體內血管發育過程,分化形成周細胞與內皮細胞,通過體內移植可以整合到病變的血管上,發揮修復血管的作用[5];此外,也可以體外培養形成3D血管類器官,用于篩選糖尿病微血管病變的治療藥物[6]。目前誘導干細胞分化為周細胞與內皮細胞的經典方案為先向中胚層方向誘導,再向血管方向誘導的“兩步法”[7]。但該方法耗時長、效率較低,且具體機制尚不明確。本研究對hiPSC與hESC分化為周細胞和內皮細胞過程中的差異表達基因(DEG)進行分析,分析調控分化進程的關鍵分子與信號通路,旨在為優化干細胞分化方案、揭示血管發育機制提供依據。
1 材料和方法
1.1 材料
人尿源性hiPSC細胞系(中山大學中山眼科中心劉春巧教授惠贈);hESC H9細胞系(中國科學院細胞庫)。Matrigel基質膠(美國BioCoat公司);mTeSR培養基、dispase消化液(加拿大StemCell公司);內皮細胞生長培養基-2套裝(EGM-2 kit培養基,瑞士LONZA公司);重組人Activin A蛋白、抗人CD31藻藍蛋白偶聯抗體(CD31-APC)、多聚甲醛(PFA)組織固定液、TRIzol裂解液(美國Thermo Fisher公司);重組人骨形成蛋白(BMP4)、Anti-Caldesmon抗體(英國Abcam公司);血管內皮生長因子(VEGF)、SB431542、CHIR因子、抗人血小板源性生長因子受體β抗體(PDGFR-β)(美國R&D公司);Gelatin基質膠(德國Sigma公司);測序試劑盒(美國Illumina公司)。
1.2 方法
hiPSC、hESC擴增培養和誘導分化。hiPSC、hESC分別接種于Matrigel基質膠包被的6孔板中,加入mTeSR培養基,37 ℃、5%CO2培養箱中進行擴增培養,細胞生長至40%~50%融合時記錄為0 d。加入中胚層分化培養液(mTeSR+25 μg/ml Activin A蛋白+30 μg/ml BMP4蛋白+50 μg/ml VEGF+4 mmol/L CHIR因子)培養3 d,其后更換為血管分化培養液(mTeSR+50 μg/ml VEGF+20 mmol/L SB431542因子)培養7 d。倒置顯微鏡觀察細胞生長狀況與形態變化,拍照記錄。
分離鑒定內皮細胞和周細胞。分別取hiPSC、hESC分化10 d時的細胞制成細胞懸液,加入CD31-APC抗體避光孵育45 min,流式細胞儀分離鑒定內皮細胞,FlowJo軟件分析獲得數據。將含有周細胞的剩余細胞懸液轉移至Gelatin包被的6孔板中,EGM-2 kit培養基繼續培養至周細胞融合。4% PFA組織固定液固定細胞爬片10 min,滴加含周細胞特征性標志物Caldesmon與PDGFR-β的一抗,4 ℃孵育過夜,滴加相應二抗,室溫避光孵育1 h,洗滌封片。熒光顯微鏡下觀察并拍照記錄。
1.3 RNA提取、轉錄組測序
分別收集hiPSC、hESC分化0、4、7、10 d的細胞,TRIzol裂解液裂解細胞,收集上清液,加入0.5 ml異丙醇沉淀RNA,10 000×g離心10 min,75%乙醇洗滌RNA沉淀,10 000×g離心10 min,棄上清,室溫干燥。加入無RNase的溶液溶解RNA,-80 ℃保存。不同細胞系實驗獨立重復3次。
誘導培養0、4、7、10 d(分化記作分化前、早期、中期、晚期)d分別收集hiPSC、hESC進行轉錄組測序。采用帶有Oligo的磁珠富集真核生物mRNA,打斷形成短片段,反轉錄合成cDNA鏈,末端修復后連接測序的接頭,瓊脂糖凝膠電泳對不同大小片段進行選擇,聚合酶鏈反應(PCR)擴增富集cDNA,完成文庫制備。應用Illumina HiSeq2500進行雙端測序。不同細胞系分別獨立重復測序3次。
1.4 DEG篩選和生物信息學分析
測序數據過濾,去除接頭、污染和低質量序列,采用hisat2將高質量序列匹配到參考基因組上。利用FeatureCounts軟件進行基因水平的定量分析,計算每個基因在各樣本中的讀段計數(read count)。標準化后,根據模型進行假設檢驗概率(P值)計算,多重假設檢驗校正,得到調整后的P值。以|log2[差異表達倍數(FC)]|>1且P<0.05為條件篩選DEG[8]。
應用R軟件中pheatmap函數繪制聚類熱圖,分析DEG變化,紅色越深表示DEG表達水平越高,藍色越深表示DEG表達水平越低;VENNY系統(http://bioinfogp.cnb.csic.es/tools/venny/index.html)繪制韋恩圖;R軟件中的phyper函數對分化過程中持續存在差異的DEG進行基因本體(GO,http://www.geneontology.org/)和京都基因與基因組百科全書(KEGG,https://www.kegg.jp/)富集分析,富集顯著性標準為P<0.05。
2 結果
2.1 hiPSC、hESC分化為周細胞和內皮細胞過程中形態變化與標志物表達情況
未分化hiPSC、hESC呈圓形克隆狀生長,邊界清晰,細胞排列緊密。誘導培養4 d時,可見分化的梭形細胞向外爬行生長;10 d時,梭形細胞數量明顯增多,部分區域呈融合生長,且hESC較hiPSC組分化細胞數量更多(圖1)。
圖1
hiPSC、hESC分化過程中形態變化光學顯微鏡像(標尺:500 μm) 1A~1C分別示hiPSC誘導培養0、4、10 d,0 d時,hiPSC呈圓形克隆狀生長,邊界清晰;4 d時,分化細胞向外爬行生長呈梭形;10 d時,梭形細胞數量增多,局部呈融合生長。1D~1F分別示hESC誘導培養0、4、10 d,0、4 d時,形態變化與hiPSC一致;10 d時,分化細胞數量較hiPSC更多 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞
流式細胞儀檢測結果顯示,誘導培養10 d時,hiPSC、hESC中均存在CD31+的內皮細胞(圖2);免疫熒光染色結果顯示,hiPSC、hESC誘導培養的周細胞中均高表達特征性標志物Caldesmon、PDGFR-β(圖3)。
圖2
hiPSC、hESC誘導培養10 d時流式細胞儀檢測像 2A、2B分別示hiPSC、hESC,兩個細胞系均可見CD31+內皮細胞存在 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞;CD31-APC:抗人CD31藻藍蛋白偶聯抗體
圖3
hiPSC、hESC誘導培養10 d時細胞熒光顯微鏡像(標尺:50 μm) 3A、3B分別示hiPSC、hESC,Caldesmon標記的周細胞呈紅色熒光,表達于細胞骨架;PDGFR-β標記的周細胞呈綠色熒光,表達于細胞膜;DAPI標記的細胞核呈藍色熒光 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞;PDGFR-β:血小板衍生生長因子受體-β:DAPI:4',6-二脒基-2-苯基吲哚
2.2 測序數據的分組與質量評估
測序結果顯示,hiPSC、hESC各有平均4.3107的過濾后讀段計數,原始數據Q30比例為93.93%,匹配率均>95.9%(表1)。
2.3 不同分化時間DEG表達譜分析
hiPSC、hESC隨分化時間延長,DEG顯著上調或下調且變化趨勢較為一致(圖4A)。分化中期,GDF3、NANOG、DPPA等干細胞標志性基因顯著下調;分化中期及晚期,周細胞標志基因PDGFR-β、ACTA2、ANGPT2、NDUFA4L2、DLK1,內皮細胞標志基因PECAM1、CLDN5、EMP1、VCAM1、VWF顯著上調(圖4B)。
圖4
hiPSC、hESC不同分化時間DEG分析聚類熱圖(紅色越深表示FC越高,藍色越深表示FC越低) 4A示hiPSC、hESC中大量DEG隨分化時間顯著上調或下調,變化趨勢較為一致;4B示分化中期干細胞標志性基因顯著下調,分化中期及晚期周細胞與內皮細胞標志性基因顯著上調 DEG:差異表達基因;hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞;FC:差異表達倍數
2.4 持續DEG的韋恩分析和GO富集分析
韋恩分析結果顯示,hiPSC與hESC在分化早期、中期、晚期共檢測到持續DEG 491個,其中hiPSC、hESC分別為164、335個(圖5)。hiPSC、hESC同時表達的基因為8個。其中,SLC30A3、LCK、TNFRSF8、PRDM14、GLB1L3持續表達下調;CLEC18C、CLEC18B、F2RL2持續表達上調(表2)。CLEC18C、CLEC18B為C型凝集素結構域家族;F2RL2為蛋白酶激活受體家族;SLC30A3為溶質載體家族;LCK為酪氨酸蛋白激酶家族;TNFRSF8為腫瘤壞死因子受體超家族;PRDM14為轉錄調節子家族;GLB1L3為編碼β-半乳糖樣蛋白。
圖5
hiPSC、hESC不同分化時間的差異表達基因韋恩圖 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞
GO富集分析結果顯示,持續上調表達的DEG主要富集在神經發生、分化、發育蛋白等條目中;持續下調表達的DEG主要富集在膜結構、磷脂代謝過程等條目中(圖6)。
圖6
hiPSC、hESC持續DEG的GO富集分析圖 條柱代表富集GO條目的P值,柱線越高表示P值越小,富集越顯著 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞; DEG:差異表達基因;GO:基因本體
2.5 持續DEG的KEGG通路分析
KEGG通路顯著性富集分析結果顯示,受分化進程影響,hiPSC、hESC中上調表達的基因主要富集于癌癥相關通路、干細胞多能性調節通路、Wnt信號通路、Hippo信號通路;下調表達的基因主要富集于代謝相關通路(圖7)。
圖7
hiPSC、hESC持續DEG的KEGG富集排名前10的信號通路氣泡圖 7A、7B和7C、7D分別示hiPSC、hESC分化過程中排名前10的上調、下調信號通路 hiPSC:人誘導多能干細胞;hESC:人胚胎干細胞;DEG:差異表達基因;KEGG:京都基因與基因組百科全書;ECM:細胞外基質;PI3K-Akt:磷脂酰肌醇3激酶-絲裂原活化蛋白激酶;cAMP:環腺苷酸
3 討論
本研究通過經典的“兩步法”成功誘導hiPSC和hESC分化為血管周細胞和內皮細胞,利用轉錄組測序技術分析識別了該分化過程中關鍵的DEG與相關通路,并分析參與調控分化的關鍵分子,為優化干細胞分化流程提供了科學依據。DEG聚類分析結果顯示,隨分化進程的進行,干細胞標志性基因(如NANOG、GDF3)的表達逐漸下調,表明多能性逐步喪失,而周細胞與內皮細胞標志性基因(如PDGFRB、PECAM1、VWF)則在中晚期顯著上調,支持hiPSC與hESC向特定血管相關類型分化的趨勢。在hiPSC與hESC的分化過程中,共檢測到8個具有持續特異性表達變化的DEG,包括表達上調的CLEC18C、CLEC18B、F2RL2,以及表達下調的SLC30A3、LCK、TNFRSF8、PRDM14、GLB1L3,這些DEG可能通過激活Wnt通路、Hippo通路,抑制代謝通路,解除對干細胞多能性的維持以及影響細胞周期、抑制細胞增殖等作用,促進周細胞與內皮細胞的分化。
CLEC18C、CLEC18B、F2RL2基因的持續上調表達增加了對細胞分化的支持作用。CLEC18C與CLEC18B屬于人C型凝集素家族,在多種細胞類型中參與調控細胞遷移和分化[9]。研究表明,CLEC18C在腫瘤和神經組織中高表達,通過激活Wnt信號通路促進細胞的增殖和遷移[10],提示其在分化過程中可能通過類似途徑支持干細胞向特定細胞類型的分化。F2RL2屬于蛋白酶激活受體家族,其高表達與膠質瘤、口腔鱗癌等腫瘤的預后不良有密切聯系[11],提示該基因在干細胞增殖和維持中也扮演類似關鍵角色。在本研究中,CLEC18C、CLEC18B和F2RL2的上調表達可能通過增強干細胞的增殖和遷移能力,為分化過程中細胞數量的增加提供支持,同時為特定細胞類型的分化奠定基礎。近年癌癥相關通路在干細胞研究中逐漸受到關注,因其在干細胞和癌細胞的自我更新和分化過程中可能共享一些調控機制,如Wnt和Notch通路既參與干細胞的自我更新,也在許多癌癥中被異常激活[12],癌癥相關信號通路的上調可幫助干細胞維持適當的增殖狀態,有助于維持細胞增殖和分化的動態平衡[13]。本研究KEGG通路富集性分析結果顯示,上調的DEG在癌癥通路存在顯著富集,可能反映了干細胞在分化過程中的增殖和存活需求,確保分化過程中有足夠的細胞數量,同時維持分化潛力。
Wnt與Hippo信號通路在調控hiPSC和hESC的分化中也發揮關鍵調控作用。在經典Wnt/β-連環蛋白(β-catenin)依賴型通路中,當Wnt配體與細胞膜上的Frizzled受體結合時,β-catenin得以穩定化并累積在細胞質中,隨后進入細胞核,激活與分化相關的目標基因[14]。這一過程不僅有助于干細胞向中胚層細胞的轉化,還通過上調PECAM1和VWF等標志性基因促進周細胞和內皮細胞的生成[15-17]。同時,Hippo信號通路通過調控YAP/TAZ轉錄共激活因子的活性,確保分化過程中細胞的增殖平衡,當Hippo通路被激活時,YAP/TAZ被磷酸化并保留在細胞質中,從而抑制細胞的增殖[18]。這種調控機制在確保分化過程中細胞數量適度增加的同時,避免了過度增殖帶來的異常增生風險。此外,Hippo通路與Wnt通路在內皮細胞的分化成熟階段具有協同作用,共同調控內皮細胞的生成和血管新生[19-21]。在本研究中,Wnt與Hippo信號通路在hiPSC和hESC分化過程中顯著上調,進一步提示其在血管周細胞和內皮細胞形成中的關鍵作用,我們推測Wnt信號通路的激活使干細胞逐步向血管相關細胞類型轉化,為周細胞和內皮細胞的形成奠定基礎,而Hippo通路的激活確保了細胞在分化過程中不會發生過度增殖,從而維持了分化進程的平衡與穩定。
持續特異性下調表達的LCK、PRDM14、GLB1L3基因在干細胞的多能性喪失中起到關鍵作用。LCK作為非受體酪氨酸激酶,在多種干細胞和造血細胞中高表達,與細胞增殖和存活密切相關[22-23]。LCK高表達于成年小鼠的造血細胞與hESC,且在胚胎干細胞分化為擬胚體過程中LCK表達水平顯著下降[24]。推測在干細胞分化過程中LCK的下調有助于降低細胞的增殖信號,促使細胞從增殖狀態進入分化狀態。PRDM14是維持胚胎干細胞多能性的特異性轉錄調節因子[25],在人原始生殖細胞中敲除該基因后會導致干細胞相關基因顯著下調,并促進分化相關基因上調[26]。其在分化過程中的下調,提示干細胞多能性基因逐漸關閉,為特異性分化路徑的開啟提供了條件。GLB1L3與胚胎干細胞的多能性密切相關[27],推測其下調表達可能通過解除干細胞多能性的維持,從而有利于分化的進行。
TNFRSF8、PRDM14、SLC30A3基因的持續特異性下調有助于抑制細胞周期,防止過度增殖。TNFRSF8編碼的蛋白CD30屬于腫瘤壞死因子受體家族,其下調表達可引起細胞周期蛋白激酶抑制因子CDKN2A、CDKN1A的表達上調,導致細胞阻滯于G1/S與G2/M周期,進而抑制細胞的過度增殖[28]。在293T細胞中,PRDM14通過抑制細胞周期G1/S期轉變,也可影響細胞增殖與遷移[29]。推測TNFRSF8、PRDM14的下調可能通過影響細胞周期,抑制干細胞的過度增殖達到促進分化的作用。SLC30A3編碼鋅轉運蛋白,參與調控細胞內鋅離子平衡,研究表明,鋅離子水平在細胞增殖和分化中具有重要調控作用,胚胎發育早期鋅水平的不足會抑制細胞周期與轉錄組水平,導致細胞增殖減少和分化受阻[30-31]。SLC30A3的下調可能通過減少細胞內鋅離子濃度,影響細胞周期和分裂速度,從而推動分化進程。
GO富集分析結果顯示,下調表達的DEG主要富集于膜結構、磷脂代謝相關生物過程。磷脂作為細胞膜的基本構成成分,參與細胞形態、遷移和增殖的調控[32]。分化過程中,磷脂代謝的下調可能通過調控細胞膜的流動性和穩定性,減少不必要的細胞遷移和增殖,從而增強分化過程的專一性和穩定性。此外,磷脂代謝的下調也可能通過減少細胞內能量的過度消耗,優化分化過程中對能量的需求。代謝通路在干細胞的多能性維持和分化中也扮演重要角色,代謝狀態的改變往往伴隨著干細胞狀態變化,研究表明,糖酵解和氧化磷酸化的平衡以及脂質代謝水平對干細胞分化具有深遠影響[33]。在本研究中,代謝相關通路的顯著下調可能與細胞代謝活動的降低及能量利用效率的提升有關,以滿足分化過程中對代謝的不同需求。
本研究存在的局限性包括:首先,研究結果主要依賴于轉錄組學數據分析,關鍵基因的功能未能通過基因敲除或過表達實驗進行驗證;其次,盡管Wnt通路、Hippo通路和代謝通路在分析結果中表現出重要性,但缺乏相關研究驗證其在分化過程中的實際分子機制;此外,本研究采用的干細胞系類型較為單一,樣本的多樣性較為局限。未來相關研究可進一步驗證關鍵基因和信號通路的功能,并通過多樣化的分析方法和更廣泛的樣本類型,深入探討干細胞分化的機制,揭示其在臨床應用中的潛力。
本研究使用經典的“兩步法”成功誘導hiPSC和hESC分化為血管周細胞和內皮細胞,通過轉錄組測序技術分析識別了該分化過程中關鍵的DEG與相關通路,為進一步優化干細胞分化流程、提高分化效率提供了科學依據,為血管性疾病的治療提供了新的研究方向和潛在的策略。