靶向p21活化激酶1(PAK1)是胰腺癌治療的新策略。復方苦參注射液中含有多種抗胰腺癌成分,但具體作用靶點未知。本研究基于分子對接的虛擬篩選發現,復方苦參注射液中的14α-羥基苦參堿與PAK1的別構調節位點具有較高的結合自由能。分子動力學模擬發現,14α-羥基苦參堿使PAK1的α1和α2螺旋向外延伸形成一個較深的別構調節口袋,同時誘導三磷酸腺苷(ATP)結合口袋處的β-折疊向內關閉,導致ATP結合口袋處于“半閉合”狀態而失活。當去除14α-羥基苦參堿后,PAK1從失活構象向活性構象轉變,因此推測,14α-羥基苦參堿可能是PAK1的可逆別構調節抑制劑。本研究采用現代技術方法對中藥活性成分進行研究,為天然產物的開發利用及尋找新的胰腺癌治療方案奠定基礎。
引用本文: 林琳, 常靜杰, 田耀洲, 陳姣. 計算機輔助復方苦參注射液中抑制p21活化激酶1的活性成分預測及分子機制研究. 生物醫學工程學雜志, 2024, 41(2): 313-320. doi: 10.7507/1001-5515.202306011 復制
版權信息: ?四川大學華西醫院華西期刊社《生物醫學工程學雜志》版權所有,未經授權不得轉載、改編
0 引言
胰腺癌作為“癌中之王”,發現難、進展快、致死率高[1]。現有藥物的局限性以及逐漸升高的死亡率促使人們不斷尋求更有效的胰腺癌治療方案。p21活化激酶1(p21-activated kinase 1,PAK1)是一類非受體型絲氨酸/蘇氨酸蛋白激酶,研究發現其在多種癌癥組織(如胰腺癌、乳腺癌、肺癌、皮膚癌等)中均出現高表達及活化[2-5]。活化的PAK1參與調控多條信號通路,如通過磷酸化B細胞淋巴瘤2(B cell lymphoma-2,Bcl-2)蛋白相關死亡促進因子(Bcl-2–associated agonist of cell death,BAD)阻止其與Bcl-2結合從而抑制細胞凋亡;通過磷酸化膜突蛋白—埃茲蛋白—根蛋白樣蛋白(moesin-ezrin-radixin–like protein,Merlin)使其從細胞膜表面移位而抑制其功能,同時也會促進細胞外調節蛋白激酶信號通路的激活;通過磷酸化β-連環蛋白(β-catenin)促進其進入細胞核并發揮轉錄活性而調控無翅和整合-1(Wingless and integration-1,WNT)蛋白/β?catenin信號通路,上調髓細胞增生蛋白和細胞周期蛋白D的表達;PAK1還能激活磷脂酰肌醇3-激酶信號通路,上調缺氧誘導因子1α的表達等[6-8]。由此可見,PAK1在細胞的增殖、存活、遷移及血管生成等相關信號通路中發揮中樞作用,這給靶向PAK1治療相關癌癥提供了科學依據。
前期研究發現,臨床上胰腺癌患者的腫瘤組織中PAK1的表達水平明顯高于相鄰的正常胰腺組織;而且,低表達PAK1的胰腺癌患者總生存期明顯長于高表達PAK1的患者,說明PAK1的表達水平影響胰腺癌的發展、預后及生存周期[9]。其他多項研究也證實,PAK1在胰腺癌組織樣本及細胞株中均出現高表達[2-3, 10-11];抑制PAK1的活性能夠抑制胰腺癌細胞的生長及存活,促進細胞凋亡,并且能夠提高胰腺癌細胞對吉西他濱的敏感性[12-13]。以上研究均說明,靶向PAK1可以作為胰腺癌治療的新手段,將給胰腺癌患者帶來新的曙光。
復方苦參注射液是一種從苦參和土茯苓中提取的活性物質組成的復方制劑,臨床上用于輔助治療各種癌癥,包括胰腺癌、肝癌、食管癌等[14-17],與常規化療藥聯用能增強抗腫瘤療效,降低化療毒性,提高患者的生活質量。體外實驗表明,復方苦參注射液的多種有效成分能抑制胰腺癌細胞增殖、侵襲和轉移,促進細胞凋亡并誘導自噬[18-19],還能增強胰腺癌耐藥細胞株對化療藥的敏感性[20],但具體作用靶點是否為PAK1仍然未知。鑒于PAK1在胰腺癌細胞的增殖、遷移和凋亡等相關信號通路中發揮的中樞作用,本研究將基于分子對接的虛擬篩選和分子動力學(molecular dynamics, MD)模擬預測復方苦參注射液中靶向PAK1的抑制劑并研究其機制,為天然活性成分的開發利用及尋找新的胰腺癌治療方案奠定基礎。
1 材料與方法
本研究采用計算機輔助藥物設計軟件Schr?dinger 2015-3(Schr?dinger,LLC,美國)進行分子對接、中藥活性數據庫的構建和靶標蛋白結構的準備,采用MD模擬軟件Amber 14(University of California,美國)進行體系準備、MD模擬及數據分析,采用分子可視化軟件PyMOL 1.7.4(Schr?dinger,LLC,美國)作圖。
1.1 復方苦參注射液活性成分數據庫的構建
西北農林科技大學開發和維護的公開中藥系統藥理學(traditional Chinese medicine systems pharmacology,TCMSP)分析平臺收集了499種中藥、29 384種成分。本研究通過TCMSP平臺檢索苦參和土茯苓的活性成分,建立復方苦參注射液活性成分數據庫。在Schr?dinger 2015-3軟件的配體準備模塊中對數據庫的小分子進行質子化及能量優化。
1.2 靶標蛋白PAK1結構的獲取和預處理
本研究使用的人源PAK1與別構調節抑制劑結合的復合物結構下載自美國羅格斯大學和加州大學圣地亞哥分校–圣地亞哥超級計算機中心創建的公開蛋白質數據庫(protein data bank,PDB),PDB編號為4ZJJ。該數據庫是國際上最完整的蛋白質三維結構數據庫,包含了214 226種實驗測定的三維結構和1 068 577種計算預測的結構模型。將PAK1的結構導入Schr?dinger 2015-3軟件對其進行質子化及能量優化。定義結構中已知抑制劑(S)-N-(叔丁基)-3-((2-氯-5-乙基-8-氟-二苯二氮卓-11-基)氨基)吡咯烷-1-甲酰胺的結合位點為虛擬篩選位點,生成格點文件,用于復方苦參注射液中靶向PAK1抑制劑的虛擬篩選。
1.3 基于分子對接的虛擬篩選
在Schr?dinger 2015-3軟件的分子對接模塊Glide(Schr?dinger, LLC,美國)中進行PAK1抑制劑的虛擬篩選,對接打分采用標準精度,對接后進行能量優化。計算苦參和土茯苓中對接打分排前10名的分子與PAK1的廣義玻恩和表面積相結合的分子力學能量(molecular mechanics energies combined with the generalized Born and surface area,MM/GBSA)。選取與PAK1的MM/GBSA結合自由能最高的分子,采用MD模擬方法研究其抑制PAK1的機制。
1.4 MD模擬
結構準備:將以下三個體系進行MD模擬:① 篩選到的抑制劑與PAK1結合的結構;② PAK1的結構;③ 三磷酸腺苷(adenosine triphosphate,ATP)與PAK1結合的活性構象結構(PDB編號: 3Q53)。體系準備方法參考本課題組以前發表的文獻[21]。
MD模擬:在Amber 14軟件中,首先基于最陡下降法和共軛梯度法對各體系進行兩步能量最小化,以減少原子間的不良接觸。之后在恒容條件下,控制時間于450 ps內,將體系溫度由? 273 ℃升至27 ℃,限制常數設為10.0 kcal/(mol·?-2)。然后解除限制,在恒溫(27 ℃)恒壓(100 000 Pa)下進行500 ps的MD模擬以平衡體系。最后,設定步長為2 fs進行300 ns的MD模擬,其他參數設置參考文獻[21]。
數據分析:使用Amber 14軟件的軌跡數據分析模塊cpptraj(University of California,美國)計算均方根偏差(root-mean-square deviation,RMSD)、RMSD矩陣和原子之間的距離。使用PyMOL 1.7.4軟件的模式向量分析腳本modevectors.py(Schr?dinger,LLC,美國)可視化兩個指定體系之間的運動方向。
2 結果
2.1 構建復方苦參注射液活性成分數據庫
復方苦參注射液是由苦參和土茯苓按照7∶3比例提取的活性成分組成。苦參的主要化學成分是生物堿類和黃酮類,土茯苓的主要化學成分包括甾醇類、黃酮(苷)類和揮發油類等。通過TCMSP平臺檢索苦參和土茯苓中已報道的化學結構,其中苦參中檢索到113個分子,土茯苓中檢索到74個分子,主要成分類型如表1所示。
2.2 14α-羥基苦參堿與PAK1具有較高的結合自由能
PAK1別構調節抑制劑具有高選擇性、高活性的特點,本研究基于分子對接方法從復方苦參注射液中虛擬篩選出能別構調節PAK1的活性成分。從苦參和土茯苓中分別篩選出對接打分排前10名的分子,如表2所示。計算這些分子與PAK1的結合自由能MM/GBSA,發現苦參中與PAK1結合自由能最高的分子是14α-羥基苦參堿[TCMSP編號:分子(molecule,MOL)006561],MM/GBSA為? 49.13 kcal/mol;土茯苓中與PAK1結合自由能最高的分子是異黃杞苷(TCMSP編號:MOL004567),MM/GBSA為? 49.24 kcal/mol,如表2所示。
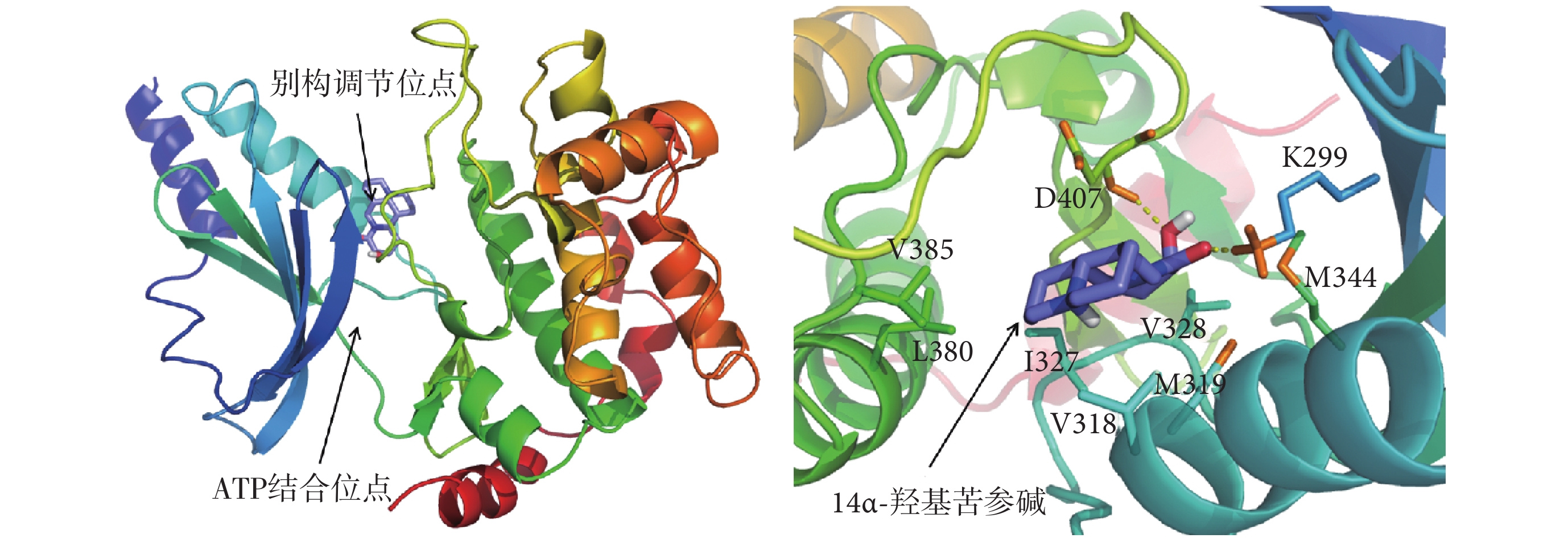
由于苦參生物堿類分子是復方苦參注射液質量控制的主要成分,且含量較高[22],因此選取苦參中與PAK1結合自由能最高的分子—14α-羥基苦參堿,研究其抑制PAK1的結構生物學機制。如圖1所示,PAK1的別構調節位點位于ATP結合位點的背對位置,14α-羥基苦參堿結合于PAK1的別構調節位點,與第299位的賴氨酸(lysine299,K299)和407位的天冬氨酸(aspartic acid407,D407)形成氫鍵作用,與第318位的纈氨酸(valine318,V318)、319位的甲硫氨酸(methionine319,M319)、327位的異亮氨酸(isoleucine327,I327)、328位的纈氨酸(valine328,V328)、344位的甲硫氨酸(methionine344,M344)、380位的亮氨酸(leucine380,L380)和385位的纈氨酸(valine385,V385)等氨基酸形成疏水作用。14α-羥基苦參堿和用于定義虛擬篩選位點的別構調節抑制劑(S)-N-(叔丁基)-3-((2-氯-5-乙基-8-氟-二苯二氮卓-11-基)氨基)吡咯烷-1-甲酰胺(對PAK1具有高抑制活性和選擇性)與PAK1的結合模式在氫鍵作用上略有差異,表現在后者與PAK1的K299和第315位的谷氨酸(glutamic acid315,E315)形成氫鍵作用,其他作用模式類似[23]。
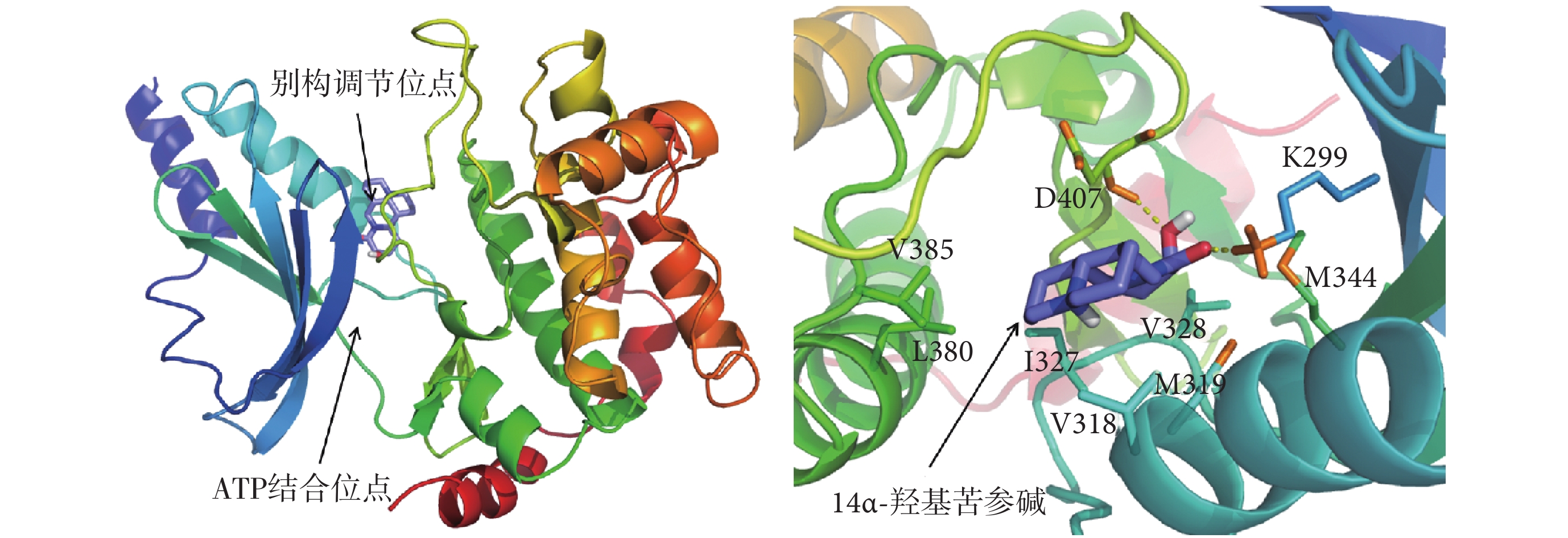 圖1
14α-羥基苦參堿與PAK1的結合模式
Figure1.
The binding mode of 14α-hydroxymatrine with PAK1
圖1
14α-羥基苦參堿與PAK1的結合模式
Figure1.
The binding mode of 14α-hydroxymatrine with PAK1
2.3 14α-羥基苦參堿將PAK1鎖定在失活的“半閉合”構象
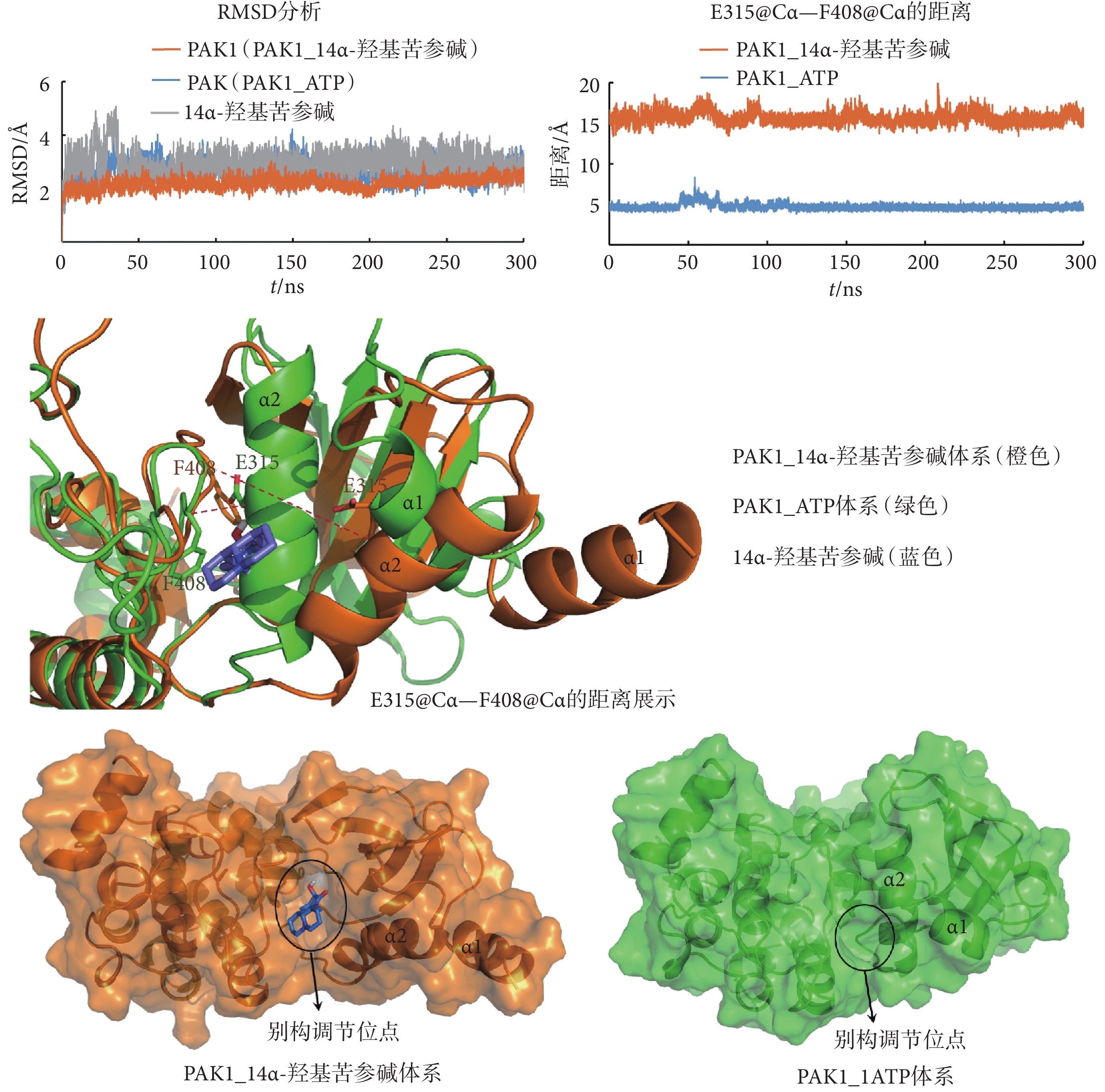
為研究14α-羥基苦參堿抑制PAK1的分子機制,將14α-羥基苦參堿與PAK1結合的復合物結構(PAK1_14α-羥基苦參堿)進行300 ns的MD模擬,以PAK1與ATP結合的活性PAK1體系(PAK1_ATP)為對照。RMSD結果如圖2所示,50 ns之后14α-羥基苦參堿和兩個體系中的PAK1均達到平衡狀態。第408位的苯丙氨酸(phenylalanine408,F408)和E315是PAK1的別構調節口袋中分別靠內、外兩側的關鍵氨基酸,二者之間的距離可以反映別構調節口袋的大小和深度。通過MD模擬發現,14α-羥基苦參堿與PAK1結合之后,形成被14α-羥基苦參堿抑制的PAK1,其中PAK1的α1和α2螺旋向外延伸,使得E315和F408的α碳原子(Cα)之間(E315@Cα—F408@Cα)的距離(15.53 ?)明顯大于PAK1_ATP活性構象中E315@Cα—F408@Cα的距離(4.60 ?),如圖2、表3所示。并且,PAK1_14α-羥基苦參堿結構中F408的側鏈苯環發生轉向,由伸向別構調節位點一側轉到伸向ATP結合位點一側,說明14α-羥基苦參堿誘導PAK1形成一個較深的別構調節口袋以便更好地與之結合,而PAK1_ATP的活性構象中別構調節口袋則較淺,不易結合小分子藥物。
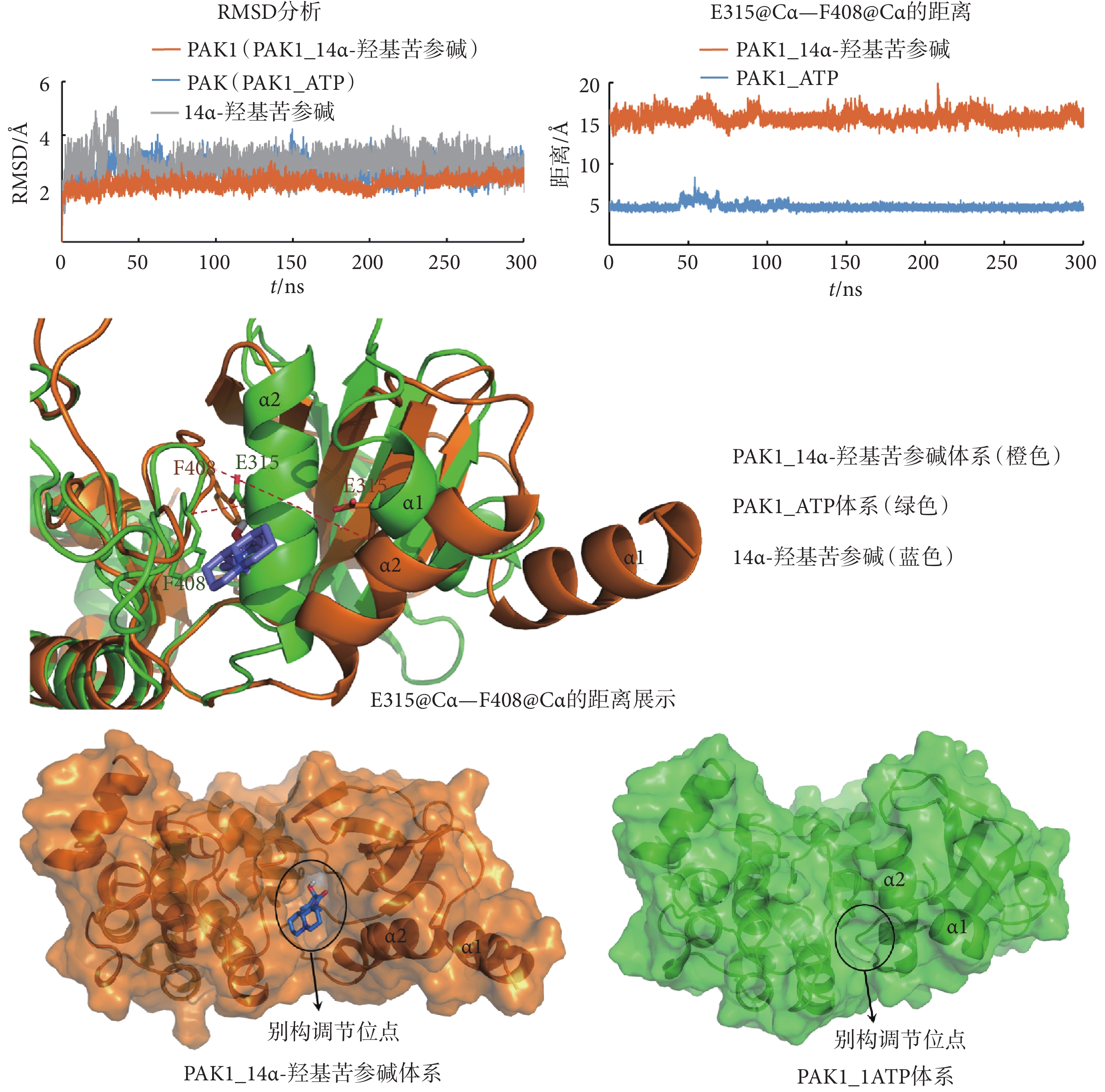 圖2
MD模擬分析PAK1_14α-羥基苦參堿和PAK1_ATP體系的別構調節位點
Figure2.
Analysis of the allosteric site in PAK1_14α-hydroxymatrine and PAK1_ATP systems by MD simulations
圖2
MD模擬分析PAK1_14α-羥基苦參堿和PAK1_ATP體系的別構調節位點
Figure2.
Analysis of the allosteric site in PAK1_14α-hydroxymatrine and PAK1_ATP systems by MD simulations
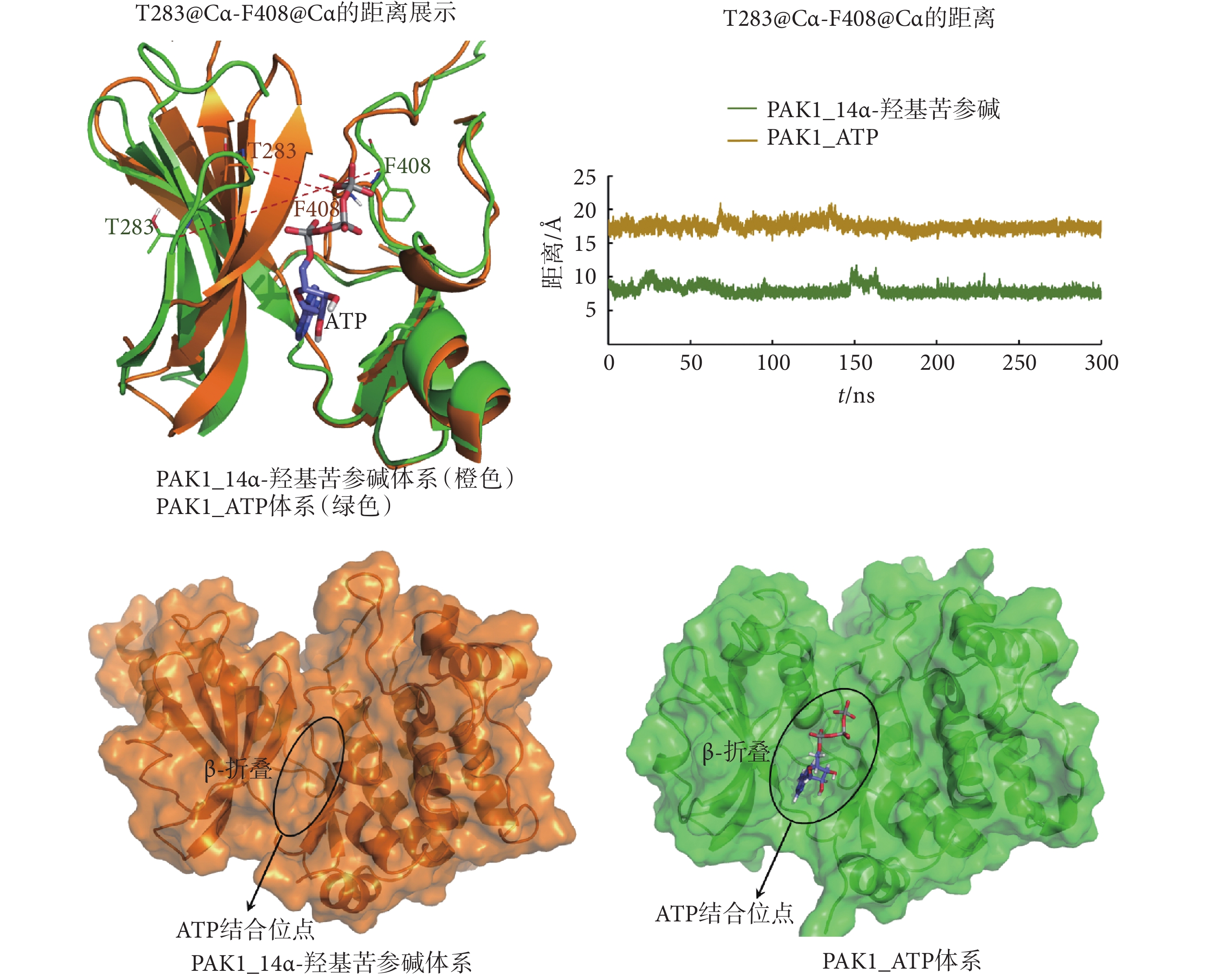
第283位的蘇氨酸(threonine283,T283)和F408是PAK1的ATP口袋中的兩個關鍵氨基酸,它們之間的距離能反映ATP結合口袋的開放程度。通過MD模擬發現,14α-羥基苦參堿的結合使PAK1的ATP結合口袋處的β-折疊向內關閉,導致T283@Cα—F408@Cα的距離(7.69 ?)明顯小于PAK1_ATP活性構象中T283@Cα—F408@Cα的距離(17.32 ?),如圖3、表3所示,使ATP結合口袋處于“半閉合”狀態。此外,14α-羥基苦參堿的結合使PAK1的F408的側鏈苯環轉向ATP結合口袋一側,導致ATP結合口袋變淺,不易與ATP結合而使PAK1失活,如圖3所示。
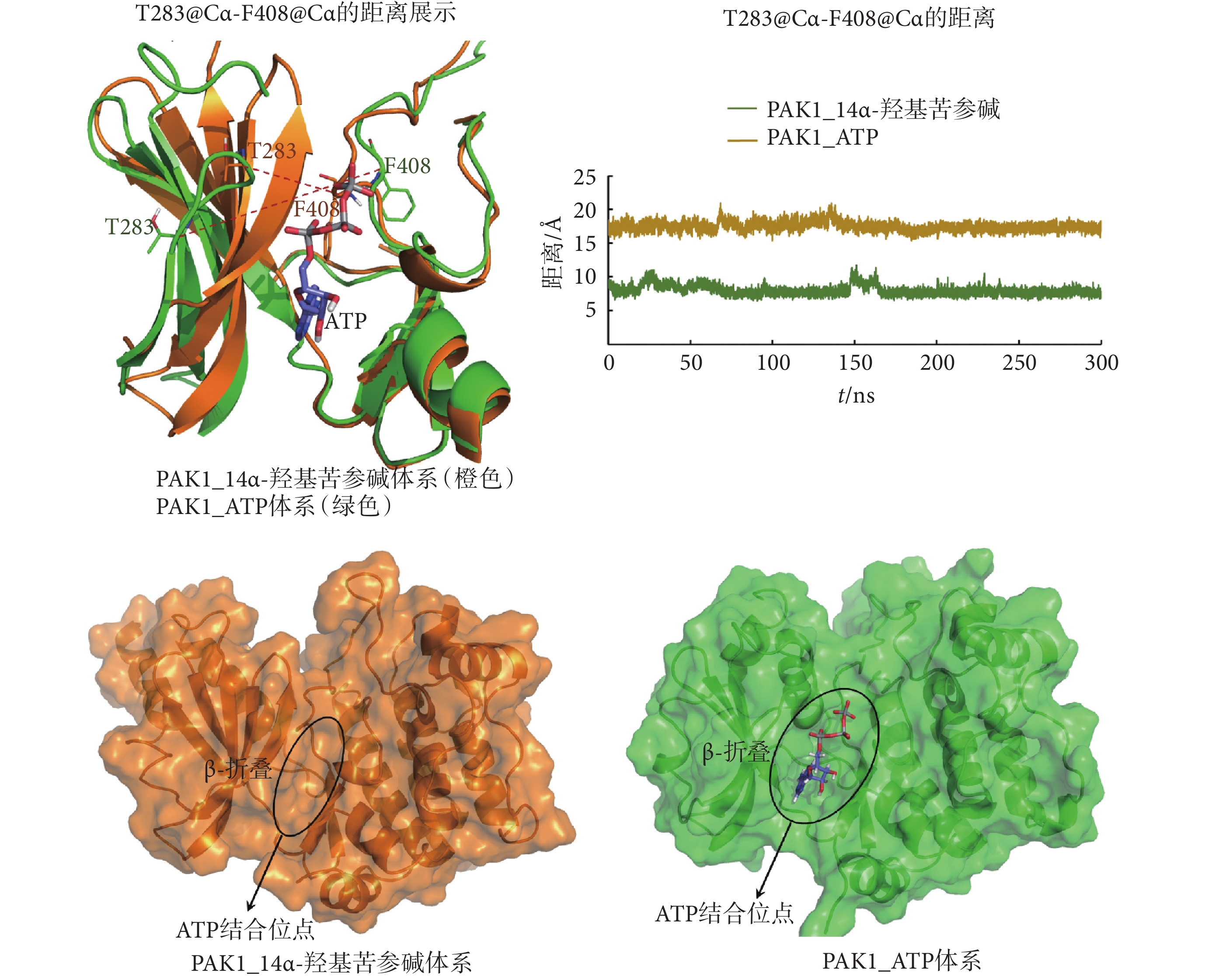 圖3
MD分析PAK1_14α-羥基苦參堿和PAK1_ATP體系中的ATP結合位點
Figure3.
Analysis of the ATP binding site in PAK1_14α-hydroxymatrine and PAK1_ATP systems by MD simulations
圖3
MD分析PAK1_14α-羥基苦參堿和PAK1_ATP體系中的ATP結合位點
Figure3.
Analysis of the ATP binding site in PAK1_14α-hydroxymatrine and PAK1_ATP systems by MD simulations
2.4 去除14α-羥基苦參堿使PAK1向活性構象轉變
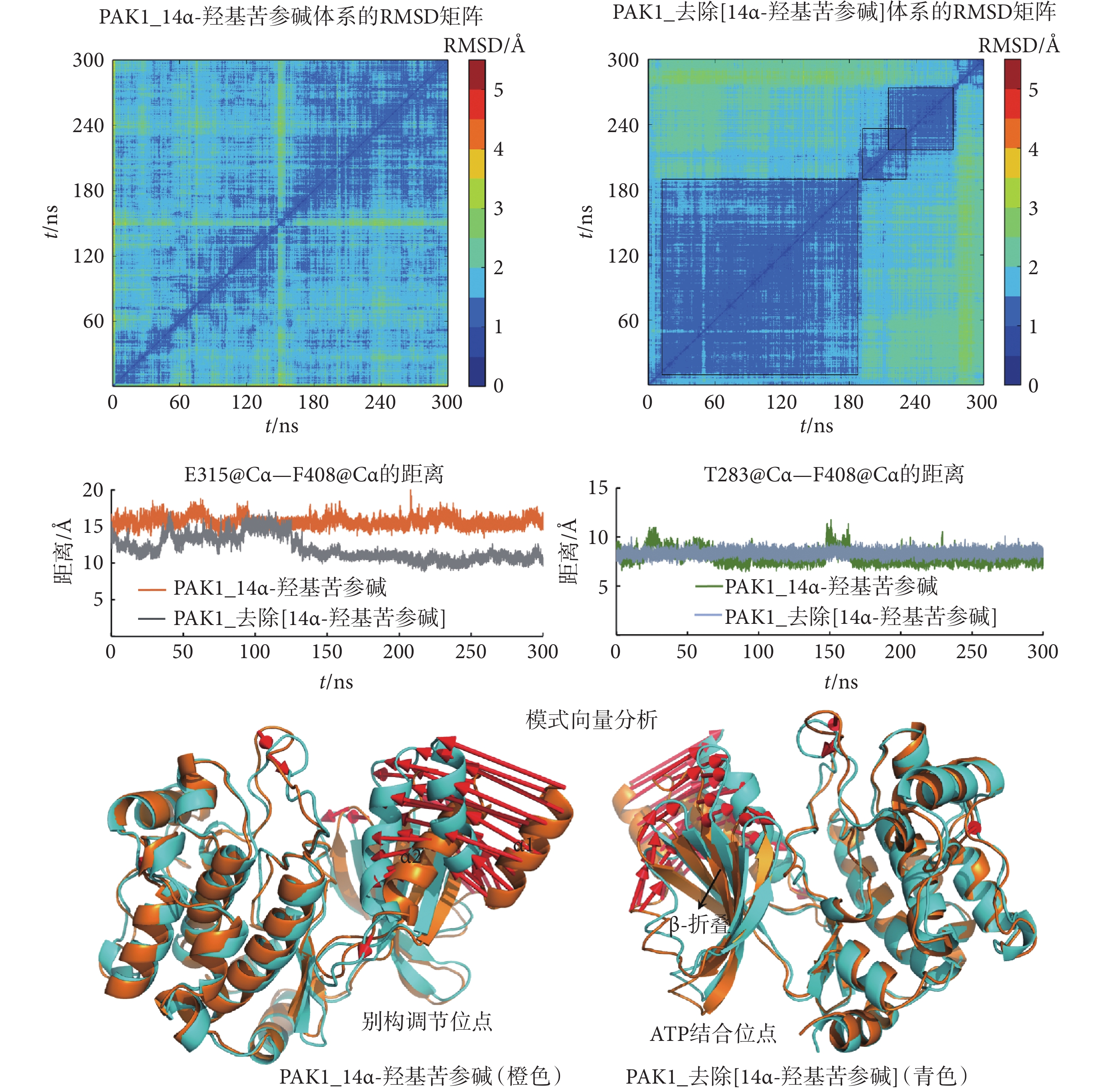
為了驗證以上預測,本研究將14α-羥基苦參堿從PAK1_14α-羥基苦參堿復合物中去除(PAK1_去除[14α-羥基苦參堿]),通過MD模擬研究PAK1的構象變化。如圖4的RMSD矩陣分析所示,去除14α-羥基苦參堿之后,PAK1的構象發生了一系列變化。分析關鍵氨基酸之間的距離發現,去除14α-羥基苦參堿之后,PAK1的E315@Cα—F408@Cα的距離明顯減小(10.81 ?),而T283@Cα—F408@Cα的距離有所增加(8.40 ?),如圖4、表3所示,說明PAK1的別構調節口袋變小,而ATP結合口袋變大。將PAK1_去除[14α-羥基苦參堿]和PAK1_14α-羥基苦參堿的結構進行疊合,采用模式向量分析發現,去除14α-羥基苦參堿之后,別構調節位點處的α1和α2螺旋明顯向酶的中心結構靠攏,而ATP結合位點處的β-折疊有向外開放的趨勢,如圖4所示。這些結果均說明,去除14α-羥基苦參堿之后,PAK1逐漸從失活構象向活性構象轉變。
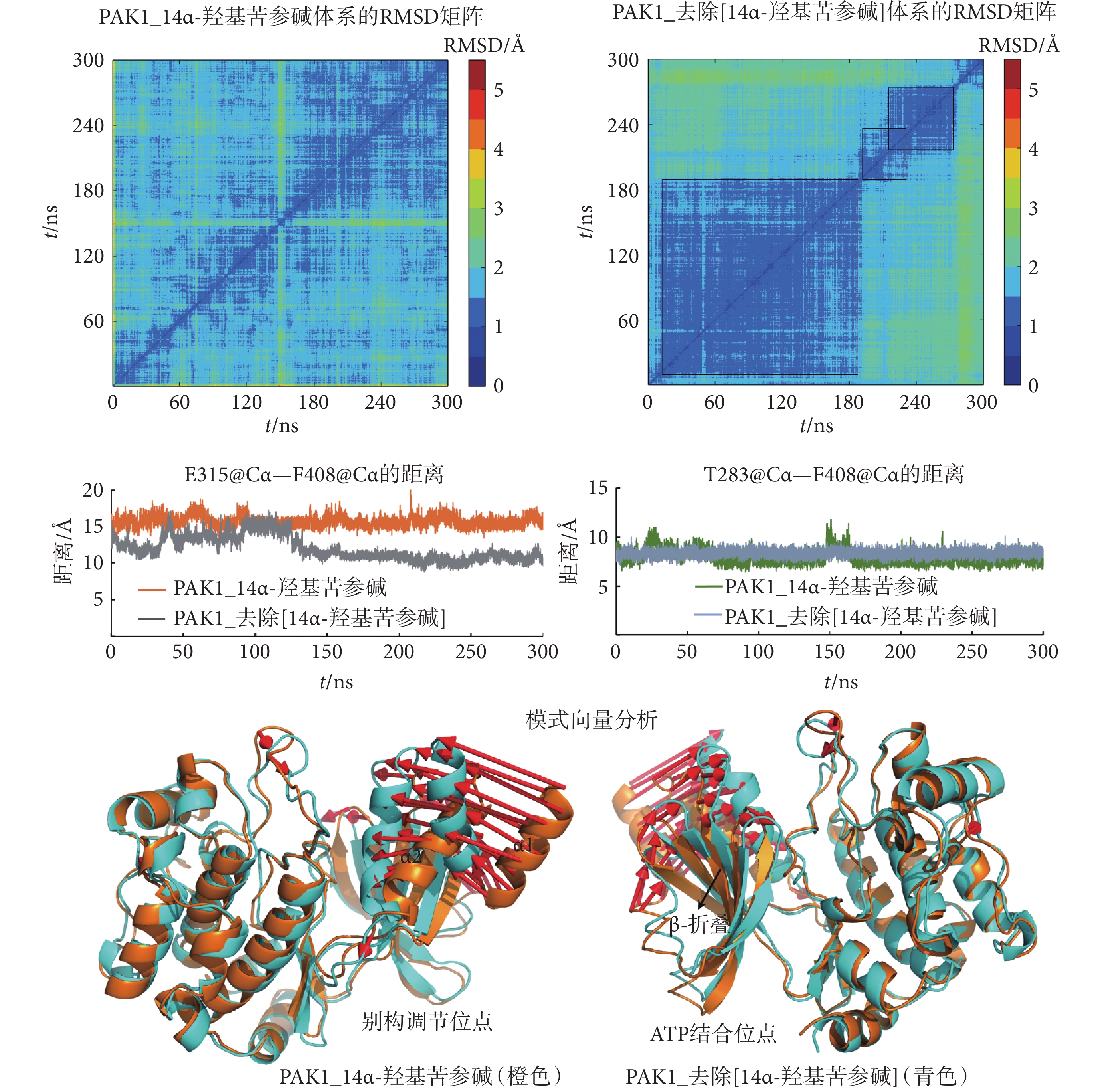 圖4
MD模擬PAK1_去除[14α-羥基苦參堿]體系的別構調節位點和ATP結合位點的變化
Figure4.
The changes of the allosteric site and ATP binding site of PAK1_remove[14α-hydroxymatrine] system by MD simulations
圖4
MD模擬PAK1_去除[14α-羥基苦參堿]體系的別構調節位點和ATP結合位點的變化
Figure4.
The changes of the allosteric site and ATP binding site of PAK1_remove[14α-hydroxymatrine] system by MD simulations
3 討論
胰腺癌具有發現難、進展快、預后不良的特點,臨床診療迫切需要更好的生物標志物和潛在的藥物靶點。PAK1在多種人類腫瘤組織中過度表達和(或)過度激活[24],調節復雜的信號轉導網絡。多項研究表明,PAK1是調控胰腺癌細胞生長的關鍵因子,其表達水平影響胰腺癌的發展、預后及生存周期;抑制PAK1的活性能夠抑制胰腺癌細胞的生長及存活,促進細胞凋亡[3-7]。因此,PAK1被認為是一個潛在的胰腺癌治療靶點,目前已有一些抑制劑正在研發。
復方苦參注射液在臨床上被廣泛用于輔助治療惡性腫瘤及腹腔積液等并發癥,其活性成分苦參生物堿已被證實具有抗胰腺癌活性,能抑制多種胰腺癌細胞的增殖、遷移和侵襲,誘導細胞凋亡[25-27],還能增強胰腺癌耐藥細胞株對化療藥的敏感性[20],但具體作用靶點未知。鑒于PAK1在胰腺癌發生、發展中的重要作用,本研究篩選了復方苦參注射液中可能靶向PAK1的活性成分。結果發現,14α-羥基苦參堿與PAK1的別構調節位點具有較高的結合自由能,作用的關鍵氨基酸主要有K299、D407、V318、M319、I327、M344、L380和V385等。
通常,別構調節抑制劑具有高選擇性、高活性的特點,它們結合于酶的非催化位點,引起酶的構象發生可逆改變,進而改變酶的活性狀態。本研究通過MD模擬進一步研究了14α-羥基苦參堿抑制PAK1的結構生物學機制。發現14α-羥基苦參堿與PAK1結合之后,別構調節位點的α1和α2螺旋向外延伸,F408和E315之間的距離增大,并且F408的側鏈苯環由別構調節位點一側轉向ATP結合位點一側,使得PAK1形成一個較深的別構調節口袋,從而更好地結合小分子藥物。同時,14α-羥基苦參堿誘導PAK1的ATP結合口袋處的β-折疊向內關閉,使得T283和F408之間的距離減小,導致PAK1的ATP結合口袋處于“半閉合”狀態。此外,F408的側鏈苯環轉向ATP結合口袋一側,使ATP結合口袋變淺而不易結合ATP,導致PAK1失活。
將14α-羥基苦參堿從PAK1中去除,通過MD模擬發現PAK1的構象發生了一系列變化。別構調節位點處F408和E315之間的距離明顯減小,而ATP結合位點處T283和F408之間的距離有所增加。結構疊合后的模式向量分析發現,去除14α-羥基苦參堿之后,別構調節位點處的α1和α2螺旋向PAK1的中心結構靠攏,而ATP結合位點處的β-折疊有向外開放的趨勢。這些結果均說明,去除14α-羥基苦參堿之后,PAK1從失活構象向活性構象轉變,提示14α-羥基苦參堿可能是一個可逆的PAK1別構調節抑制劑。
綜上所述,本研究通過虛擬篩選和MD模擬發現,復方苦參注射液中的14α-羥基苦參堿可能是PAK1的別構調節抑制劑,能誘導ATP結合口袋處于“半閉合”狀態而使PAK1失去酶活性。本研究可為天然活性成分的開發利用及尋找新的胰腺癌治療方案奠定基礎。
重要聲明
利益沖突:所有作者均聲明不存在利益沖突。
作者貢獻聲明:林琳、常靜杰和陳姣共同負責本研究中數據庫的構建、分子對接和分子動力學模擬,陳姣和田耀洲負責數據分析指導,林琳、陳姣負責論文寫作與修改。
0 引言
胰腺癌作為“癌中之王”,發現難、進展快、致死率高[1]。現有藥物的局限性以及逐漸升高的死亡率促使人們不斷尋求更有效的胰腺癌治療方案。p21活化激酶1(p21-activated kinase 1,PAK1)是一類非受體型絲氨酸/蘇氨酸蛋白激酶,研究發現其在多種癌癥組織(如胰腺癌、乳腺癌、肺癌、皮膚癌等)中均出現高表達及活化[2-5]。活化的PAK1參與調控多條信號通路,如通過磷酸化B細胞淋巴瘤2(B cell lymphoma-2,Bcl-2)蛋白相關死亡促進因子(Bcl-2–associated agonist of cell death,BAD)阻止其與Bcl-2結合從而抑制細胞凋亡;通過磷酸化膜突蛋白—埃茲蛋白—根蛋白樣蛋白(moesin-ezrin-radixin–like protein,Merlin)使其從細胞膜表面移位而抑制其功能,同時也會促進細胞外調節蛋白激酶信號通路的激活;通過磷酸化β-連環蛋白(β-catenin)促進其進入細胞核并發揮轉錄活性而調控無翅和整合-1(Wingless and integration-1,WNT)蛋白/β?catenin信號通路,上調髓細胞增生蛋白和細胞周期蛋白D的表達;PAK1還能激活磷脂酰肌醇3-激酶信號通路,上調缺氧誘導因子1α的表達等[6-8]。由此可見,PAK1在細胞的增殖、存活、遷移及血管生成等相關信號通路中發揮中樞作用,這給靶向PAK1治療相關癌癥提供了科學依據。
前期研究發現,臨床上胰腺癌患者的腫瘤組織中PAK1的表達水平明顯高于相鄰的正常胰腺組織;而且,低表達PAK1的胰腺癌患者總生存期明顯長于高表達PAK1的患者,說明PAK1的表達水平影響胰腺癌的發展、預后及生存周期[9]。其他多項研究也證實,PAK1在胰腺癌組織樣本及細胞株中均出現高表達[2-3, 10-11];抑制PAK1的活性能夠抑制胰腺癌細胞的生長及存活,促進細胞凋亡,并且能夠提高胰腺癌細胞對吉西他濱的敏感性[12-13]。以上研究均說明,靶向PAK1可以作為胰腺癌治療的新手段,將給胰腺癌患者帶來新的曙光。
復方苦參注射液是一種從苦參和土茯苓中提取的活性物質組成的復方制劑,臨床上用于輔助治療各種癌癥,包括胰腺癌、肝癌、食管癌等[14-17],與常規化療藥聯用能增強抗腫瘤療效,降低化療毒性,提高患者的生活質量。體外實驗表明,復方苦參注射液的多種有效成分能抑制胰腺癌細胞增殖、侵襲和轉移,促進細胞凋亡并誘導自噬[18-19],還能增強胰腺癌耐藥細胞株對化療藥的敏感性[20],但具體作用靶點是否為PAK1仍然未知。鑒于PAK1在胰腺癌細胞的增殖、遷移和凋亡等相關信號通路中發揮的中樞作用,本研究將基于分子對接的虛擬篩選和分子動力學(molecular dynamics, MD)模擬預測復方苦參注射液中靶向PAK1的抑制劑并研究其機制,為天然活性成分的開發利用及尋找新的胰腺癌治療方案奠定基礎。
1 材料與方法
本研究采用計算機輔助藥物設計軟件Schr?dinger 2015-3(Schr?dinger,LLC,美國)進行分子對接、中藥活性數據庫的構建和靶標蛋白結構的準備,采用MD模擬軟件Amber 14(University of California,美國)進行體系準備、MD模擬及數據分析,采用分子可視化軟件PyMOL 1.7.4(Schr?dinger,LLC,美國)作圖。
1.1 復方苦參注射液活性成分數據庫的構建
西北農林科技大學開發和維護的公開中藥系統藥理學(traditional Chinese medicine systems pharmacology,TCMSP)分析平臺收集了499種中藥、29 384種成分。本研究通過TCMSP平臺檢索苦參和土茯苓的活性成分,建立復方苦參注射液活性成分數據庫。在Schr?dinger 2015-3軟件的配體準備模塊中對數據庫的小分子進行質子化及能量優化。
1.2 靶標蛋白PAK1結構的獲取和預處理
本研究使用的人源PAK1與別構調節抑制劑結合的復合物結構下載自美國羅格斯大學和加州大學圣地亞哥分校–圣地亞哥超級計算機中心創建的公開蛋白質數據庫(protein data bank,PDB),PDB編號為4ZJJ。該數據庫是國際上最完整的蛋白質三維結構數據庫,包含了214 226種實驗測定的三維結構和1 068 577種計算預測的結構模型。將PAK1的結構導入Schr?dinger 2015-3軟件對其進行質子化及能量優化。定義結構中已知抑制劑(S)-N-(叔丁基)-3-((2-氯-5-乙基-8-氟-二苯二氮卓-11-基)氨基)吡咯烷-1-甲酰胺的結合位點為虛擬篩選位點,生成格點文件,用于復方苦參注射液中靶向PAK1抑制劑的虛擬篩選。
1.3 基于分子對接的虛擬篩選
在Schr?dinger 2015-3軟件的分子對接模塊Glide(Schr?dinger, LLC,美國)中進行PAK1抑制劑的虛擬篩選,對接打分采用標準精度,對接后進行能量優化。計算苦參和土茯苓中對接打分排前10名的分子與PAK1的廣義玻恩和表面積相結合的分子力學能量(molecular mechanics energies combined with the generalized Born and surface area,MM/GBSA)。選取與PAK1的MM/GBSA結合自由能最高的分子,采用MD模擬方法研究其抑制PAK1的機制。
1.4 MD模擬
結構準備:將以下三個體系進行MD模擬:① 篩選到的抑制劑與PAK1結合的結構;② PAK1的結構;③ 三磷酸腺苷(adenosine triphosphate,ATP)與PAK1結合的活性構象結構(PDB編號: 3Q53)。體系準備方法參考本課題組以前發表的文獻[21]。
MD模擬:在Amber 14軟件中,首先基于最陡下降法和共軛梯度法對各體系進行兩步能量最小化,以減少原子間的不良接觸。之后在恒容條件下,控制時間于450 ps內,將體系溫度由? 273 ℃升至27 ℃,限制常數設為10.0 kcal/(mol·?-2)。然后解除限制,在恒溫(27 ℃)恒壓(100 000 Pa)下進行500 ps的MD模擬以平衡體系。最后,設定步長為2 fs進行300 ns的MD模擬,其他參數設置參考文獻[21]。
數據分析:使用Amber 14軟件的軌跡數據分析模塊cpptraj(University of California,美國)計算均方根偏差(root-mean-square deviation,RMSD)、RMSD矩陣和原子之間的距離。使用PyMOL 1.7.4軟件的模式向量分析腳本modevectors.py(Schr?dinger,LLC,美國)可視化兩個指定體系之間的運動方向。
2 結果
2.1 構建復方苦參注射液活性成分數據庫
復方苦參注射液是由苦參和土茯苓按照7∶3比例提取的活性成分組成。苦參的主要化學成分是生物堿類和黃酮類,土茯苓的主要化學成分包括甾醇類、黃酮(苷)類和揮發油類等。通過TCMSP平臺檢索苦參和土茯苓中已報道的化學結構,其中苦參中檢索到113個分子,土茯苓中檢索到74個分子,主要成分類型如表1所示。
2.2 14α-羥基苦參堿與PAK1具有較高的結合自由能
PAK1別構調節抑制劑具有高選擇性、高活性的特點,本研究基于分子對接方法從復方苦參注射液中虛擬篩選出能別構調節PAK1的活性成分。從苦參和土茯苓中分別篩選出對接打分排前10名的分子,如表2所示。計算這些分子與PAK1的結合自由能MM/GBSA,發現苦參中與PAK1結合自由能最高的分子是14α-羥基苦參堿[TCMSP編號:分子(molecule,MOL)006561],MM/GBSA為? 49.13 kcal/mol;土茯苓中與PAK1結合自由能最高的分子是異黃杞苷(TCMSP編號:MOL004567),MM/GBSA為? 49.24 kcal/mol,如表2所示。
由于苦參生物堿類分子是復方苦參注射液質量控制的主要成分,且含量較高[22],因此選取苦參中與PAK1結合自由能最高的分子—14α-羥基苦參堿,研究其抑制PAK1的結構生物學機制。如圖1所示,PAK1的別構調節位點位于ATP結合位點的背對位置,14α-羥基苦參堿結合于PAK1的別構調節位點,與第299位的賴氨酸(lysine299,K299)和407位的天冬氨酸(aspartic acid407,D407)形成氫鍵作用,與第318位的纈氨酸(valine318,V318)、319位的甲硫氨酸(methionine319,M319)、327位的異亮氨酸(isoleucine327,I327)、328位的纈氨酸(valine328,V328)、344位的甲硫氨酸(methionine344,M344)、380位的亮氨酸(leucine380,L380)和385位的纈氨酸(valine385,V385)等氨基酸形成疏水作用。14α-羥基苦參堿和用于定義虛擬篩選位點的別構調節抑制劑(S)-N-(叔丁基)-3-((2-氯-5-乙基-8-氟-二苯二氮卓-11-基)氨基)吡咯烷-1-甲酰胺(對PAK1具有高抑制活性和選擇性)與PAK1的結合模式在氫鍵作用上略有差異,表現在后者與PAK1的K299和第315位的谷氨酸(glutamic acid315,E315)形成氫鍵作用,其他作用模式類似[23]。
圖1
14α-羥基苦參堿與PAK1的結合模式
Figure1.
The binding mode of 14α-hydroxymatrine with PAK1
2.3 14α-羥基苦參堿將PAK1鎖定在失活的“半閉合”構象
為研究14α-羥基苦參堿抑制PAK1的分子機制,將14α-羥基苦參堿與PAK1結合的復合物結構(PAK1_14α-羥基苦參堿)進行300 ns的MD模擬,以PAK1與ATP結合的活性PAK1體系(PAK1_ATP)為對照。RMSD結果如圖2所示,50 ns之后14α-羥基苦參堿和兩個體系中的PAK1均達到平衡狀態。第408位的苯丙氨酸(phenylalanine408,F408)和E315是PAK1的別構調節口袋中分別靠內、外兩側的關鍵氨基酸,二者之間的距離可以反映別構調節口袋的大小和深度。通過MD模擬發現,14α-羥基苦參堿與PAK1結合之后,形成被14α-羥基苦參堿抑制的PAK1,其中PAK1的α1和α2螺旋向外延伸,使得E315和F408的α碳原子(Cα)之間(E315@Cα—F408@Cα)的距離(15.53 ?)明顯大于PAK1_ATP活性構象中E315@Cα—F408@Cα的距離(4.60 ?),如圖2、表3所示。并且,PAK1_14α-羥基苦參堿結構中F408的側鏈苯環發生轉向,由伸向別構調節位點一側轉到伸向ATP結合位點一側,說明14α-羥基苦參堿誘導PAK1形成一個較深的別構調節口袋以便更好地與之結合,而PAK1_ATP的活性構象中別構調節口袋則較淺,不易結合小分子藥物。
圖2
MD模擬分析PAK1_14α-羥基苦參堿和PAK1_ATP體系的別構調節位點
Figure2.
Analysis of the allosteric site in PAK1_14α-hydroxymatrine and PAK1_ATP systems by MD simulations
第283位的蘇氨酸(threonine283,T283)和F408是PAK1的ATP口袋中的兩個關鍵氨基酸,它們之間的距離能反映ATP結合口袋的開放程度。通過MD模擬發現,14α-羥基苦參堿的結合使PAK1的ATP結合口袋處的β-折疊向內關閉,導致T283@Cα—F408@Cα的距離(7.69 ?)明顯小于PAK1_ATP活性構象中T283@Cα—F408@Cα的距離(17.32 ?),如圖3、表3所示,使ATP結合口袋處于“半閉合”狀態。此外,14α-羥基苦參堿的結合使PAK1的F408的側鏈苯環轉向ATP結合口袋一側,導致ATP結合口袋變淺,不易與ATP結合而使PAK1失活,如圖3所示。
圖3
MD分析PAK1_14α-羥基苦參堿和PAK1_ATP體系中的ATP結合位點
Figure3.
Analysis of the ATP binding site in PAK1_14α-hydroxymatrine and PAK1_ATP systems by MD simulations
2.4 去除14α-羥基苦參堿使PAK1向活性構象轉變
為了驗證以上預測,本研究將14α-羥基苦參堿從PAK1_14α-羥基苦參堿復合物中去除(PAK1_去除[14α-羥基苦參堿]),通過MD模擬研究PAK1的構象變化。如圖4的RMSD矩陣分析所示,去除14α-羥基苦參堿之后,PAK1的構象發生了一系列變化。分析關鍵氨基酸之間的距離發現,去除14α-羥基苦參堿之后,PAK1的E315@Cα—F408@Cα的距離明顯減小(10.81 ?),而T283@Cα—F408@Cα的距離有所增加(8.40 ?),如圖4、表3所示,說明PAK1的別構調節口袋變小,而ATP結合口袋變大。將PAK1_去除[14α-羥基苦參堿]和PAK1_14α-羥基苦參堿的結構進行疊合,采用模式向量分析發現,去除14α-羥基苦參堿之后,別構調節位點處的α1和α2螺旋明顯向酶的中心結構靠攏,而ATP結合位點處的β-折疊有向外開放的趨勢,如圖4所示。這些結果均說明,去除14α-羥基苦參堿之后,PAK1逐漸從失活構象向活性構象轉變。
圖4
MD模擬PAK1_去除[14α-羥基苦參堿]體系的別構調節位點和ATP結合位點的變化
Figure4.
The changes of the allosteric site and ATP binding site of PAK1_remove[14α-hydroxymatrine] system by MD simulations
3 討論
胰腺癌具有發現難、進展快、預后不良的特點,臨床診療迫切需要更好的生物標志物和潛在的藥物靶點。PAK1在多種人類腫瘤組織中過度表達和(或)過度激活[24],調節復雜的信號轉導網絡。多項研究表明,PAK1是調控胰腺癌細胞生長的關鍵因子,其表達水平影響胰腺癌的發展、預后及生存周期;抑制PAK1的活性能夠抑制胰腺癌細胞的生長及存活,促進細胞凋亡[3-7]。因此,PAK1被認為是一個潛在的胰腺癌治療靶點,目前已有一些抑制劑正在研發。
復方苦參注射液在臨床上被廣泛用于輔助治療惡性腫瘤及腹腔積液等并發癥,其活性成分苦參生物堿已被證實具有抗胰腺癌活性,能抑制多種胰腺癌細胞的增殖、遷移和侵襲,誘導細胞凋亡[25-27],還能增強胰腺癌耐藥細胞株對化療藥的敏感性[20],但具體作用靶點未知。鑒于PAK1在胰腺癌發生、發展中的重要作用,本研究篩選了復方苦參注射液中可能靶向PAK1的活性成分。結果發現,14α-羥基苦參堿與PAK1的別構調節位點具有較高的結合自由能,作用的關鍵氨基酸主要有K299、D407、V318、M319、I327、M344、L380和V385等。
通常,別構調節抑制劑具有高選擇性、高活性的特點,它們結合于酶的非催化位點,引起酶的構象發生可逆改變,進而改變酶的活性狀態。本研究通過MD模擬進一步研究了14α-羥基苦參堿抑制PAK1的結構生物學機制。發現14α-羥基苦參堿與PAK1結合之后,別構調節位點的α1和α2螺旋向外延伸,F408和E315之間的距離增大,并且F408的側鏈苯環由別構調節位點一側轉向ATP結合位點一側,使得PAK1形成一個較深的別構調節口袋,從而更好地結合小分子藥物。同時,14α-羥基苦參堿誘導PAK1的ATP結合口袋處的β-折疊向內關閉,使得T283和F408之間的距離減小,導致PAK1的ATP結合口袋處于“半閉合”狀態。此外,F408的側鏈苯環轉向ATP結合口袋一側,使ATP結合口袋變淺而不易結合ATP,導致PAK1失活。
將14α-羥基苦參堿從PAK1中去除,通過MD模擬發現PAK1的構象發生了一系列變化。別構調節位點處F408和E315之間的距離明顯減小,而ATP結合位點處T283和F408之間的距離有所增加。結構疊合后的模式向量分析發現,去除14α-羥基苦參堿之后,別構調節位點處的α1和α2螺旋向PAK1的中心結構靠攏,而ATP結合位點處的β-折疊有向外開放的趨勢。這些結果均說明,去除14α-羥基苦參堿之后,PAK1從失活構象向活性構象轉變,提示14α-羥基苦參堿可能是一個可逆的PAK1別構調節抑制劑。
綜上所述,本研究通過虛擬篩選和MD模擬發現,復方苦參注射液中的14α-羥基苦參堿可能是PAK1的別構調節抑制劑,能誘導ATP結合口袋處于“半閉合”狀態而使PAK1失去酶活性。本研究可為天然活性成分的開發利用及尋找新的胰腺癌治療方案奠定基礎。
重要聲明
利益沖突:所有作者均聲明不存在利益沖突。
作者貢獻聲明:林琳、常靜杰和陳姣共同負責本研究中數據庫的構建、分子對接和分子動力學模擬,陳姣和田耀洲負責數據分析指導,林琳、陳姣負責論文寫作與修改。