引用本文: 吳勝, 符順丹, 秦亞丹. 兒童支氣管哮喘患者鼻咽微生態特征分析. 華西醫學, 2024, 39(4): 561-566. doi: 10.7507/1002-0179.202309171 復制
版權信息: ?四川大學華西醫院華西期刊社《華西醫學》版權所有,未經授權不得轉載、改編
微生態失調是局部微生物組成的失衡,定義為促進健康或疾病的細菌的減少或增加[1]。目前認為,微生態失調會增加慢性炎癥性疾病的風險,包括支氣管哮喘(以下簡稱“哮喘”)[2]。鼻咽位于鼻子、耳朵、鼻竇和下呼吸道之間,是包含多種微生物群落的分泌物的主要來源[3]。各種內部因素(纖維化、MUC5B 基因的遺傳缺陷)和外部因素(抗菌藥物、吸煙或飲食變化)會導致鼻咽微生物群生態失調[2]。研究發現,鼻咽微生物群生態失調會誘發局部和全身免疫反應,從而形成反饋回路,使局部免疫細胞和微生物群相互交流。這種串擾可能是在兒童中觀察到的腸道微生物群和哮喘之間關聯的免疫學背景[4]。有研究在氣道中發現了失調的微生物群導致的不同先天性和適應性免疫變化,其中包括白細胞介素(interleukin, IL)-17 過量產生、輔助性 T 細胞 17(T helper cell 17, Th17)細胞反應、恒定的自然殺傷 T 細胞和肺部駐留的 γδT 細胞[5]。然而,目前關于兒童哮喘患者鼻咽微生態的研究較少。因此,本研究重點關注哮喘兒童的鼻咽微生態的特征,并進一步分析了鼻咽微生態失調和局部炎癥反應之間的相互作用。
1 對象與方法
1.1 研究對象
回顧性納入 2020 年 11 月—2023 年 3 月在海南省中醫院接受治療的 41 例哮喘兒童。納入標準:由肺病醫生根據陽性哮喘預測指數診斷為哮喘,并通過抗炎治療期間的臨床改善和/或陽性支氣管可逆性試驗進行確認[6]。排除標準:① 伴有影響呼吸道癥狀的食物過敏;② 鼻內鏡檢查禁忌證或鼻黏膜活檢禁忌證;③ 扁桃體三度肥大(通過上呼吸道內鏡檢查確認);④ 免疫缺陷;⑤ 哮喘惡化需要全身施用糖皮質激素;⑥ 肥胖;⑦ 研究人員認為可能影響評估和研究過程的接觸煙草煙霧或其他慢性疾病和臨床狀況;⑧ 就診前 4 周內接受鼻內皮質類固醇治療;⑨ 就診前 2 周內急性呼吸道感染。同時,選擇同期行腺樣體檢查的 26 例健康兒童作為對照組。對照組納入標準:不具有哮喘病史且未被診斷為哮喘。對照組排除標準同哮喘組。本研究由海南省中醫院倫理委員會批準(批準號:2019015),并獲得了所有哮喘患兒或健康體檢兒童父母和法定監護人的書面知情同意。
1.2 鼻黏膜取樣、mRNA 分離和定量聚合酶鏈反應(polymerase chain reaction,PCR)
1.2.1 鼻黏膜取樣
在患兒入院 24 h 內使用塑料刮匙(美國 Rhino-probe 公司)從下鼻甲的前內側采集鼻黏膜樣本。將帶有鼻上皮樣本的刮匙放置在含有 2 mL 冷無菌 RPMI 1640 培養基(美國 Gibco 公司)的 15 mL 管中,并在收集后 3 h 內運輸至生物實驗室。用力搖動刮匙,將組織樣本移入培養基中。然后用無菌鑷子取下刮匙,將樣品渦旋約 30 s。然后將培養基離心(100×g,10 min,室溫),細胞和組織沉淀進一步如下處理。
1.2.2 mRNA 分離
通過輕輕抽吸小心除去 RMPI 1640 殘留物,使用 TRIzol 試劑(日本 Takara 公司)提取總 RNA。使用 NanoDrop ND-1000 分光光度計(美國賽默飛世爾科技公司)測量 RNA 濃度。樣本立即被逆轉錄。
1.2.3 逆轉錄和定量 PCR
使用隨機引物和 Maxima 逆轉錄酶試劑盒(美國賽默飛世爾科技公司)進行逆轉錄反應,一式三份。得到的互補 DNA 樣品儲存在–20℃直到使用。使用 TB Green Reagent Premix Ex Taq Ⅱ(日本 Takara 公司)和 Prism 7500 SDS(美國賽默飛世爾科技公司)進行 PCR。記錄靶基因的 CT 值,使用 2–△△CT 方法計算指示基因的相對表達。將特定基因的表達與作為參照基因的 18S 核糖體 RNA(ribosomal RNA, rRNA)進行比較,并表示為△△CT。引物序列見表1。
1.3 高通量測序文庫制備和測序
使用 PowerSoil DNA 分離試劑盒(美國 Mo Bio Laboratories 公司)從鼻拭子中提取 DNA。用體積為 50 μL 的無核酸酶水洗脫樣品。16S rRNA 作為陰性對照。使用 Qubit 高靈敏度分析試劑盒(美國賽默飛世爾科技公司)測定 DNA 濃度。靶向 16S rRNA 基因 V3-V4 可變區的特異性正向引物(5’-TCGTCGGCAGCGTCAGATGTATAAGACAGCCTACGGGNGGCWGCAG-3’)和反向引物(5’-GTCTCGGGGCTCGGAGATGTATTGTATATGATCC-3’)用于擴增基因組模板 DNA。使用 Illumina 的 NEBNext Library Quant 試劑盒(美國 New England Biolabs 公司)對文庫進行精確定量。然后,將樣品以等摩爾濃度合并,并使用 Illumina MiSeq 平臺(美國 Illumina 公司)作為成對末端測序。
1.4 測序數據的處理和分析
測序后,用 BaseSpace 在線平臺(美國 Illumina 公司)解析 67 個樣品的原始數據。每個樣本的 Fastq 文件使用 trim_galore v.1.18 進行微調。使用 FastQC (v0.11.8)評估預處理前后的讀數質量。使用 Qiime 2(v.2019.10)平臺執行進一步的分析步驟。通過使用 DADA2 插件進行成對末端讀取生成擴增子序列變體(amplicon sequence variant, ASV)。去除所有樣本中頻率低于 118 的 ASV,以對數據進行去噪。基于阿爾法稀疏分析,用于多樣性度量計算的采樣深度設置為 3651。通過 classify-learn 插件使用默認參數完成 ASV 的分類。然后將群落組成與數據庫 Silva 132 進行比較。通過 Shannon 指數評估生物多樣性[5]。
1.5 統計學方法
使用 SPSS 24.0 軟件進行統計分析。通過 Kolmogorov-Smirnov 檢驗確定連續變量的正態性。符合正態分布的計量數據以均數±標準差描述,不符合正態分布的計量數據以中位數(下四分位數,上四分位數)描述。正態分布數據組間比較采用 t 檢驗,非正態分布數據組間比較采用 Mann-Whitney U 檢驗。分類變量以頻數和/或百分比表示,組間比較采用 χ2 檢驗。使用受試者操作特征(receiver operating characteristic, ROC)曲線分析確定生物多樣性降低(由 Shannon 指數反映)最佳分界點,以對納入兒童進行亞組分析。最后,使用帶有 Hodges-Lehmann 多重比較校正的 Kruskal-Wallis H 檢驗用于確定不同 Shannon 指數亞組兒童的鼻部炎癥特征。雙側檢驗水準 α=0.05。
2 結果
2.1 研究對象的臨床特征
67 例兒童被納入分析。參與者的臨床特征如表2 所示。與對照組相比,哮喘組存在特應癥的兒童比例更高,差異有統計學意義(P<0.05)。
2.2 鼻咽細菌群落組成和豐度
在所有參與者中總共檢測到 1700 個分類操作單元。變形菌門具有高度普遍性(占所獲序列總數的 45.7%),其次是厚壁菌門(29.3%)、放線菌門(15.3%)、擬桿菌門(4.4%)。前五大優勢屬分別為假單胞菌屬(22.3%)、棒狀桿菌屬(12.7%)、類芽孢桿菌屬(11.8%)、鏈球菌屬(3.6%)和短桿菌屬(3.2%)。
在門的水平上,與對照組相比,在哮喘組兒童中綠彎菌門、髕骨細菌門、軟壁菌門和硝化螺旋菌門占比增加(P<0.05),迷蹤菌門占比降低(P<0.05),見表3。表4 顯示了兩組中屬成員的豐度比較。與對照組相比,哮喘組兒童中芽孢桿菌屬(厚壁菌門)、瘤胃球菌屬(厚壁菌門)、紅球菌屬(放線菌門)、不動桿菌屬(變形菌門)、莫拉菌屬(變形菌門)和亞細亞菌屬(變形菌門)的成員明顯增加(P<0.05),腸球菌屬(厚壁菌門)、需烷烴型菌屬(變形菌門)、立克次體屬(變形菌門)、根瘤桿菌屬(變形菌門)的成員減少(P<0.05)。
2.3 按疾病分類的細菌多樣性分析
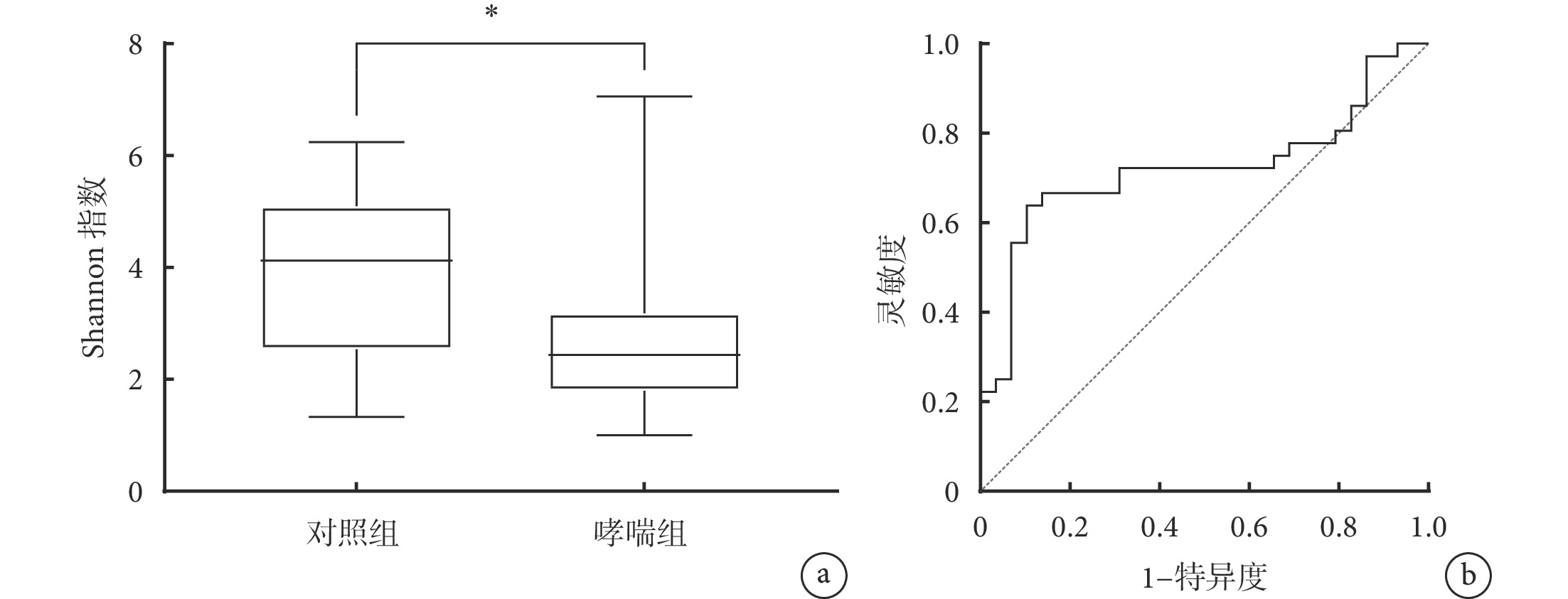
與對照組相比,哮喘組的 Shannon 指數降低(2.63±1.45 vs. 3.90±1.44;t=2.708,P=0.010),見圖1a。根據 ROC 曲線分析確定的 Shannon 指數最佳分界點值為 3.10,并根據分界點對研究對象進行二分法定義,其中 47 例 Shannon 指數≤分界點,另外 20 例高于分界點(圖1b)。
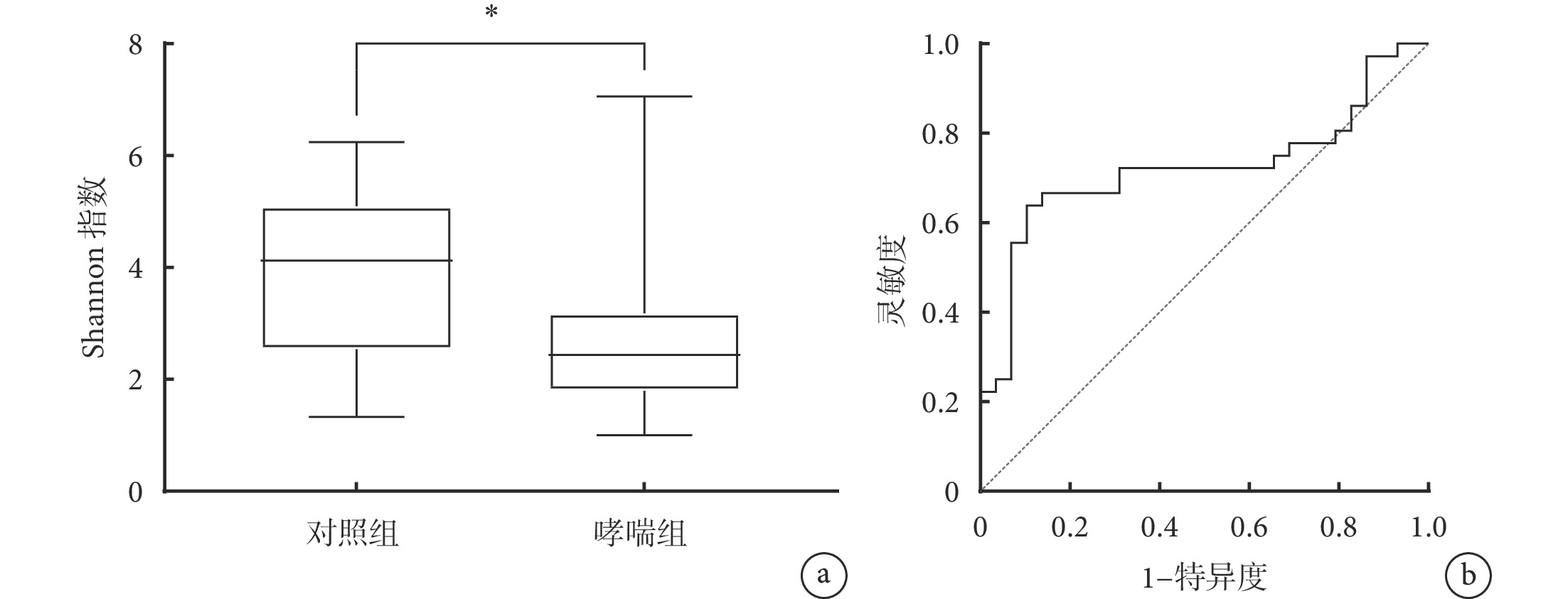 圖1
哮喘組和對照組兒童的細菌多樣性分析
圖1
哮喘組和對照組兒童的細菌多樣性分析
a. 哮喘組和對照組兒童之間的 Shannon 指數比較,*表示
2.4 根據 Shannon 指數二分法比較鼻部炎癥基因表達水平
與 Shannon 指數高于分界點兒童相比,Shannon 指數低于分界點的兒童鼻腔炎癥的特征是 IL17A、T1R3 表達增加(P<0.05),TSLP 表達降低(P<0.05),見表5。
3 討論
通過應用基于 16S rRNA 分子的測序方法,本研究顯示了哮喘組兒童和健康對照組兒童的鼻咽微生物組成的差異,表明細菌群落的變化可能有助于哮喘疾病的發展。此外,本研究中哮喘組在特應癥患兒比例方面顯著高于健康對照組,表明哮喘組兒童對變態反應性疾病的易感性。研究顯示,細菌定植和哮喘之間存在關聯[7]。一項基于傳統培養的鑒定方法研究表明,下咽部被肺炎鏈球菌、流感嗜血桿菌或卡他莫拉菌定植的新生兒在兒童期出現反復喘息和哮喘的風險增加[8]。然而,這些細菌在哮喘發病機制中的作用仍不清楚。
氣道微生物組的組成與兒童哮喘的發展高度相關,因此氣道內特定細菌成員的生態失調可能會加強呼吸道過敏反應的可能性[9]。之前對哮喘患者隊列的研究從下氣道獲得了 5 個主要門:變形菌門、厚壁菌門、放線菌門、擬桿菌門和梭桿菌門,變形菌門的成員包括莫拉菌屬、嗜血菌屬和奈瑟菌屬,在哮喘患者的氣道中占主導地位[10-11]。在本研究中,我們在哮喘兒童中發現了 2 個主要的門,即變形菌門和厚壁菌門,并且變形菌門的莫拉菌屬占主導地位,這與之前的研究一致。該過程的詳細機制仍然需要闡明,但是用莫拉菌屬物種進行的體外測試揭示了這種細菌可誘導肺上皮細胞損傷和炎性細胞因子表達(IL-8、IL-13 和 IL-17),從而導致哮喘的發展[12]。此外,幾份報告揭示了不動桿菌屬在患有氣道過敏癥的成人中比較豐富,而需烷烴型菌屬和立克次體屬不太豐富[13]。在本研究中,哮喘組不動桿菌更豐富,而需烷烴型菌屬和立克次體屬則比健康對照組更少見。這一發現與上文提到的先前研究相一致,這意味著某些細菌屬(即不動桿菌屬)可能在過敏性呼吸道疾病中起重要作用,尤其是對于過敏性體質的人。
如上所述,哮喘可能是通過呼吸道內細菌壁或細菌產物(如內毒素)的成分激活免疫系統而引發的[14]。研究表明,早期暴露于細菌內毒素可防止日后出現特應癥。此外,高水平的細菌內毒素可能會減少過敏和特應性哮喘[15]。這種潛在的保護作用與“衛生假說”一致,該假說認為,在更衛生的環境(即細菌含量較少的環境)中長大的兒童可能會增加患呼吸道過敏的風險[7]。許多當前模型中的微生態失調與上下呼吸道的慢性炎癥有關[16]。這種現象可以從 2 個方面進行解釋。首先,這表明與微生態失調相關的炎癥可能是由于缺乏能夠調節免疫調節機制的特定腐生細菌的保護性菌株[17]。在本研究中,哮喘與由低豐度的細菌定義的微生態失調相關。在另一項針對兒科人群的研究中,哮喘發作與前鼻孔中彎曲桿菌和懶惰狡詐菌的豐度較低有關[18]。這些差異可以通過微生物生物分布來解釋。在本研究中,我們評估了鼻咽部的微生物群,與前鼻孔相比,鼻咽部菌群的健康組成明顯不同[19]。其次,炎癥可能是對致病菌群的自然和明顯反應[20]。因此,鼻咽部微生態失調可影響宿主免疫力,導致肺部炎癥和免疫功能障礙增加,誘發哮喘。
哮喘特異性微生物組成也與較低的生物多樣性有關,這在本研究中通過 Shannon 指數降低反映出來。在這些患者中,我們觀察到了鼻分泌物中 IL17A、T1R3 表達增加。此外,Shannon 指數的降低與頻繁使用抗菌藥物有關,并與鼻咽部芽孢桿菌增加有關。這一發現與之前公布的數據一致[21]。最近研究表明,口服抗菌藥物抑制了微生物群加工的腸道代謝物,并上調了與 3 型先天性淋巴細胞和 IL-17+ 細胞相關的炎癥[22]。TIR3 被發現是微生物群和肥胖之間的聯系[23]。根據上述數據,我們可以推測,IL-17 和 T1R3 的局部表達較高是上呼吸道微生物群生物多樣性減少的免疫學標志。重要的是,在生物多樣性減少的兒童中,鼻腔較高的 IL-17 水平表明,這種現象主要與亞急性炎癥有關。這一發現提示可能存在嚴重的臨床后果,因為我們不能通過局部皮質類固醇對亞急性炎癥有最佳反應。
目前的研究存在一些局限性。首先,樣本量小可能影響結果的統計解釋。其次,研究存在一些其他限制,包括在樣本收集和處理過程中樣本的潛在污染、缺乏對潛在細菌-宿主反應的評估以及缺乏對潛在病毒病原體的識別。未來的微生物組分析應使用更大的樣本量進行,并應包括對細菌和/或病毒與宿主之間潛在相互作用的評估。隨著測序技術的不斷發展,在物種水平上鑒定微生物組成分有望很快成為可能。
總之,本研究數據指出哮喘兒童和健康對照兒童的鼻咽微生態組成和豐度明顯不同。未來需要通過開展相關的功能分析來進一步研究特定微生物組和過敏性呼吸道疾病之間的聯系,并通過臨床和科學研究,以期找到新的治療和預防微生態失調的策略。
利益沖突:所有作者聲明不存在利益沖突。
微生態失調是局部微生物組成的失衡,定義為促進健康或疾病的細菌的減少或增加[1]。目前認為,微生態失調會增加慢性炎癥性疾病的風險,包括支氣管哮喘(以下簡稱“哮喘”)[2]。鼻咽位于鼻子、耳朵、鼻竇和下呼吸道之間,是包含多種微生物群落的分泌物的主要來源[3]。各種內部因素(纖維化、MUC5B 基因的遺傳缺陷)和外部因素(抗菌藥物、吸煙或飲食變化)會導致鼻咽微生物群生態失調[2]。研究發現,鼻咽微生物群生態失調會誘發局部和全身免疫反應,從而形成反饋回路,使局部免疫細胞和微生物群相互交流。這種串擾可能是在兒童中觀察到的腸道微生物群和哮喘之間關聯的免疫學背景[4]。有研究在氣道中發現了失調的微生物群導致的不同先天性和適應性免疫變化,其中包括白細胞介素(interleukin, IL)-17 過量產生、輔助性 T 細胞 17(T helper cell 17, Th17)細胞反應、恒定的自然殺傷 T 細胞和肺部駐留的 γδT 細胞[5]。然而,目前關于兒童哮喘患者鼻咽微生態的研究較少。因此,本研究重點關注哮喘兒童的鼻咽微生態的特征,并進一步分析了鼻咽微生態失調和局部炎癥反應之間的相互作用。
1 對象與方法
1.1 研究對象
回顧性納入 2020 年 11 月—2023 年 3 月在海南省中醫院接受治療的 41 例哮喘兒童。納入標準:由肺病醫生根據陽性哮喘預測指數診斷為哮喘,并通過抗炎治療期間的臨床改善和/或陽性支氣管可逆性試驗進行確認[6]。排除標準:① 伴有影響呼吸道癥狀的食物過敏;② 鼻內鏡檢查禁忌證或鼻黏膜活檢禁忌證;③ 扁桃體三度肥大(通過上呼吸道內鏡檢查確認);④ 免疫缺陷;⑤ 哮喘惡化需要全身施用糖皮質激素;⑥ 肥胖;⑦ 研究人員認為可能影響評估和研究過程的接觸煙草煙霧或其他慢性疾病和臨床狀況;⑧ 就診前 4 周內接受鼻內皮質類固醇治療;⑨ 就診前 2 周內急性呼吸道感染。同時,選擇同期行腺樣體檢查的 26 例健康兒童作為對照組。對照組納入標準:不具有哮喘病史且未被診斷為哮喘。對照組排除標準同哮喘組。本研究由海南省中醫院倫理委員會批準(批準號:2019015),并獲得了所有哮喘患兒或健康體檢兒童父母和法定監護人的書面知情同意。
1.2 鼻黏膜取樣、mRNA 分離和定量聚合酶鏈反應(polymerase chain reaction,PCR)
1.2.1 鼻黏膜取樣
在患兒入院 24 h 內使用塑料刮匙(美國 Rhino-probe 公司)從下鼻甲的前內側采集鼻黏膜樣本。將帶有鼻上皮樣本的刮匙放置在含有 2 mL 冷無菌 RPMI 1640 培養基(美國 Gibco 公司)的 15 mL 管中,并在收集后 3 h 內運輸至生物實驗室。用力搖動刮匙,將組織樣本移入培養基中。然后用無菌鑷子取下刮匙,將樣品渦旋約 30 s。然后將培養基離心(100×g,10 min,室溫),細胞和組織沉淀進一步如下處理。
1.2.2 mRNA 分離
通過輕輕抽吸小心除去 RMPI 1640 殘留物,使用 TRIzol 試劑(日本 Takara 公司)提取總 RNA。使用 NanoDrop ND-1000 分光光度計(美國賽默飛世爾科技公司)測量 RNA 濃度。樣本立即被逆轉錄。
1.2.3 逆轉錄和定量 PCR
使用隨機引物和 Maxima 逆轉錄酶試劑盒(美國賽默飛世爾科技公司)進行逆轉錄反應,一式三份。得到的互補 DNA 樣品儲存在–20℃直到使用。使用 TB Green Reagent Premix Ex Taq Ⅱ(日本 Takara 公司)和 Prism 7500 SDS(美國賽默飛世爾科技公司)進行 PCR。記錄靶基因的 CT 值,使用 2–△△CT 方法計算指示基因的相對表達。將特定基因的表達與作為參照基因的 18S 核糖體 RNA(ribosomal RNA, rRNA)進行比較,并表示為△△CT。引物序列見表1。
1.3 高通量測序文庫制備和測序
使用 PowerSoil DNA 分離試劑盒(美國 Mo Bio Laboratories 公司)從鼻拭子中提取 DNA。用體積為 50 μL 的無核酸酶水洗脫樣品。16S rRNA 作為陰性對照。使用 Qubit 高靈敏度分析試劑盒(美國賽默飛世爾科技公司)測定 DNA 濃度。靶向 16S rRNA 基因 V3-V4 可變區的特異性正向引物(5’-TCGTCGGCAGCGTCAGATGTATAAGACAGCCTACGGGNGGCWGCAG-3’)和反向引物(5’-GTCTCGGGGCTCGGAGATGTATTGTATATGATCC-3’)用于擴增基因組模板 DNA。使用 Illumina 的 NEBNext Library Quant 試劑盒(美國 New England Biolabs 公司)對文庫進行精確定量。然后,將樣品以等摩爾濃度合并,并使用 Illumina MiSeq 平臺(美國 Illumina 公司)作為成對末端測序。
1.4 測序數據的處理和分析
測序后,用 BaseSpace 在線平臺(美國 Illumina 公司)解析 67 個樣品的原始數據。每個樣本的 Fastq 文件使用 trim_galore v.1.18 進行微調。使用 FastQC (v0.11.8)評估預處理前后的讀數質量。使用 Qiime 2(v.2019.10)平臺執行進一步的分析步驟。通過使用 DADA2 插件進行成對末端讀取生成擴增子序列變體(amplicon sequence variant, ASV)。去除所有樣本中頻率低于 118 的 ASV,以對數據進行去噪。基于阿爾法稀疏分析,用于多樣性度量計算的采樣深度設置為 3651。通過 classify-learn 插件使用默認參數完成 ASV 的分類。然后將群落組成與數據庫 Silva 132 進行比較。通過 Shannon 指數評估生物多樣性[5]。
1.5 統計學方法
使用 SPSS 24.0 軟件進行統計分析。通過 Kolmogorov-Smirnov 檢驗確定連續變量的正態性。符合正態分布的計量數據以均數±標準差描述,不符合正態分布的計量數據以中位數(下四分位數,上四分位數)描述。正態分布數據組間比較采用 t 檢驗,非正態分布數據組間比較采用 Mann-Whitney U 檢驗。分類變量以頻數和/或百分比表示,組間比較采用 χ2 檢驗。使用受試者操作特征(receiver operating characteristic, ROC)曲線分析確定生物多樣性降低(由 Shannon 指數反映)最佳分界點,以對納入兒童進行亞組分析。最后,使用帶有 Hodges-Lehmann 多重比較校正的 Kruskal-Wallis H 檢驗用于確定不同 Shannon 指數亞組兒童的鼻部炎癥特征。雙側檢驗水準 α=0.05。
2 結果
2.1 研究對象的臨床特征
67 例兒童被納入分析。參與者的臨床特征如表2 所示。與對照組相比,哮喘組存在特應癥的兒童比例更高,差異有統計學意義(P<0.05)。
2.2 鼻咽細菌群落組成和豐度
在所有參與者中總共檢測到 1700 個分類操作單元。變形菌門具有高度普遍性(占所獲序列總數的 45.7%),其次是厚壁菌門(29.3%)、放線菌門(15.3%)、擬桿菌門(4.4%)。前五大優勢屬分別為假單胞菌屬(22.3%)、棒狀桿菌屬(12.7%)、類芽孢桿菌屬(11.8%)、鏈球菌屬(3.6%)和短桿菌屬(3.2%)。
在門的水平上,與對照組相比,在哮喘組兒童中綠彎菌門、髕骨細菌門、軟壁菌門和硝化螺旋菌門占比增加(P<0.05),迷蹤菌門占比降低(P<0.05),見表3。表4 顯示了兩組中屬成員的豐度比較。與對照組相比,哮喘組兒童中芽孢桿菌屬(厚壁菌門)、瘤胃球菌屬(厚壁菌門)、紅球菌屬(放線菌門)、不動桿菌屬(變形菌門)、莫拉菌屬(變形菌門)和亞細亞菌屬(變形菌門)的成員明顯增加(P<0.05),腸球菌屬(厚壁菌門)、需烷烴型菌屬(變形菌門)、立克次體屬(變形菌門)、根瘤桿菌屬(變形菌門)的成員減少(P<0.05)。
2.3 按疾病分類的細菌多樣性分析
與對照組相比,哮喘組的 Shannon 指數降低(2.63±1.45 vs. 3.90±1.44;t=2.708,P=0.010),見圖1a。根據 ROC 曲線分析確定的 Shannon 指數最佳分界點值為 3.10,并根據分界點對研究對象進行二分法定義,其中 47 例 Shannon 指數≤分界點,另外 20 例高于分界點(圖1b)。
圖1
哮喘組和對照組兒童的細菌多樣性分析
a. 哮喘組和對照組兒童之間的 Shannon 指數比較,*表示
2.4 根據 Shannon 指數二分法比較鼻部炎癥基因表達水平
與 Shannon 指數高于分界點兒童相比,Shannon 指數低于分界點的兒童鼻腔炎癥的特征是 IL17A、T1R3 表達增加(P<0.05),TSLP 表達降低(P<0.05),見表5。
3 討論
通過應用基于 16S rRNA 分子的測序方法,本研究顯示了哮喘組兒童和健康對照組兒童的鼻咽微生物組成的差異,表明細菌群落的變化可能有助于哮喘疾病的發展。此外,本研究中哮喘組在特應癥患兒比例方面顯著高于健康對照組,表明哮喘組兒童對變態反應性疾病的易感性。研究顯示,細菌定植和哮喘之間存在關聯[7]。一項基于傳統培養的鑒定方法研究表明,下咽部被肺炎鏈球菌、流感嗜血桿菌或卡他莫拉菌定植的新生兒在兒童期出現反復喘息和哮喘的風險增加[8]。然而,這些細菌在哮喘發病機制中的作用仍不清楚。
氣道微生物組的組成與兒童哮喘的發展高度相關,因此氣道內特定細菌成員的生態失調可能會加強呼吸道過敏反應的可能性[9]。之前對哮喘患者隊列的研究從下氣道獲得了 5 個主要門:變形菌門、厚壁菌門、放線菌門、擬桿菌門和梭桿菌門,變形菌門的成員包括莫拉菌屬、嗜血菌屬和奈瑟菌屬,在哮喘患者的氣道中占主導地位[10-11]。在本研究中,我們在哮喘兒童中發現了 2 個主要的門,即變形菌門和厚壁菌門,并且變形菌門的莫拉菌屬占主導地位,這與之前的研究一致。該過程的詳細機制仍然需要闡明,但是用莫拉菌屬物種進行的體外測試揭示了這種細菌可誘導肺上皮細胞損傷和炎性細胞因子表達(IL-8、IL-13 和 IL-17),從而導致哮喘的發展[12]。此外,幾份報告揭示了不動桿菌屬在患有氣道過敏癥的成人中比較豐富,而需烷烴型菌屬和立克次體屬不太豐富[13]。在本研究中,哮喘組不動桿菌更豐富,而需烷烴型菌屬和立克次體屬則比健康對照組更少見。這一發現與上文提到的先前研究相一致,這意味著某些細菌屬(即不動桿菌屬)可能在過敏性呼吸道疾病中起重要作用,尤其是對于過敏性體質的人。
如上所述,哮喘可能是通過呼吸道內細菌壁或細菌產物(如內毒素)的成分激活免疫系統而引發的[14]。研究表明,早期暴露于細菌內毒素可防止日后出現特應癥。此外,高水平的細菌內毒素可能會減少過敏和特應性哮喘[15]。這種潛在的保護作用與“衛生假說”一致,該假說認為,在更衛生的環境(即細菌含量較少的環境)中長大的兒童可能會增加患呼吸道過敏的風險[7]。許多當前模型中的微生態失調與上下呼吸道的慢性炎癥有關[16]。這種現象可以從 2 個方面進行解釋。首先,這表明與微生態失調相關的炎癥可能是由于缺乏能夠調節免疫調節機制的特定腐生細菌的保護性菌株[17]。在本研究中,哮喘與由低豐度的細菌定義的微生態失調相關。在另一項針對兒科人群的研究中,哮喘發作與前鼻孔中彎曲桿菌和懶惰狡詐菌的豐度較低有關[18]。這些差異可以通過微生物生物分布來解釋。在本研究中,我們評估了鼻咽部的微生物群,與前鼻孔相比,鼻咽部菌群的健康組成明顯不同[19]。其次,炎癥可能是對致病菌群的自然和明顯反應[20]。因此,鼻咽部微生態失調可影響宿主免疫力,導致肺部炎癥和免疫功能障礙增加,誘發哮喘。
哮喘特異性微生物組成也與較低的生物多樣性有關,這在本研究中通過 Shannon 指數降低反映出來。在這些患者中,我們觀察到了鼻分泌物中 IL17A、T1R3 表達增加。此外,Shannon 指數的降低與頻繁使用抗菌藥物有關,并與鼻咽部芽孢桿菌增加有關。這一發現與之前公布的數據一致[21]。最近研究表明,口服抗菌藥物抑制了微生物群加工的腸道代謝物,并上調了與 3 型先天性淋巴細胞和 IL-17+ 細胞相關的炎癥[22]。TIR3 被發現是微生物群和肥胖之間的聯系[23]。根據上述數據,我們可以推測,IL-17 和 T1R3 的局部表達較高是上呼吸道微生物群生物多樣性減少的免疫學標志。重要的是,在生物多樣性減少的兒童中,鼻腔較高的 IL-17 水平表明,這種現象主要與亞急性炎癥有關。這一發現提示可能存在嚴重的臨床后果,因為我們不能通過局部皮質類固醇對亞急性炎癥有最佳反應。
目前的研究存在一些局限性。首先,樣本量小可能影響結果的統計解釋。其次,研究存在一些其他限制,包括在樣本收集和處理過程中樣本的潛在污染、缺乏對潛在細菌-宿主反應的評估以及缺乏對潛在病毒病原體的識別。未來的微生物組分析應使用更大的樣本量進行,并應包括對細菌和/或病毒與宿主之間潛在相互作用的評估。隨著測序技術的不斷發展,在物種水平上鑒定微生物組成分有望很快成為可能。
總之,本研究數據指出哮喘兒童和健康對照兒童的鼻咽微生態組成和豐度明顯不同。未來需要通過開展相關的功能分析來進一步研究特定微生物組和過敏性呼吸道疾病之間的聯系,并通過臨床和科學研究,以期找到新的治療和預防微生態失調的策略。
利益沖突:所有作者聲明不存在利益沖突。