高通量染色質構象捕獲(Hi-C)技術的快速發展為染色質結構分析提供了豐富的基因組位點間交互作用數據,但目前基于Hi-C數據的已有拓撲相關結構域(TAD)識別方法存在準確率低、參數敏感等問題。在此背景下,本文設計并實現了一種基于空間密度聚類的TAD識別方法。該方法首先對原始Hi-C數據進行預處理,得到歸一化后的Hi-C接觸矩陣數據;然后計算位點之間的距離矩陣,基于位點的核心距離和可達距離生成可達性圖,并提取聚類簇;最后基于聚類結果和TAD提取邊界。該方法能夠識別出內聚性更高的TAD結構,且TAD邊界處富集了更多的ChIP-seq因子。實驗結果表明,本文方法在TAD識別中更準確,更具有現實意義。
引用本文: 龔海燕, 張司臣, 張曉彤. 基于密度聚類的染色質拓撲相關結構域識別方法. 生物醫學工程學雜志, 2024, 41(3): 552-559. doi: 10.7507/1001-5515.202311059 復制
版權信息: ?四川大學華西醫院華西期刊社《生物醫學工程學雜志》版權所有,未經授權不得轉載、改編
0 引言
在人類和其他高等真核生物中,基因組被折疊成大規模和動態的三維構象,大約2 m長的基因組DNA被折疊,以適應直徑約10 μm的細胞核的大小[1],不同的基因位點之間交互作用,作為基因表達的藍圖發揮作用。生物化學技術通過消化和重連物理空間上距離相近的染色質片段來確定不同位點的空間接近性。作為代表的高通量染色質構象捕獲(high-throughput chromatin conformation capture,Hi-C)技術是一種染色質構象捕獲技術,所需的細胞數較少,并且文庫制備步驟相對簡單,因此在研究三維基因組結構中得到了廣泛應用。Hi-C技術[2]首先對染色質進行甲醛交聯固定,然后使用限制性內切酶對DNA進行切割,并將末端使用生物素標記。接著,DNA片段通過生物素的親和性純化和連接制備成嵌合分子,并進行中斷和生物素標記篩選。最后,將篩選得到的DNA片段進行雙端測序,以計算在基因組中任意兩個染色質區域之間的嵌合分子數,從而獲取染色質交互作用數據。
拓撲相關結構域(topologically associated domains,TAD)[3]是構成染色質三維結構的重要組成部分,被認為是基因調控的功能域,對于細胞分化、發育以及疾病治療具有重要意義[4]。TAD是染色質區域內部比染色質區域之間更頻繁交互作用的結構域。這些結構域大小通常介于200 kb~5 Mb之間[3],并在不同物種和細胞類型中具有高度的保守性。大多數TAD識別方法使用直接從Hi-C接觸矩陣導出的度量來檢測TAD邊界。其中,Dixon等[4]在2012年提出了基于方向性指數DI的方法,該方法通過隨機化方向計算方向性指數來量化基因組區域上下游交互作用的偏置程度,偏置程度越高,其方向性指數越大。通過確定基因組中的DI,可以確定TAD邊界在基因組中的位置。Armatus方法[5]將TAD識別抽象為一個動態規劃問題,其他類似方法有TopDom[6]、CaTCH[7]、GMAP[8]和HiTAD[9]。一些TAD識別方法依賴統計學方法,包括HiCseg[10]、TADtree[11]、PSYCHIC[12]、TADbit[13]和HiCKey[14]。例如,Levy-Leduc等[10]提出了HiCseg,該方法為Hi-C數據定義了一個通用的統計模型,可處理原始數據和標準化數據,然后,利用最大似然方法,將二維分割問題轉化為一維分割問題,通過動態規劃算法解決。除此之外,還有一些基于聚類的方法,包括ClusterTAD[15]、IC-Finder[16]、CASPIAN[17]和TOAST[18]。ClusterTAD方法利用接觸矩陣創建特征,然后在Hi-C數據集上應用層次聚類和K-均值聚類,并應用多種距離度量方式。有一些基于網絡的方法可用于TAD的識別,包括MrTADFinder[19]和3DNetMod[20]。MrTADFinder方法利用染色質接觸圖中的TAD與網絡中密集連接的模塊之間的相似性,將TAD的識別視為一個網絡優化問題。然而,現有的TAD識別方法主要基于染色質物理交互頻率的變化、染色質分割和機器學習,但是這些方法存在一定的局限性,例如識別的TAD質量低、計算成本高、參數較多或參數敏感問題。本文提出一種考慮TAD區域內基因組序列片段的特定密度,利用聚類識別TAD的邊界和內部結構,提高所識別TAD質量,從而進一步推動對染色質空間結構和功能的理解。
1 方法
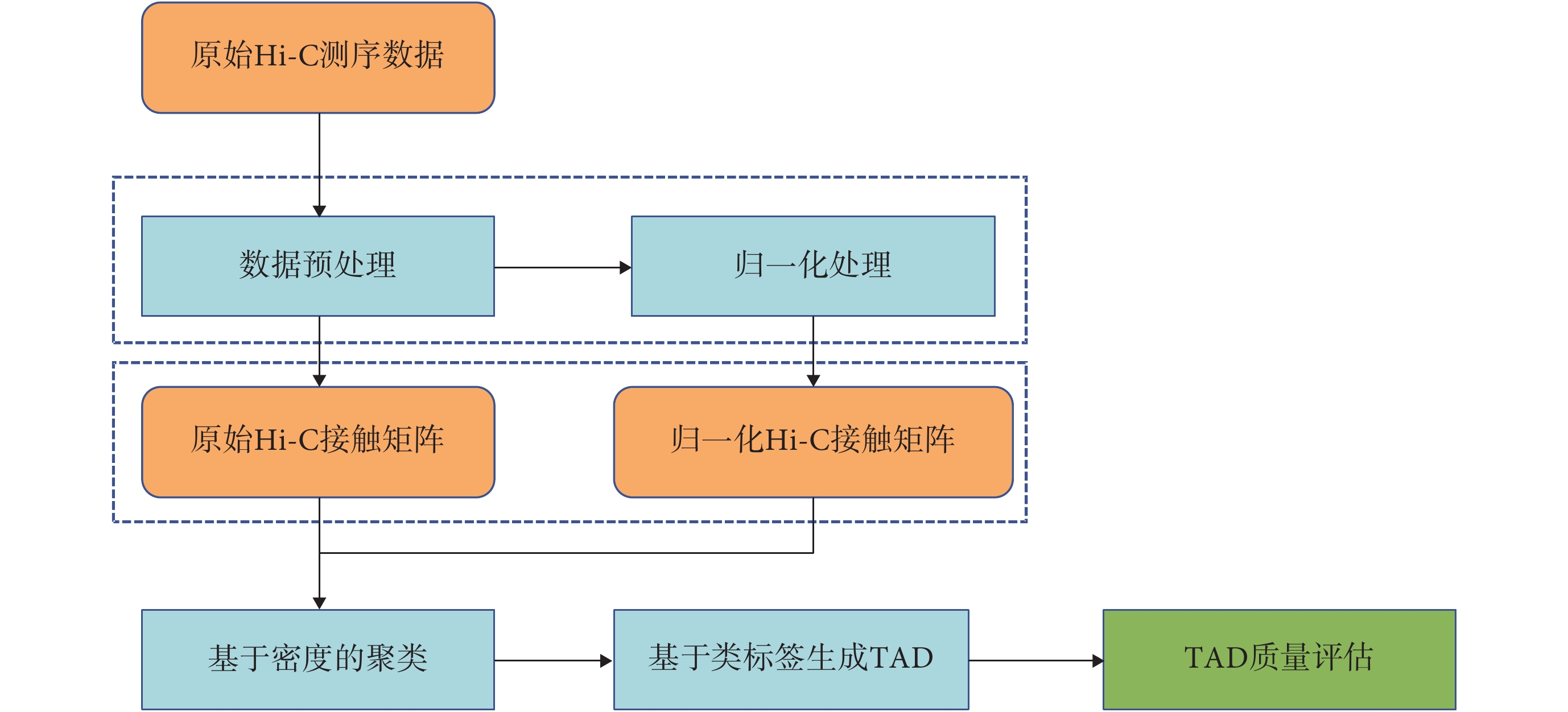
本文提出一種基于密度聚類的TAD識別算法ATLANTIC,從而從Hi-C接觸矩陣中準確識別TAD邊界。算法總體流程圖如圖1所示,ATLANTIC首先將預處理后的Hi-C矩陣數據作為輸入,利用基于密度的無監督聚類算法根據基因組節點間的交互作用將染色質劃分為不同簇,并從簇中提取TAD邊界。
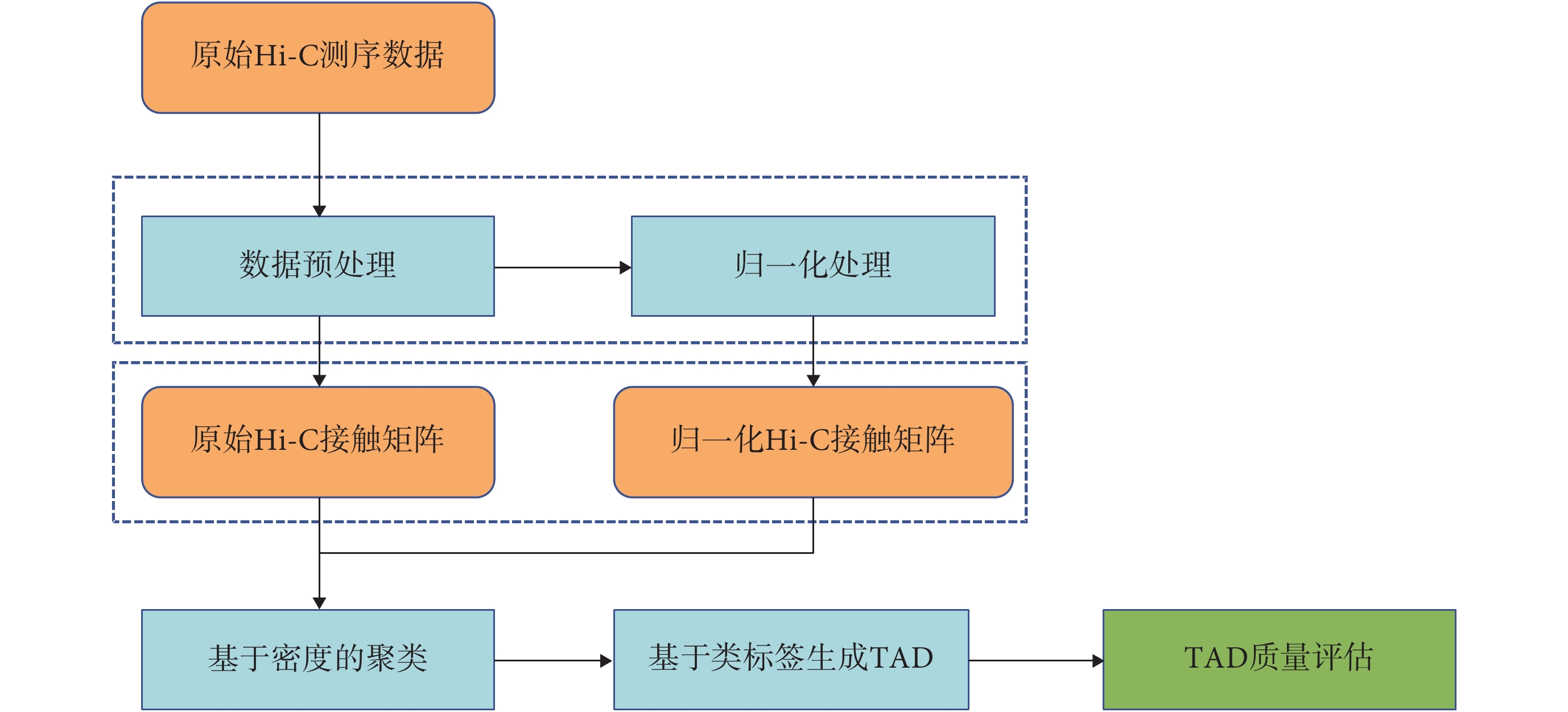 圖1
采用ATLANTIC方法進行拓撲相關結構域識別的流程圖
Figure1.
TAD identification flow chart of ATLANTIC method
圖1
采用ATLANTIC方法進行拓撲相關結構域識別的流程圖
Figure1.
TAD identification flow chart of ATLANTIC method
1.1 數據預處理模塊
Hi-C數據標準化是一項重要的數據預處理步驟,用于消除在實驗過程中由于各種非隨機因素引入的樣本間系統偏差。這些偏差可能會導致Hi-C接觸矩陣中的一些區域具有較高或較低的讀數,從而影響后續的數據分析和解釋。因此,對Hi-C數據進行標準化處理是保證數據質量和準確性的重要前提。近年來,諸多標準化方法被提出,如ICE[21]、KR[22]等。其中,KR方法是一種被廣泛使用的數學方法,它是一種矩陣平衡的方法,通過調整每個基因組區域的總Hi-C讀數來消除全局偏差。首先計算原始的Hi-C接觸矩陣每行和每列的總讀數,然后使用這些總讀數計算每個基因組區域的尺度向量;接著,使用非精確牛頓迭代法計算尺度向量,通過多次迭代來不斷調整每個區域的尺度因子,使得每個區域的總讀數與整個基因組的平均總讀數相等;最后,使用對角尺度法將矩陣轉換為雙隨機矩陣,以消除樣本間的系統偏差。KR方法在Hi-C數據標準化中具有明顯的優勢,可以消除測量偏差、保留染色質結構信息并且計算速度相對較快。而ICE[21]通過迭代矯正和特征分解的方法進行偏差消除,但是不僅需要Hi-C接觸矩陣作為輸入,還需要依賴于各種附加信息,例如限制位點、基因組序列和可映射性評分。因此,本文選擇KR方法作為Hi-C接觸矩陣歸一化的方法。
1.2 聚類模塊
聚類模塊使用預處理后的Hi-C接觸矩陣作為輸入,目的是從中提取TAD結構特征。該模塊使用了一種基于密度的空間聚類方法(ordering points to identify the clustering structure,OPTICS)[23]。該算法基于數據集的密度和距離信息來刻畫聚類結構,而無需指定簇的數量。因此,可以自適應地確定簇的數量,從而識別TAD結構,即使在不確定TAD數量的情況下也能做到這一點。具體來說,對于給定的一組Hi-C數據集 ,基于密度的聚類方法通過以下幾個步驟來獲取聚類結果。
,基于密度的聚類方法通過以下幾個步驟來獲取聚類結果。
(1)計算距離矩陣。計算距離矩陣時,算法會首先根據輸入的數據點構建一個k-d樹。k-d樹[24]是一種用于快速查找k維空間中最近鄰點的二叉樹結構,可以將多維空間劃分為多個子空間,并在每個子空間中遞歸地構建k-d樹,從而實現高效的最近鄰查詢。距離矩陣被用于確定兩個樣本之間是否是密集連接的關系,可以使用不同的距離度量方式,包括曼哈頓距離、歐幾里得距離、切比雪夫距離。
(2)計算核心距離和核心對象。使用不同的距離度量方式計算樣本點 的核心距離,記作
的核心距離,記作 。遍歷
。遍歷 中的基因位點,如果其核心距離小于鄰域半徑
中的基因位點,如果其核心距離小于鄰域半徑 ,則將其加入到核心對象集合
,則將其加入到核心對象集合 中。
中。
(3)生成最小生成樹。將距離矩陣視為一個帶權無向圖,每個數據點代表一個節點,距離代表邊的權重,然后使用Prim方法生成最小生成樹。從一個任意節點出發,選擇一個到該節點距離最近的并且未訪問的節點,將其加入生成樹中,并將該節點到其它未訪問節點的距離更新為更小的值,重復此過程,直到生成樹包含所有節點為止。
(4)計算可達距離和可達性圖。隨機選取核心對象集合 中一個核心點
中一個核心點 出發,首先將其標記為已處理;然后,基于最小生成樹找到
出發,首先將其標記為已處理;然后,基于最小生成樹找到 所有密度可達對象,將對象
所有密度可達對象,將對象 的可達距離定義為
的可達距離定義為 的核心距離與
的核心距離與 之間距離的最大值;最后,根據可達距離依次遍歷未處理過的樣本點,生成可達性圖。
之間距離的最大值;最后,根據可達距離依次遍歷未處理過的樣本點,生成可達性圖。
(5)識別聚類簇。根據上個步驟生成排序后的可達有序列表,通過引入坡度的思想來表征可達列表中相鄰點之間可達距離差距的大小。首先計算連續點的可達性距離的坡度。在形成聚類時有兩種情況,即陡降或陡升區域,其中劃分聚類的可達性距離差為負值或正值,即當第一個觀察點的可達性分別高于或低于第二個觀察點。對于一個樣本點p,當其滿足  時,說明p的可達距離比它的下一個樣本點低不止
時,說明p的可達距離比它的下一個樣本點低不止  的陡度,則稱其為
的陡度,則稱其為  上升點,點p在
上升點,點p在  上升區域。其中
上升區域。其中  表示p的可達距離。同樣地,當其滿足
表示p的可達距離。同樣地,當其滿足  時,表示p在
時,表示p在  下降區域。對于一個區間
下降區域。對于一個區間  ,它被劃分為一個簇的標準是:簇的起點被包含在
,它被劃分為一個簇的標準是:簇的起點被包含在  陡峭下降區域D中,簇的終點被包含在
陡峭下降區域D中,簇的終點被包含在  陡峭上升區域U中。同時集群數量不小于MinPts個點,群集中所有點的可達值必須至少比D第一點的可達性值和U結束后第一點的可達性值低
陡峭上升區域U中。同時集群數量不小于MinPts個點,群集中所有點的可達值必須至少比D第一點的可達性值和U結束后第一點的可達性值低  。基于以上標準將可達性圖提取出不同類別的簇結構。
。基于以上標準將可達性圖提取出不同類別的簇結構。
1.3 邊界提取模塊
本節介紹了聚類模塊用于標記基因組位點的方法。在這個過程中,每個基因組bin都被賦予了一個類別標簽。研究表明,TAD的區間長度通常在200 kb~5 Mb之間[3]。因此可使用這些標簽來確定TAD的位置,具體而言,將擁有相同標簽的基因組位點組成的連續區域看作是一個TAD;接著,利用染色質位點數量和分辨率等因素來確定這個連續區域是否應該被劃分為一個TAD,如果連續區域的長度超過200 kb,則可以被認定為一個TAD。例如,對50 kb分辨率下的Hi-C接觸矩陣數據進行分析,那么必須至少包含四個具有相同標簽的基因組bin的一段連續區域才被認為是一個TAD。這可以確保識別出長度足夠的TAD,并避免將過短的連續區域識別為TAD。總之,該模塊使用聚類模塊中的標簽信息來幫助識別TAD的位置。通過考慮染色質位點數量和分辨率,可以確保只識別出足夠長的TAD,從而提高TAD識別的準確性。
2 實驗結果與分析
2.1 數據集
2.1.1 模擬Hi-C數據
實驗使用了兩個模擬數據集,一個是Wang等[25]在實驗中生成的模擬接觸矩陣數據集,是使用DI方法[4]在人胚肺成纖維細胞系IMR-90中生成的結構域模擬矩陣。另一個是Trussart等[26]實驗生成的模擬數據集。使用聚合物建模,模擬了具有不同結構的單個染色質的6個人工生成的基因組(稱為“玩具基因組”),從中提取了168個模擬交互作用矩陣,它們的噪聲水平和結構多樣性不斷增加。本章使用了噪聲水平分別為50和100,線性密度分別為40、75、150 bp/nm的模擬Hi-C矩陣數據。
2.1.2 真實Hi-C數據
實驗使用了來自人類B淋巴細胞GM12878的真實數據集。為了獲取原始數據,首先從4dnucleome平臺獲取登錄號為4DNFI1UEG1HD的.hic格式文件;接著,使用Jucier工具[27]對該文件進行處理,得到分辨率分別為25 kb和50 kb的原始Hi-C接觸矩陣,并對它進行了KR歸一化處理。這些數據將被用于后續的實驗分析和結果展示。
2.1.3 染色質免疫共沉淀(chromatin immunoprecipitation,ChIP)數據
實驗分析使用來自DNA元件百科全書(Encyclopedia of DNA Elements,ENCODE)項目的ChIP-Seq數據來分析基因組位點CTCF以及其他組蛋白修飾的富集。其中CTCF、H3K4me3、SMC3和POLR2A ChIP-seq數據在ENCODE平臺的登錄號分別為ENCSR000EAD、ENCSR057BWO、ENCSR000DZP和ENCSR000EFK。主要使用bed narrowpeak格式的ChIP-seq數據,這些文件包含了染色體號、起始位點和終止位點等信息,可用于確定各種蛋白質結合位點在基因組上的位置。通過對這些數據進行分析,可以識別各種轉錄因子的peak值,并計算TAD結構邊界處各種轉錄因子的peak數量和密度。
2.2 評價指標
TAD識別的準確性對于深入理解基因組結構和功能具有重要意義,為了對TAD的識別結果進行評價和對比,設定TAD質量分數、聚類效果評價指標Davies-Bouldin指數和輪廓系數三種評價指標。
TAD結構的一個顯著特征是,對于給定TAD內的區域,它們具有更高的接觸頻率分布,而TAD外的區域之間接觸頻率很少。這意味著TAD內部的區域應該具有更高的相似性和一致性,而TAD之間的區域應該具有較小的相似性。基于這一性質,將識別出的第 個TAD內部各個bin之間的接觸頻率平均值記作
個TAD內部各個bin之間的接觸頻率平均值記作 ,將
,將 與相鄰
與相鄰 之間的接觸頻率平均值記作
之間的接觸頻率平均值記作 ,則TAD質量分數可用公式(1)計算得到。定義每個接觸矩陣得到的一組TAD的TAD Quality分數是這組TAD的質量分數平均值。
,則TAD質量分數可用公式(1)計算得到。定義每個接觸矩陣得到的一組TAD的TAD Quality分數是這組TAD的質量分數平均值。
 |
對于聚類效果,使用Davies-Bouldin指數(Davies-Bouldin index,DBI)和輪廓系數(Silhouette Index,SI)進行評估。DBI是一種常用的用于評估聚類效果的指標,它通過比較類內距離和類間距離來衡量聚類質量。DBI值越接近于0,表示聚類結果越好。
SI也經常被用來評估聚類的優劣,它通過比較每個樣本與所屬簇內部其他樣本的相似度和與其他簇中樣本的相似度,來度量聚類結果的內聚性和分離性。SI取值范圍在?1~1之間。SI越接近1,說明數據點聚類得越好,不同簇之間的距離越遠,同一簇內部的數據點之間的距離越近。反之,亦然。當SI接近0時,說明數據點處于聚類邊界附近,聚類效果不明顯。
為了評價算法識別的域邊界的準確性,研究人員還經常使用ChIP技術結合高通量測序來確定染色質上的特定修飾標記和結合蛋白的富集情況。其中,CTCF是一種常見的染色質結合蛋白,在真核生物基因組的三維組織中扮演著重要的角色。作為核心結構蛋白之一,CTCF幫助建立了染色質的組織結構[28],它可以與其他轉錄因子共同作用于染色質結構域的邊界處,形成DNA環,這些環的位置通常與TAD的邊界相關。因此,CTCF被認為是TAD邊界的主要因子之一。RAD21和SMC3是Cohesin復合物的兩個重要組成部分,通常出現在TAD邊界[29]。一些研究表明,POLR2A可以結合在TAD邊界區域。例如,在人類胚胎干細胞中,POLR2A的結合顯著富集在TAD邊界區域。已有研究表明,H3K4me3、H3K36me3等因子在TAD邊界處大量富集[4]。因此選擇CTCF、H3K4me3、SMC3以及POLR2A四種ChIP-seq因子在TAD邊界附近區域的富集情況來衡量算法識別的準確性。
2.3 基準模型
為了驗證ATLANTIC的性能,本文選擇了TopDom[6]、Insulation Score[30]、EAST[31]、ClusterTAD[15]幾種TAD劃分算法與ATLANTIC方法進行對比。這幾種算法識別出的TAD結構均是非分層的,其中ClusterTAD方法也是基于聚類思想的TAD識別工具,其余幾種是常見的利用Hi-C矩陣度量或者統計的方法提供的標準TAD識別工具。
2.4 算法參數
OPTICS算法是一種密度聚類算法,其核心思想是基于密度可達性來確定樣本點之間的聚類關系。在OPTICS算法中,MinPts是一個非常重要的參數,它決定了一個樣本點被視為核心點的最小鄰域樣本數。一般來說,較小的MinPts值會導致聚類更加細致,但也可能會導致過度聚類。相反,較大的MinPts值會導致聚類更加粗略,但可能會忽略掉一些重要的聚類。研究表明,TAD大小通常在200 kb~5Mb之間,因此測試MinPts取值在[3, 14]之間的影響可以有效地驗證生成的TAD質量。此外,距離度量方法也是OPTICS算法中的另一個重要參數,它可以是歐幾里得距離、曼哈頓距離、切比雪夫距離、余弦相似度等。在實踐中,應根據具體的數據集和應用場景選擇最合適的距離度量方法。
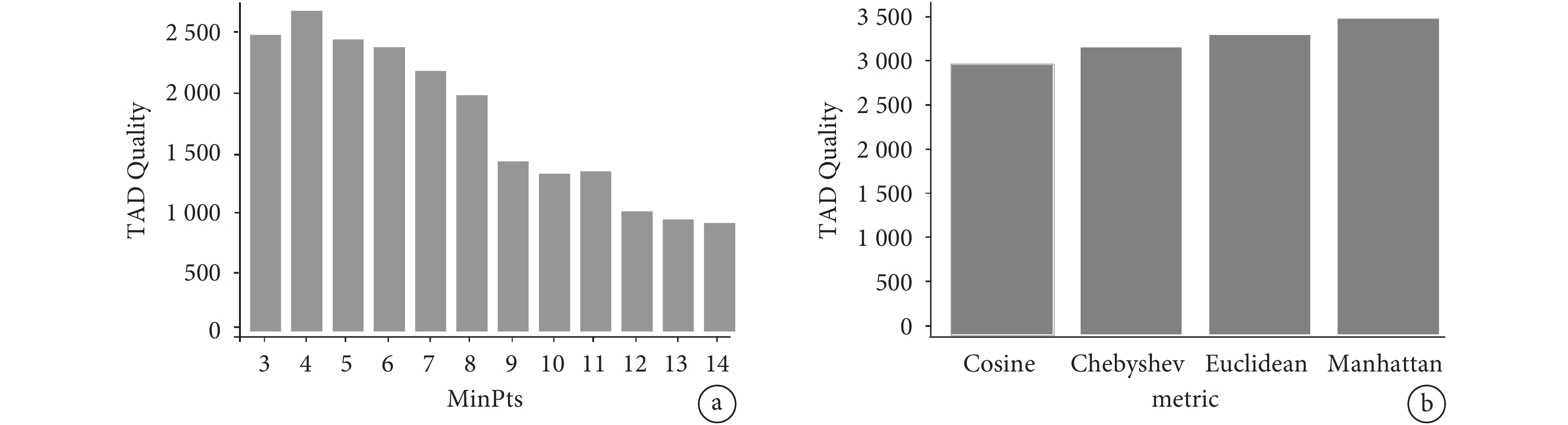
使用GM12878細胞系的19號染色質Hi-C數據作為輸入,測試不同參數對TAD的劃分結果質量的影響。如圖2a所示,當其余參數取默認值、MinPts取值為4時,識別出的TAD具有最高質量,TAD Quality達到了2 586.816。如圖2b所示,分別測試了幾種常用的距離度量方法(余弦距離、切比雪夫距離、歐氏距離以及曼哈頓距離)的TAD Quality。結果表明,該方法在距離度量的確定上具有魯棒性,不同方法均取得了較好的識別效果,其中曼哈頓距離度量方式具有最高的TAD Quality(3 474.173)。
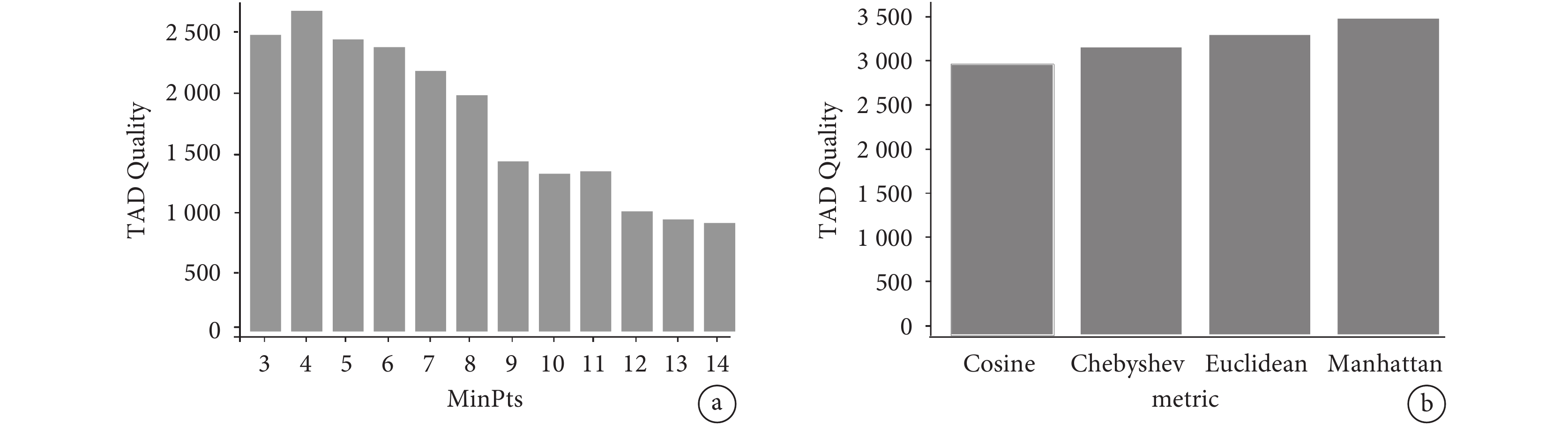 圖2
不同參數下的TAD質量對比
圖2
不同參數下的TAD質量對比
a. 不同MinPts取值的TAD Quality對比;b. 不同距離度量方式的TAD Quality對比
Figure2. Comparison of TAD quality under different parametersa. TAD Quality comparison of different MinPts values; b. TAD Quality comparison of different distance metrics
2.5 模擬數據集評估ATLANTIC
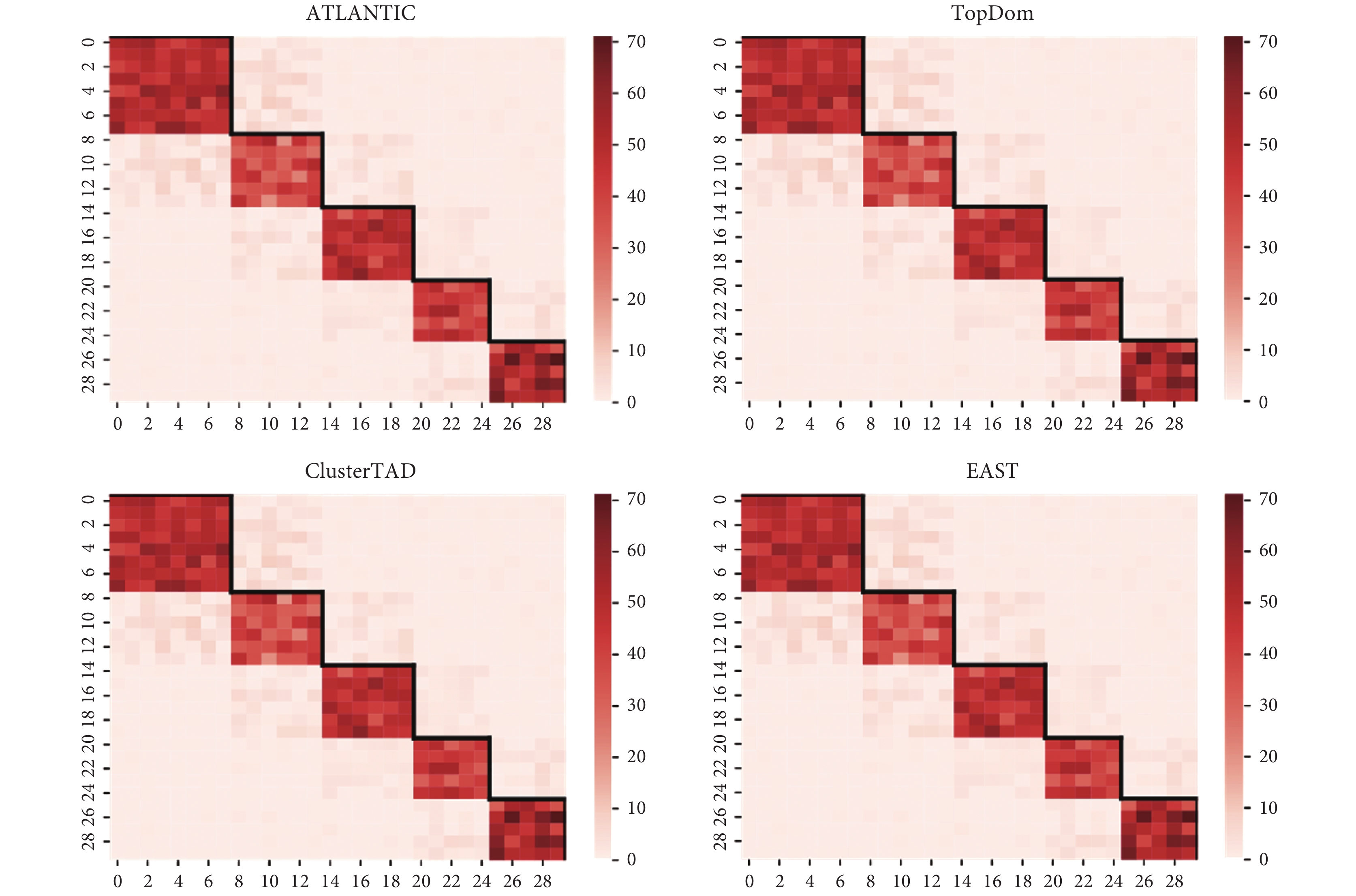
首先將ATLANTIC方法應用到了模擬Hi-C接觸矩陣數據集上,使用評價指標和方法對比來驗證方法的可用性以及可靠性。第一個模擬數據集是來自Wang等[25]發表的模擬Hi-C矩陣,該數據具有明顯的拓撲結構域特征,便于驗證方法的有效性。從圖3中可以看出,由ATLANTIC方法識別出的TAD邊界與Hi-C熱圖中顯示的TAD矩形結構緊密契合,表明該方法能夠較準確地提取Hi-C數據中的TAD邊界位置。同時,將不同方法識別出的TAD結果進行對比,發現由TopDom、ClusterTAD以及EAST方法識別出的邊界與ATLANTIC方法完全一致,側面驗證了ATLANTIC方法的有效性。
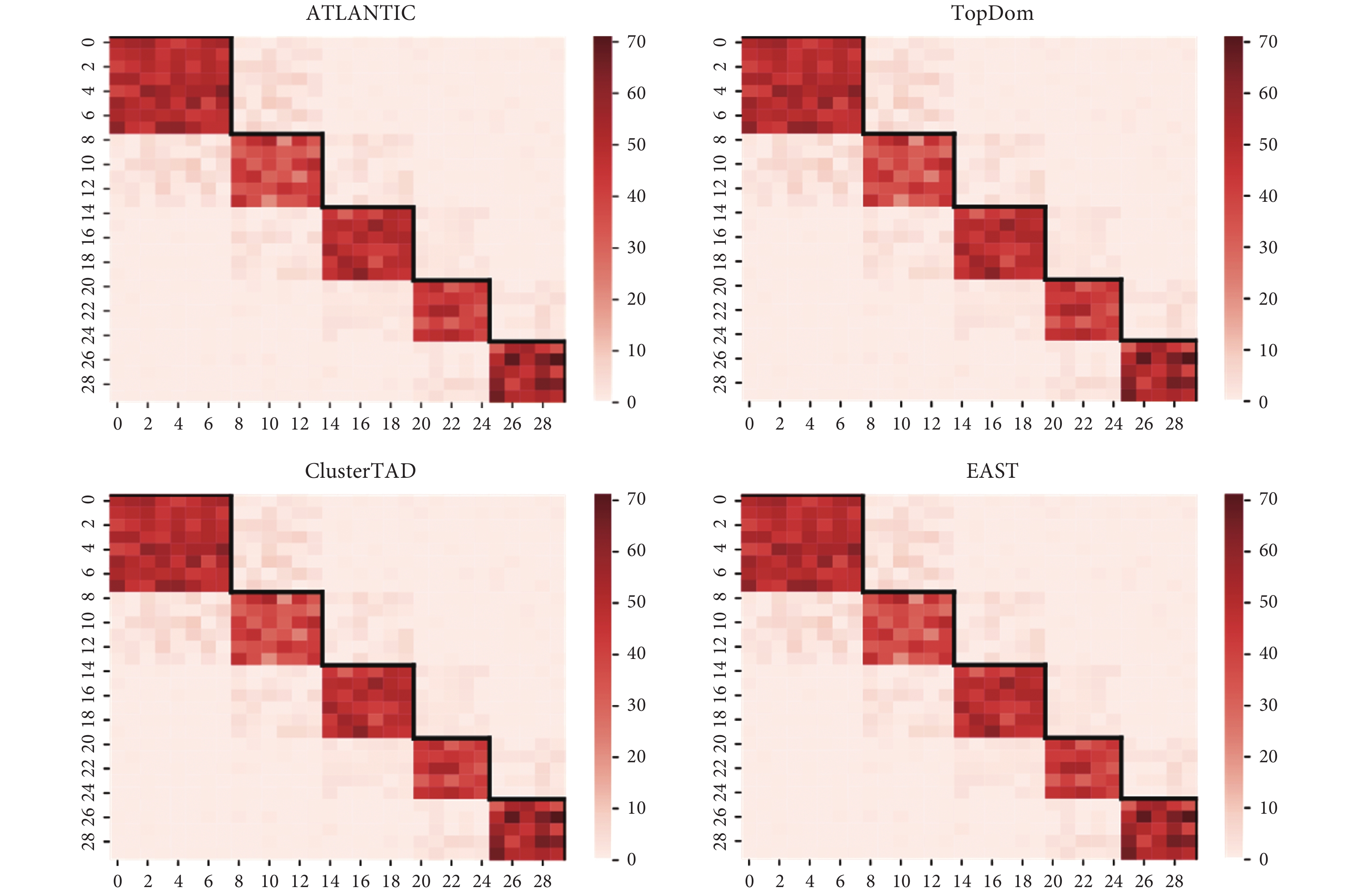 圖3
模擬數據集下不同方法識別出的TAD邊界可視化結果
Figure3.
Visualization of TAD boundaries identified by different methods under simulated data
圖3
模擬數據集下不同方法識別出的TAD邊界可視化結果
Figure3.
Visualization of TAD boundaries identified by different methods under simulated data
接著,將ATLANTIC應用到了Trussart等[26]通過實驗的模擬Hi-C數據上,并使用了不同線性密度和不同噪聲下的模擬數據集。實驗結果證明了ATLANTIC在不同噪聲水平上識別的TAD數量更加穩定,說明了該方法具有一定的魯棒性和抗噪聲能力(具體圖片參見附件1)。此外,ATLANTIC的識別結果具有較高水平的TAD Quality,證明了ATLANTIC方法的有效性(具體數據參見附件2)。
2.6 真實數據集評估ATLANTIC
除了模擬數據集,本文還將ATLANTIC方法應用到了GM12878細胞系數據集。首先使用的是1~22號染色體分辨率為50 kb的Hi-C接觸矩陣,應用曼哈頓距離度量的ATLANTIC方法進行TAD識別。識別結果較好地貼合了原始Hi-C矩陣數據(具體圖片參見附件3)。識別結果的TAD Quality分數均值達到2 301.879,在12號染色體上TAD Quality達到3 603.246(具體圖片參見附件4a)。SI值接近1,DBI值較低,處于1.5左右,意味著聚類結果具有較好的分離性和緊密性(具體圖片參見附件4b)。
同時,實驗以GM12878的19號染色體50 kb分辨率下的Hi-C接觸矩陣作為輸入,驗證了CTCF、H3K4me3、SMC3和POLR2A四種ChIP-seq信號因子在TAD邊界處的富集數量。實驗表明,由ATLANTIC識別出的TAD在邊界附近出現了大量蛋白結合位點,尤其是在TAD邊界處出現了大量促進轉錄相關因子和組蛋白修飾的富集(具體圖片參見附件5)。其中,CTCF結合位點在TAD邊界出現大量富集,表明它們可能在染色質空間中起到隔離相鄰TAD的絕緣體的作用[4]。啟動子標記(H3K4me3)在邊界區域的高度富集表明部分TAD邊界區域可能是轉錄激活位點。該結果驗證了ATLANTIC方法識別TAD的準確性,同時表明本方法識別出了具有明顯生物學特征的TAD結構劃分。
此外,實驗還將ATLANTIC應用到了更高分辨率下的Hi-C數據集。以GM12878的25 kb分辨率的1~22號染色質Hi-C矩陣作為輸入,計算了識別出的TAD質量分數以及聚類效果得分。在不同染色質數據上,TAD Quality分數相對穩定,其中19號染色體識別結果的TAD Quality達到了998.917,并且驗證了聚類的準確性和方法的穩定性(具體圖片參見附件6)。
基于GM12878的50 kb分辨率Hi-C接觸矩陣,本文與其他四種方法在TAD數量、平均長度以及質量分數三個指標上進行了對比。從表1可以看出,各個方法識別出的TAD數量和大小具有相似性,其中TopDom識別出的數量較多,分析原因是由于它識別出的TAD不存在空隙區域,而ATLANTIC方法相比其他算法可得到更高的TAD Quality分數。
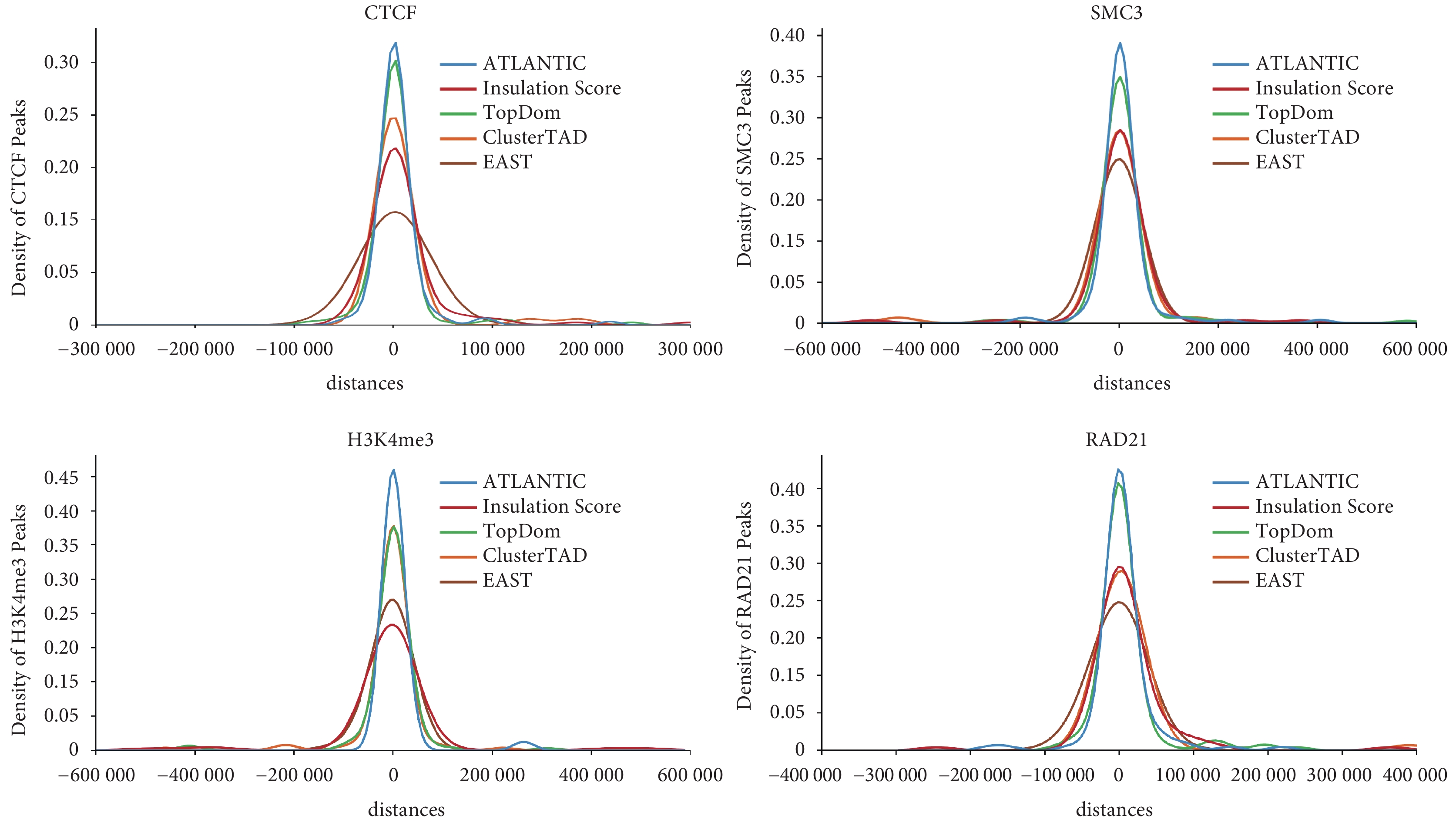
實驗還比較了由不同算法識別出的TAD邊界相關蛋白結合位點(CTCF、H3K4me3、RAD21、SMC3四種ChIP-seq信號因子)的富集情況。圖4展示了四種方法在邊界附近相關富集因子的分布核密度曲線,其中藍色曲線為ATLANTIC方法結果。從圖中可以看出,ATLANTIC能夠識別出的邊界位置相關因子富集程度要優于其余幾種方法,說明ATLANTIC方法識別出的TAD邊界更加符合生物學特征。
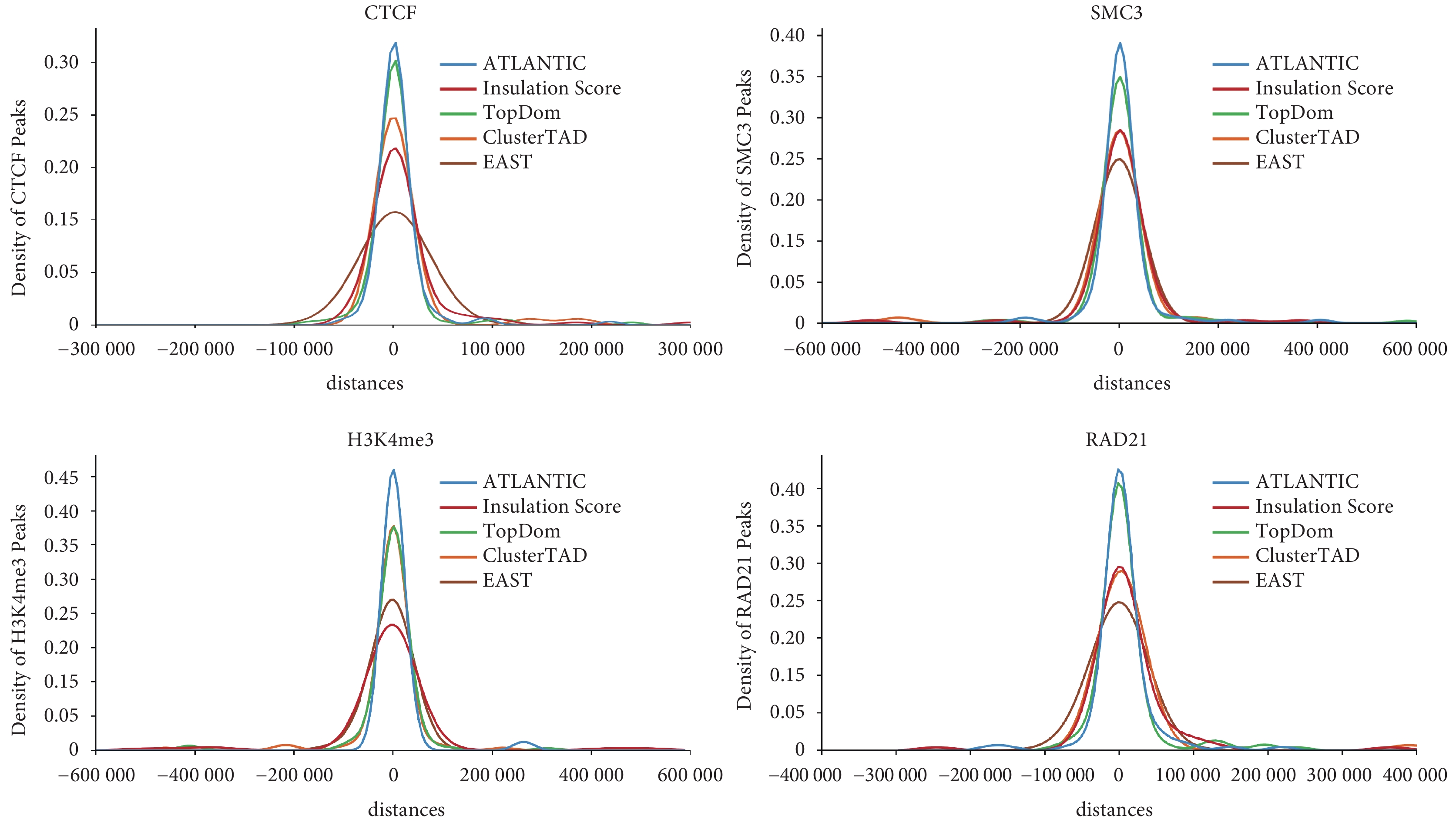 圖4
不同方法在真實數據集上識別的TAD邊界蛋白結合位點富集概率曲線
Figure4.
Enrichment probability curves of TAD boundary protein binding sites identified by different methods
圖4
不同方法在真實數據集上識別的TAD邊界蛋白結合位點富集概率曲線
Figure4.
Enrichment probability curves of TAD boundary protein binding sites identified by different methods
3 總結與展望
本文提出了一種基于空間密度聚類的染色質TAD邊界識別算法ATLANTIC。該方法通過對Hi-C接觸矩陣中的基因組位點計算可達性圖并提取聚類簇,實現對TAD邊界的識別。方法主要分為三個模塊:首先對原始Hi-C數據進行歸一化,以獲取標準化后的數據;然后將Hi-C矩陣應用于基于密度的聚類方法進行分類成簇;最后根據劃分結果識別連續區域的TAD結構。根據TAD的結構特性和聚類方法的一般特性設置了評價指標,并將該方法運用到了模擬數據集和真實數據集上,結果表明,ATLANTIC方法能夠準確、有效地識別TAD,并在不同分辨率數據集下取得較好效果。實驗還將ATLANTIC方法與已有四種方法進行了對比,結果顯示ATLANTIC方法識別的TAD邊界錨定了更多的CTCF和其他蛋白結合位點因子比例,這些發現對于進一步研究基因組結構與功能的關系具有重要的意義。但ATLANTIC仍然存在一些限制,例如使用高分辨率(25 kb及更高)Hi-C數據來識別TAD結構時,算法的運行時間較長。未來的工作可能包括通過研究并行計算的算法來提高在高分辨率Hi-C數據上的運行效率。
重要聲明
利益沖突聲明:本文全體作者均聲明不存在利益沖突。
作者貢獻聲明:龔海燕負責實驗開展和論文寫作,張司臣負責實驗開展,張曉彤負責論文指導與審閱。
本文附件見本刊網站的電子版本(biomedeng.cn)。
0 引言
在人類和其他高等真核生物中,基因組被折疊成大規模和動態的三維構象,大約2 m長的基因組DNA被折疊,以適應直徑約10 μm的細胞核的大小[1],不同的基因位點之間交互作用,作為基因表達的藍圖發揮作用。生物化學技術通過消化和重連物理空間上距離相近的染色質片段來確定不同位點的空間接近性。作為代表的高通量染色質構象捕獲(high-throughput chromatin conformation capture,Hi-C)技術是一種染色質構象捕獲技術,所需的細胞數較少,并且文庫制備步驟相對簡單,因此在研究三維基因組結構中得到了廣泛應用。Hi-C技術[2]首先對染色質進行甲醛交聯固定,然后使用限制性內切酶對DNA進行切割,并將末端使用生物素標記。接著,DNA片段通過生物素的親和性純化和連接制備成嵌合分子,并進行中斷和生物素標記篩選。最后,將篩選得到的DNA片段進行雙端測序,以計算在基因組中任意兩個染色質區域之間的嵌合分子數,從而獲取染色質交互作用數據。
拓撲相關結構域(topologically associated domains,TAD)[3]是構成染色質三維結構的重要組成部分,被認為是基因調控的功能域,對于細胞分化、發育以及疾病治療具有重要意義[4]。TAD是染色質區域內部比染色質區域之間更頻繁交互作用的結構域。這些結構域大小通常介于200 kb~5 Mb之間[3],并在不同物種和細胞類型中具有高度的保守性。大多數TAD識別方法使用直接從Hi-C接觸矩陣導出的度量來檢測TAD邊界。其中,Dixon等[4]在2012年提出了基于方向性指數DI的方法,該方法通過隨機化方向計算方向性指數來量化基因組區域上下游交互作用的偏置程度,偏置程度越高,其方向性指數越大。通過確定基因組中的DI,可以確定TAD邊界在基因組中的位置。Armatus方法[5]將TAD識別抽象為一個動態規劃問題,其他類似方法有TopDom[6]、CaTCH[7]、GMAP[8]和HiTAD[9]。一些TAD識別方法依賴統計學方法,包括HiCseg[10]、TADtree[11]、PSYCHIC[12]、TADbit[13]和HiCKey[14]。例如,Levy-Leduc等[10]提出了HiCseg,該方法為Hi-C數據定義了一個通用的統計模型,可處理原始數據和標準化數據,然后,利用最大似然方法,將二維分割問題轉化為一維分割問題,通過動態規劃算法解決。除此之外,還有一些基于聚類的方法,包括ClusterTAD[15]、IC-Finder[16]、CASPIAN[17]和TOAST[18]。ClusterTAD方法利用接觸矩陣創建特征,然后在Hi-C數據集上應用層次聚類和K-均值聚類,并應用多種距離度量方式。有一些基于網絡的方法可用于TAD的識別,包括MrTADFinder[19]和3DNetMod[20]。MrTADFinder方法利用染色質接觸圖中的TAD與網絡中密集連接的模塊之間的相似性,將TAD的識別視為一個網絡優化問題。然而,現有的TAD識別方法主要基于染色質物理交互頻率的變化、染色質分割和機器學習,但是這些方法存在一定的局限性,例如識別的TAD質量低、計算成本高、參數較多或參數敏感問題。本文提出一種考慮TAD區域內基因組序列片段的特定密度,利用聚類識別TAD的邊界和內部結構,提高所識別TAD質量,從而進一步推動對染色質空間結構和功能的理解。
1 方法
本文提出一種基于密度聚類的TAD識別算法ATLANTIC,從而從Hi-C接觸矩陣中準確識別TAD邊界。算法總體流程圖如圖1所示,ATLANTIC首先將預處理后的Hi-C矩陣數據作為輸入,利用基于密度的無監督聚類算法根據基因組節點間的交互作用將染色質劃分為不同簇,并從簇中提取TAD邊界。
圖1
采用ATLANTIC方法進行拓撲相關結構域識別的流程圖
Figure1.
TAD identification flow chart of ATLANTIC method
1.1 數據預處理模塊
Hi-C數據標準化是一項重要的數據預處理步驟,用于消除在實驗過程中由于各種非隨機因素引入的樣本間系統偏差。這些偏差可能會導致Hi-C接觸矩陣中的一些區域具有較高或較低的讀數,從而影響后續的數據分析和解釋。因此,對Hi-C數據進行標準化處理是保證數據質量和準確性的重要前提。近年來,諸多標準化方法被提出,如ICE[21]、KR[22]等。其中,KR方法是一種被廣泛使用的數學方法,它是一種矩陣平衡的方法,通過調整每個基因組區域的總Hi-C讀數來消除全局偏差。首先計算原始的Hi-C接觸矩陣每行和每列的總讀數,然后使用這些總讀數計算每個基因組區域的尺度向量;接著,使用非精確牛頓迭代法計算尺度向量,通過多次迭代來不斷調整每個區域的尺度因子,使得每個區域的總讀數與整個基因組的平均總讀數相等;最后,使用對角尺度法將矩陣轉換為雙隨機矩陣,以消除樣本間的系統偏差。KR方法在Hi-C數據標準化中具有明顯的優勢,可以消除測量偏差、保留染色質結構信息并且計算速度相對較快。而ICE[21]通過迭代矯正和特征分解的方法進行偏差消除,但是不僅需要Hi-C接觸矩陣作為輸入,還需要依賴于各種附加信息,例如限制位點、基因組序列和可映射性評分。因此,本文選擇KR方法作為Hi-C接觸矩陣歸一化的方法。
1.2 聚類模塊
聚類模塊使用預處理后的Hi-C接觸矩陣作為輸入,目的是從中提取TAD結構特征。該模塊使用了一種基于密度的空間聚類方法(ordering points to identify the clustering structure,OPTICS)[23]。該算法基于數據集的密度和距離信息來刻畫聚類結構,而無需指定簇的數量。因此,可以自適應地確定簇的數量,從而識別TAD結構,即使在不確定TAD數量的情況下也能做到這一點。具體來說,對于給定的一組Hi-C數據集,基于密度的聚類方法通過以下幾個步驟來獲取聚類結果。
(1)計算距離矩陣。計算距離矩陣時,算法會首先根據輸入的數據點構建一個k-d樹。k-d樹[24]是一種用于快速查找k維空間中最近鄰點的二叉樹結構,可以將多維空間劃分為多個子空間,并在每個子空間中遞歸地構建k-d樹,從而實現高效的最近鄰查詢。距離矩陣被用于確定兩個樣本之間是否是密集連接的關系,可以使用不同的距離度量方式,包括曼哈頓距離、歐幾里得距離、切比雪夫距離。
(2)計算核心距離和核心對象。使用不同的距離度量方式計算樣本點的核心距離,記作。遍歷中的基因位點,如果其核心距離小于鄰域半徑,則將其加入到核心對象集合中。
(3)生成最小生成樹。將距離矩陣視為一個帶權無向圖,每個數據點代表一個節點,距離代表邊的權重,然后使用Prim方法生成最小生成樹。從一個任意節點出發,選擇一個到該節點距離最近的并且未訪問的節點,將其加入生成樹中,并將該節點到其它未訪問節點的距離更新為更小的值,重復此過程,直到生成樹包含所有節點為止。
(4)計算可達距離和可達性圖。隨機選取核心對象集合中一個核心點出發,首先將其標記為已處理;然后,基于最小生成樹找到所有密度可達對象,將對象的可達距離定義為的核心距離與之間距離的最大值;最后,根據可達距離依次遍歷未處理過的樣本點,生成可達性圖。
(5)識別聚類簇。根據上個步驟生成排序后的可達有序列表,通過引入坡度的思想來表征可達列表中相鄰點之間可達距離差距的大小。首先計算連續點的可達性距離的坡度。在形成聚類時有兩種情況,即陡降或陡升區域,其中劃分聚類的可達性距離差為負值或正值,即當第一個觀察點的可達性分別高于或低于第二個觀察點。對于一個樣本點p,當其滿足 時,說明p的可達距離比它的下一個樣本點低不止 的陡度,則稱其為 上升點,點p在 上升區域。其中 表示p的可達距離。同樣地,當其滿足 時,表示p在 下降區域。對于一個區間 ,它被劃分為一個簇的標準是:簇的起點被包含在 陡峭下降區域D中,簇的終點被包含在 陡峭上升區域U中。同時集群數量不小于MinPts個點,群集中所有點的可達值必須至少比D第一點的可達性值和U結束后第一點的可達性值低 。基于以上標準將可達性圖提取出不同類別的簇結構。
1.3 邊界提取模塊
本節介紹了聚類模塊用于標記基因組位點的方法。在這個過程中,每個基因組bin都被賦予了一個類別標簽。研究表明,TAD的區間長度通常在200 kb~5 Mb之間[3]。因此可使用這些標簽來確定TAD的位置,具體而言,將擁有相同標簽的基因組位點組成的連續區域看作是一個TAD;接著,利用染色質位點數量和分辨率等因素來確定這個連續區域是否應該被劃分為一個TAD,如果連續區域的長度超過200 kb,則可以被認定為一個TAD。例如,對50 kb分辨率下的Hi-C接觸矩陣數據進行分析,那么必須至少包含四個具有相同標簽的基因組bin的一段連續區域才被認為是一個TAD。這可以確保識別出長度足夠的TAD,并避免將過短的連續區域識別為TAD。總之,該模塊使用聚類模塊中的標簽信息來幫助識別TAD的位置。通過考慮染色質位點數量和分辨率,可以確保只識別出足夠長的TAD,從而提高TAD識別的準確性。
2 實驗結果與分析
2.1 數據集
2.1.1 模擬Hi-C數據
實驗使用了兩個模擬數據集,一個是Wang等[25]在實驗中生成的模擬接觸矩陣數據集,是使用DI方法[4]在人胚肺成纖維細胞系IMR-90中生成的結構域模擬矩陣。另一個是Trussart等[26]實驗生成的模擬數據集。使用聚合物建模,模擬了具有不同結構的單個染色質的6個人工生成的基因組(稱為“玩具基因組”),從中提取了168個模擬交互作用矩陣,它們的噪聲水平和結構多樣性不斷增加。本章使用了噪聲水平分別為50和100,線性密度分別為40、75、150 bp/nm的模擬Hi-C矩陣數據。
2.1.2 真實Hi-C數據
實驗使用了來自人類B淋巴細胞GM12878的真實數據集。為了獲取原始數據,首先從4dnucleome平臺獲取登錄號為4DNFI1UEG1HD的.hic格式文件;接著,使用Jucier工具[27]對該文件進行處理,得到分辨率分別為25 kb和50 kb的原始Hi-C接觸矩陣,并對它進行了KR歸一化處理。這些數據將被用于后續的實驗分析和結果展示。
2.1.3 染色質免疫共沉淀(chromatin immunoprecipitation,ChIP)數據
實驗分析使用來自DNA元件百科全書(Encyclopedia of DNA Elements,ENCODE)項目的ChIP-Seq數據來分析基因組位點CTCF以及其他組蛋白修飾的富集。其中CTCF、H3K4me3、SMC3和POLR2A ChIP-seq數據在ENCODE平臺的登錄號分別為ENCSR000EAD、ENCSR057BWO、ENCSR000DZP和ENCSR000EFK。主要使用bed narrowpeak格式的ChIP-seq數據,這些文件包含了染色體號、起始位點和終止位點等信息,可用于確定各種蛋白質結合位點在基因組上的位置。通過對這些數據進行分析,可以識別各種轉錄因子的peak值,并計算TAD結構邊界處各種轉錄因子的peak數量和密度。
2.2 評價指標
TAD識別的準確性對于深入理解基因組結構和功能具有重要意義,為了對TAD的識別結果進行評價和對比,設定TAD質量分數、聚類效果評價指標Davies-Bouldin指數和輪廓系數三種評價指標。
TAD結構的一個顯著特征是,對于給定TAD內的區域,它們具有更高的接觸頻率分布,而TAD外的區域之間接觸頻率很少。這意味著TAD內部的區域應該具有更高的相似性和一致性,而TAD之間的區域應該具有較小的相似性。基于這一性質,將識別出的第個TAD內部各個bin之間的接觸頻率平均值記作,將與相鄰之間的接觸頻率平均值記作,則TAD質量分數可用公式(1)計算得到。定義每個接觸矩陣得到的一組TAD的TAD Quality分數是這組TAD的質量分數平均值。
|
對于聚類效果,使用Davies-Bouldin指數(Davies-Bouldin index,DBI)和輪廓系數(Silhouette Index,SI)進行評估。DBI是一種常用的用于評估聚類效果的指標,它通過比較類內距離和類間距離來衡量聚類質量。DBI值越接近于0,表示聚類結果越好。
SI也經常被用來評估聚類的優劣,它通過比較每個樣本與所屬簇內部其他樣本的相似度和與其他簇中樣本的相似度,來度量聚類結果的內聚性和分離性。SI取值范圍在?1~1之間。SI越接近1,說明數據點聚類得越好,不同簇之間的距離越遠,同一簇內部的數據點之間的距離越近。反之,亦然。當SI接近0時,說明數據點處于聚類邊界附近,聚類效果不明顯。
為了評價算法識別的域邊界的準確性,研究人員還經常使用ChIP技術結合高通量測序來確定染色質上的特定修飾標記和結合蛋白的富集情況。其中,CTCF是一種常見的染色質結合蛋白,在真核生物基因組的三維組織中扮演著重要的角色。作為核心結構蛋白之一,CTCF幫助建立了染色質的組織結構[28],它可以與其他轉錄因子共同作用于染色質結構域的邊界處,形成DNA環,這些環的位置通常與TAD的邊界相關。因此,CTCF被認為是TAD邊界的主要因子之一。RAD21和SMC3是Cohesin復合物的兩個重要組成部分,通常出現在TAD邊界[29]。一些研究表明,POLR2A可以結合在TAD邊界區域。例如,在人類胚胎干細胞中,POLR2A的結合顯著富集在TAD邊界區域。已有研究表明,H3K4me3、H3K36me3等因子在TAD邊界處大量富集[4]。因此選擇CTCF、H3K4me3、SMC3以及POLR2A四種ChIP-seq因子在TAD邊界附近區域的富集情況來衡量算法識別的準確性。
2.3 基準模型
為了驗證ATLANTIC的性能,本文選擇了TopDom[6]、Insulation Score[30]、EAST[31]、ClusterTAD[15]幾種TAD劃分算法與ATLANTIC方法進行對比。這幾種算法識別出的TAD結構均是非分層的,其中ClusterTAD方法也是基于聚類思想的TAD識別工具,其余幾種是常見的利用Hi-C矩陣度量或者統計的方法提供的標準TAD識別工具。
2.4 算法參數
OPTICS算法是一種密度聚類算法,其核心思想是基于密度可達性來確定樣本點之間的聚類關系。在OPTICS算法中,MinPts是一個非常重要的參數,它決定了一個樣本點被視為核心點的最小鄰域樣本數。一般來說,較小的MinPts值會導致聚類更加細致,但也可能會導致過度聚類。相反,較大的MinPts值會導致聚類更加粗略,但可能會忽略掉一些重要的聚類。研究表明,TAD大小通常在200 kb~5Mb之間,因此測試MinPts取值在[3, 14]之間的影響可以有效地驗證生成的TAD質量。此外,距離度量方法也是OPTICS算法中的另一個重要參數,它可以是歐幾里得距離、曼哈頓距離、切比雪夫距離、余弦相似度等。在實踐中,應根據具體的數據集和應用場景選擇最合適的距離度量方法。
使用GM12878細胞系的19號染色質Hi-C數據作為輸入,測試不同參數對TAD的劃分結果質量的影響。如圖2a所示,當其余參數取默認值、MinPts取值為4時,識別出的TAD具有最高質量,TAD Quality達到了2 586.816。如圖2b所示,分別測試了幾種常用的距離度量方法(余弦距離、切比雪夫距離、歐氏距離以及曼哈頓距離)的TAD Quality。結果表明,該方法在距離度量的確定上具有魯棒性,不同方法均取得了較好的識別效果,其中曼哈頓距離度量方式具有最高的TAD Quality(3 474.173)。
圖2
不同參數下的TAD質量對比
a. 不同MinPts取值的TAD Quality對比;b. 不同距離度量方式的TAD Quality對比
Figure2. Comparison of TAD quality under different parametersa. TAD Quality comparison of different MinPts values; b. TAD Quality comparison of different distance metrics
2.5 模擬數據集評估ATLANTIC
首先將ATLANTIC方法應用到了模擬Hi-C接觸矩陣數據集上,使用評價指標和方法對比來驗證方法的可用性以及可靠性。第一個模擬數據集是來自Wang等[25]發表的模擬Hi-C矩陣,該數據具有明顯的拓撲結構域特征,便于驗證方法的有效性。從圖3中可以看出,由ATLANTIC方法識別出的TAD邊界與Hi-C熱圖中顯示的TAD矩形結構緊密契合,表明該方法能夠較準確地提取Hi-C數據中的TAD邊界位置。同時,將不同方法識別出的TAD結果進行對比,發現由TopDom、ClusterTAD以及EAST方法識別出的邊界與ATLANTIC方法完全一致,側面驗證了ATLANTIC方法的有效性。
圖3
模擬數據集下不同方法識別出的TAD邊界可視化結果
Figure3.
Visualization of TAD boundaries identified by different methods under simulated data
接著,將ATLANTIC應用到了Trussart等[26]通過實驗的模擬Hi-C數據上,并使用了不同線性密度和不同噪聲下的模擬數據集。實驗結果證明了ATLANTIC在不同噪聲水平上識別的TAD數量更加穩定,說明了該方法具有一定的魯棒性和抗噪聲能力(具體圖片參見附件1)。此外,ATLANTIC的識別結果具有較高水平的TAD Quality,證明了ATLANTIC方法的有效性(具體數據參見附件2)。
2.6 真實數據集評估ATLANTIC
除了模擬數據集,本文還將ATLANTIC方法應用到了GM12878細胞系數據集。首先使用的是1~22號染色體分辨率為50 kb的Hi-C接觸矩陣,應用曼哈頓距離度量的ATLANTIC方法進行TAD識別。識別結果較好地貼合了原始Hi-C矩陣數據(具體圖片參見附件3)。識別結果的TAD Quality分數均值達到2 301.879,在12號染色體上TAD Quality達到3 603.246(具體圖片參見附件4a)。SI值接近1,DBI值較低,處于1.5左右,意味著聚類結果具有較好的分離性和緊密性(具體圖片參見附件4b)。
同時,實驗以GM12878的19號染色體50 kb分辨率下的Hi-C接觸矩陣作為輸入,驗證了CTCF、H3K4me3、SMC3和POLR2A四種ChIP-seq信號因子在TAD邊界處的富集數量。實驗表明,由ATLANTIC識別出的TAD在邊界附近出現了大量蛋白結合位點,尤其是在TAD邊界處出現了大量促進轉錄相關因子和組蛋白修飾的富集(具體圖片參見附件5)。其中,CTCF結合位點在TAD邊界出現大量富集,表明它們可能在染色質空間中起到隔離相鄰TAD的絕緣體的作用[4]。啟動子標記(H3K4me3)在邊界區域的高度富集表明部分TAD邊界區域可能是轉錄激活位點。該結果驗證了ATLANTIC方法識別TAD的準確性,同時表明本方法識別出了具有明顯生物學特征的TAD結構劃分。
此外,實驗還將ATLANTIC應用到了更高分辨率下的Hi-C數據集。以GM12878的25 kb分辨率的1~22號染色質Hi-C矩陣作為輸入,計算了識別出的TAD質量分數以及聚類效果得分。在不同染色質數據上,TAD Quality分數相對穩定,其中19號染色體識別結果的TAD Quality達到了998.917,并且驗證了聚類的準確性和方法的穩定性(具體圖片參見附件6)。
基于GM12878的50 kb分辨率Hi-C接觸矩陣,本文與其他四種方法在TAD數量、平均長度以及質量分數三個指標上進行了對比。從表1可以看出,各個方法識別出的TAD數量和大小具有相似性,其中TopDom識別出的數量較多,分析原因是由于它識別出的TAD不存在空隙區域,而ATLANTIC方法相比其他算法可得到更高的TAD Quality分數。
實驗還比較了由不同算法識別出的TAD邊界相關蛋白結合位點(CTCF、H3K4me3、RAD21、SMC3四種ChIP-seq信號因子)的富集情況。圖4展示了四種方法在邊界附近相關富集因子的分布核密度曲線,其中藍色曲線為ATLANTIC方法結果。從圖中可以看出,ATLANTIC能夠識別出的邊界位置相關因子富集程度要優于其余幾種方法,說明ATLANTIC方法識別出的TAD邊界更加符合生物學特征。
圖4
不同方法在真實數據集上識別的TAD邊界蛋白結合位點富集概率曲線
Figure4.
Enrichment probability curves of TAD boundary protein binding sites identified by different methods
3 總結與展望
本文提出了一種基于空間密度聚類的染色質TAD邊界識別算法ATLANTIC。該方法通過對Hi-C接觸矩陣中的基因組位點計算可達性圖并提取聚類簇,實現對TAD邊界的識別。方法主要分為三個模塊:首先對原始Hi-C數據進行歸一化,以獲取標準化后的數據;然后將Hi-C矩陣應用于基于密度的聚類方法進行分類成簇;最后根據劃分結果識別連續區域的TAD結構。根據TAD的結構特性和聚類方法的一般特性設置了評價指標,并將該方法運用到了模擬數據集和真實數據集上,結果表明,ATLANTIC方法能夠準確、有效地識別TAD,并在不同分辨率數據集下取得較好效果。實驗還將ATLANTIC方法與已有四種方法進行了對比,結果顯示ATLANTIC方法識別的TAD邊界錨定了更多的CTCF和其他蛋白結合位點因子比例,這些發現對于進一步研究基因組結構與功能的關系具有重要的意義。但ATLANTIC仍然存在一些限制,例如使用高分辨率(25 kb及更高)Hi-C數據來識別TAD結構時,算法的運行時間較長。未來的工作可能包括通過研究并行計算的算法來提高在高分辨率Hi-C數據上的運行效率。
重要聲明
利益沖突聲明:本文全體作者均聲明不存在利益沖突。
作者貢獻聲明:龔海燕負責實驗開展和論文寫作,張司臣負責實驗開展,張曉彤負責論文指導與審閱。
本文附件見本刊網站的電子版本(biomedeng.cn)。