引用本文: 汪倩, 余春玫, 楊潔, 羅蓉. Cockayne 綜合征一例. 華西醫學, 2025, 40(1): 162-164. doi: 10.7507/1002-0179.202403294 復制
版權信息: ?四川大學華西醫院華西期刊社《華西醫學》版權所有,未經授權不得轉載、改編
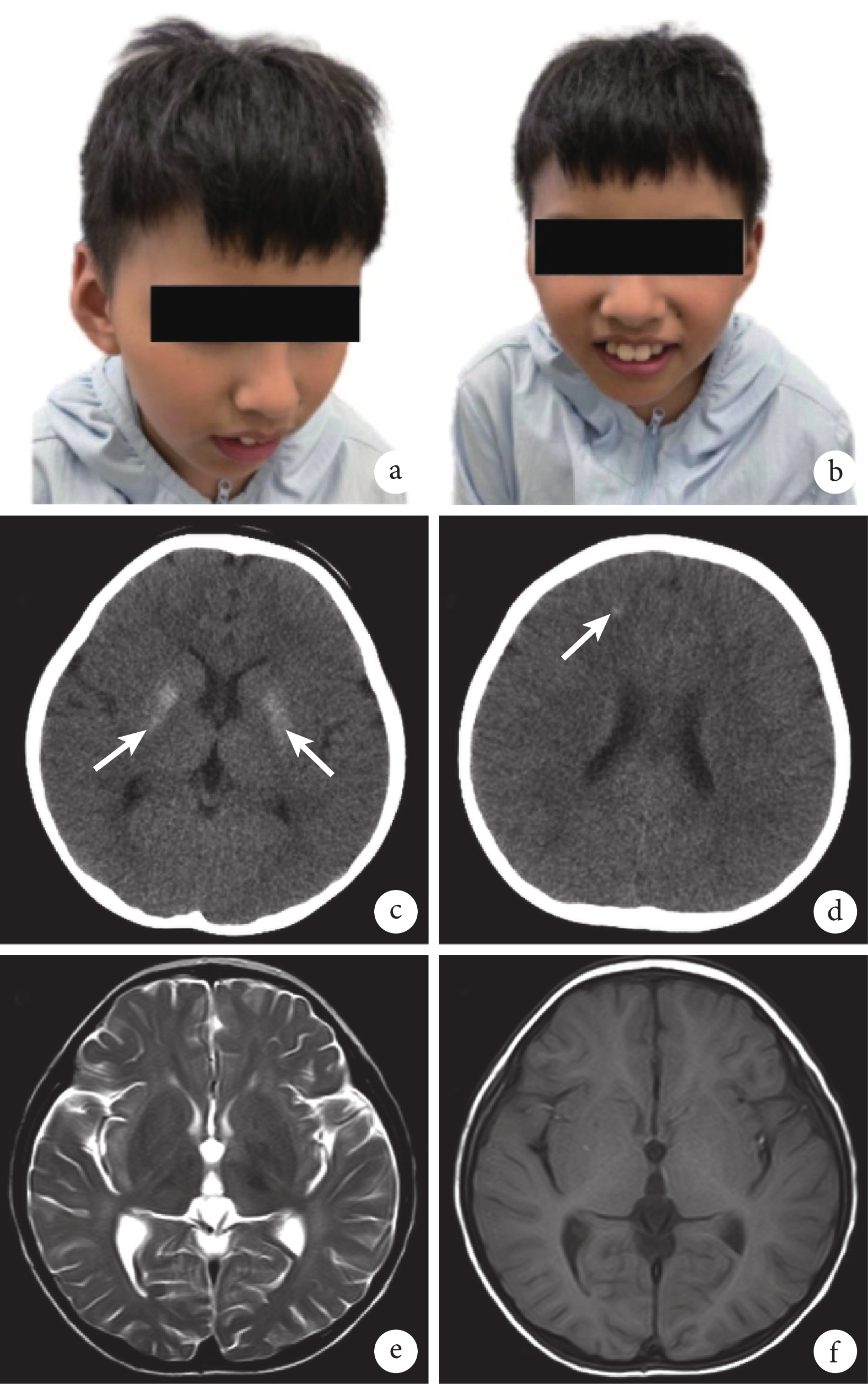
病例介紹 患兒,男,8 歲,因“精神運動發育落后 7 年”于 2023 年 7 月前往四川大學華西第二醫院兒童神經科門診就診。該男性患兒系孕 1 產 1,父母體健,非近親婚配,母親孕期無異常,足月順產,產時無窒息、搶救史。患兒 1 歲 7 個月時開始獨立行走,后逐漸出現走路姿勢異常,易摔跤,至今不可蹲、跑;1 歲會說話,現僅可說短句,后面容逐漸改變,出現鼻梁高聳、眼窩凹陷和大耳;2 歲后曬后面部皮膚出現紅腫,后出現蛻皮,伴隨少許色素沉著;3 歲時有“熱性驚厥”病史,后未再抽搐。患兒家族中無類似患者。體格檢查:神志清楚,精神可;身高 121 cm(<P3),體重 22 kg(<P10),頭圍 46 cm(<P3),體質量指數 15 kg/m2;皮膚干燥、粗糙,面部皮膚散在少許色素沉著,大耳,雙側眼窩凹陷,鼻梁高聳,小下頜(圖1a、1b);心肺腹未見異常;脊柱未見異常;雙手掌紋正常,雙手精細動作差,四肢肌力正常,雙下肢肌張力稍增高,雙側膝反射亢進,雙踝關節背屈受限,病理征陰性;肢體協調性欠佳,易摔倒,可理解并說出簡單短句。輔助檢查:2018 年患兒頭顱 CT 顯示雙側基底節區對稱性分布高密度影,雙側額葉少許小點片狀高密度影(圖1c、1d);同年頭顱 MRI 有髓鞘化成熟障礙征象(圖1e、1f);2023 年頭顱 CT 提示雙側額頂葉皮髓質交界區、豆狀核、齒狀核見對稱分布鈣化灶。聽誘發電位檢查顯示雙側Ⅲ波潛伏期可疑延長,雙側Ⅳ、Ⅴ波潛伏期延長,雙側Ⅰ-Ⅴ波間潛伏期延長,聽閾顯示雙側Ⅴ波均未引出。視覺誘發電位顯示雙側 P100 波潛伏期可疑延長。肝功能檢查顯示丙氨酸轉氨酶及天冬氨酸轉氨酶有一過性升高,后復查水平正常。下肢周圍神經功能檢測、視頻腦電圖、血尿代謝篩查、血氨、乳酸、銅藍蛋白及腎功能檢查均未見異常。患兒就診時行韋氏兒童智力量表測試,顯示其處于智力低下水平(總智商分數<41 分)。
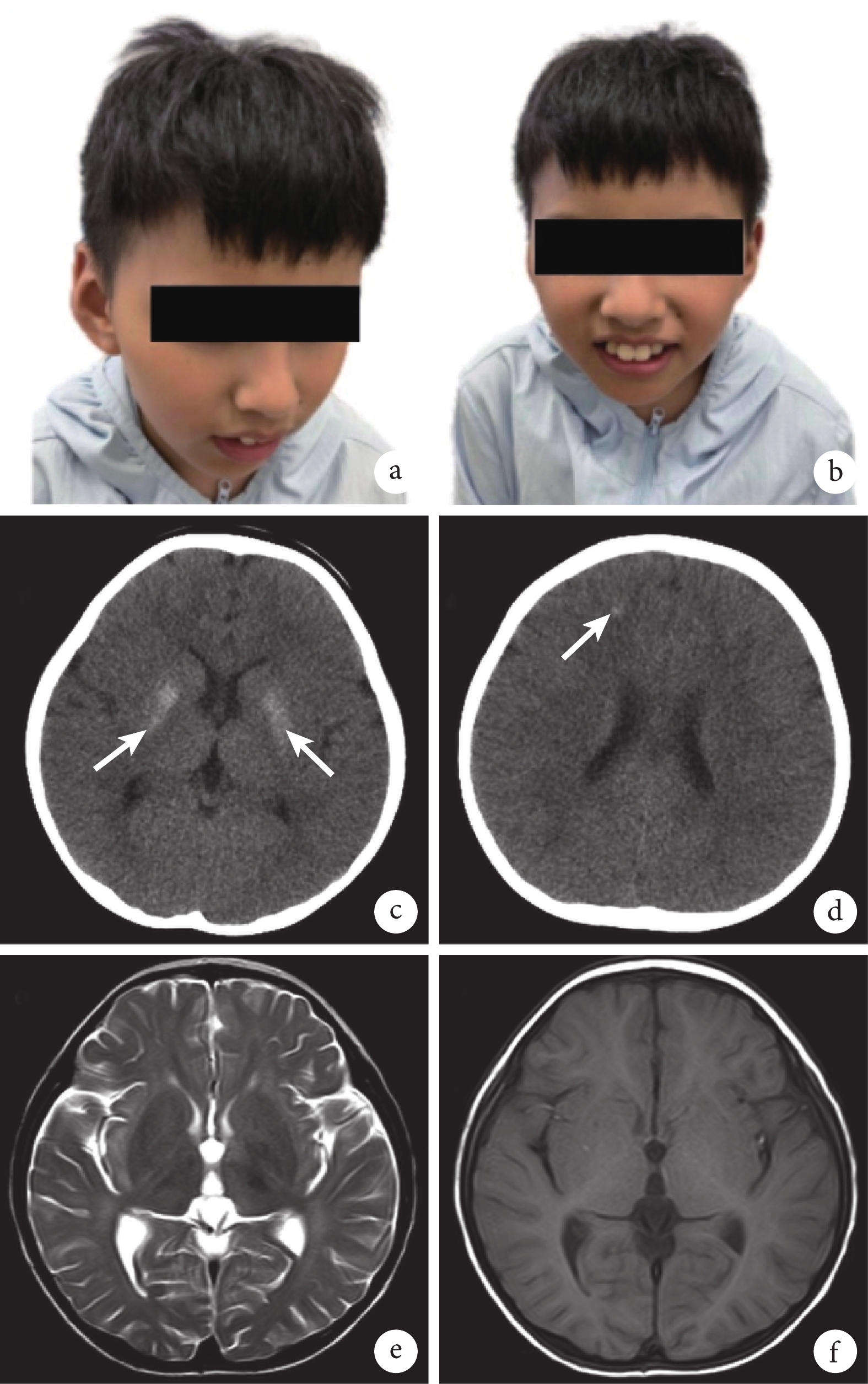 圖1
患兒面部特征及頭顱影像學檢查圖像
圖1
患兒面部特征及頭顱影像學檢查圖像
a、b. 患兒目前外貌;c、d. 2018 年患兒頭顱 CT 檢查圖像,顯示雙側基底節區對稱性分布高密度影,雙側額葉少許小點片狀高密度影(白箭);e、f. 2018 年患兒頭顱 MRI 檢查圖像,顯示髓鞘化成熟障礙
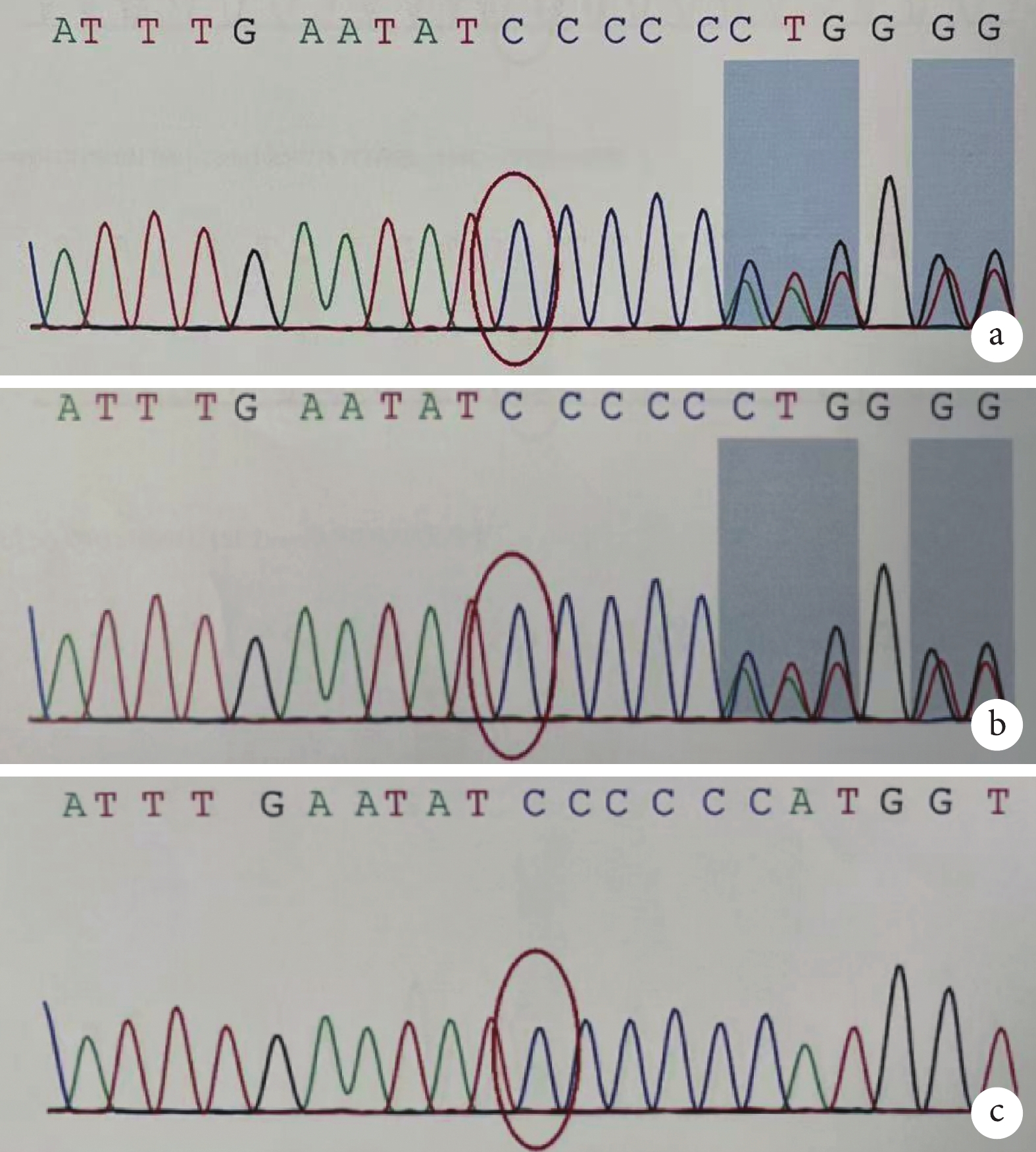
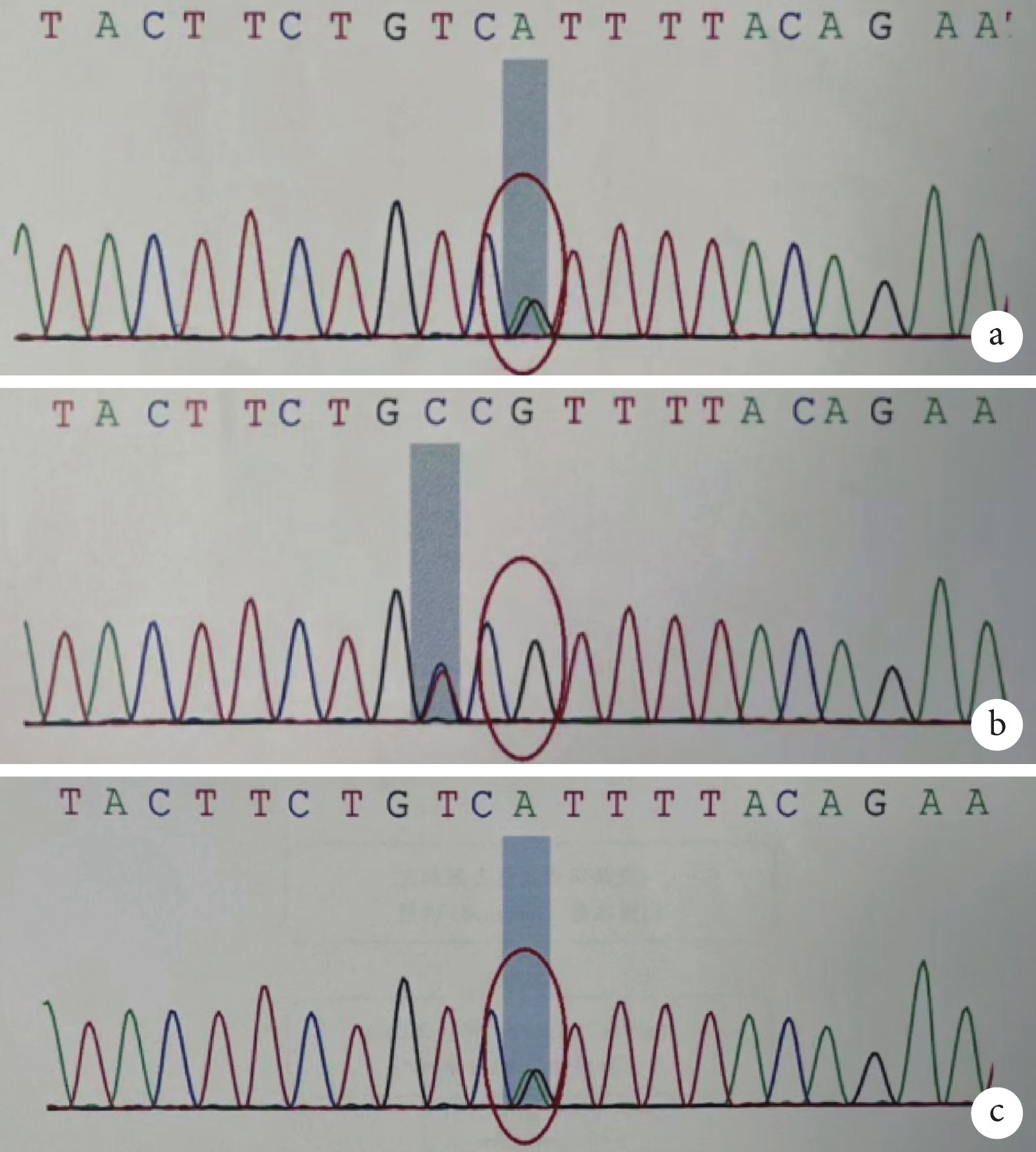
全外顯子基因測序發現,患兒 ERCC6 基因 NM_000124 轉錄本外顯子存在 2 處突變,分別來自于患兒父親和母親,為復雜雜合突變,其中 c.2144delG 為缺失突變(圖2),導致氨基酸改變 p.G715Dfs*24(移碼突變-24 位后終止);另一突變位點 c.526C>T(胞嘧啶>胸腺嘧啶)(圖3),導致氨基酸改變 p.R176*(精氨酸>終止)。2 處突變均導致蛋白翻譯提前終止,對蛋白功能的影響可能較大,可以解釋患者表型的致病性變異。
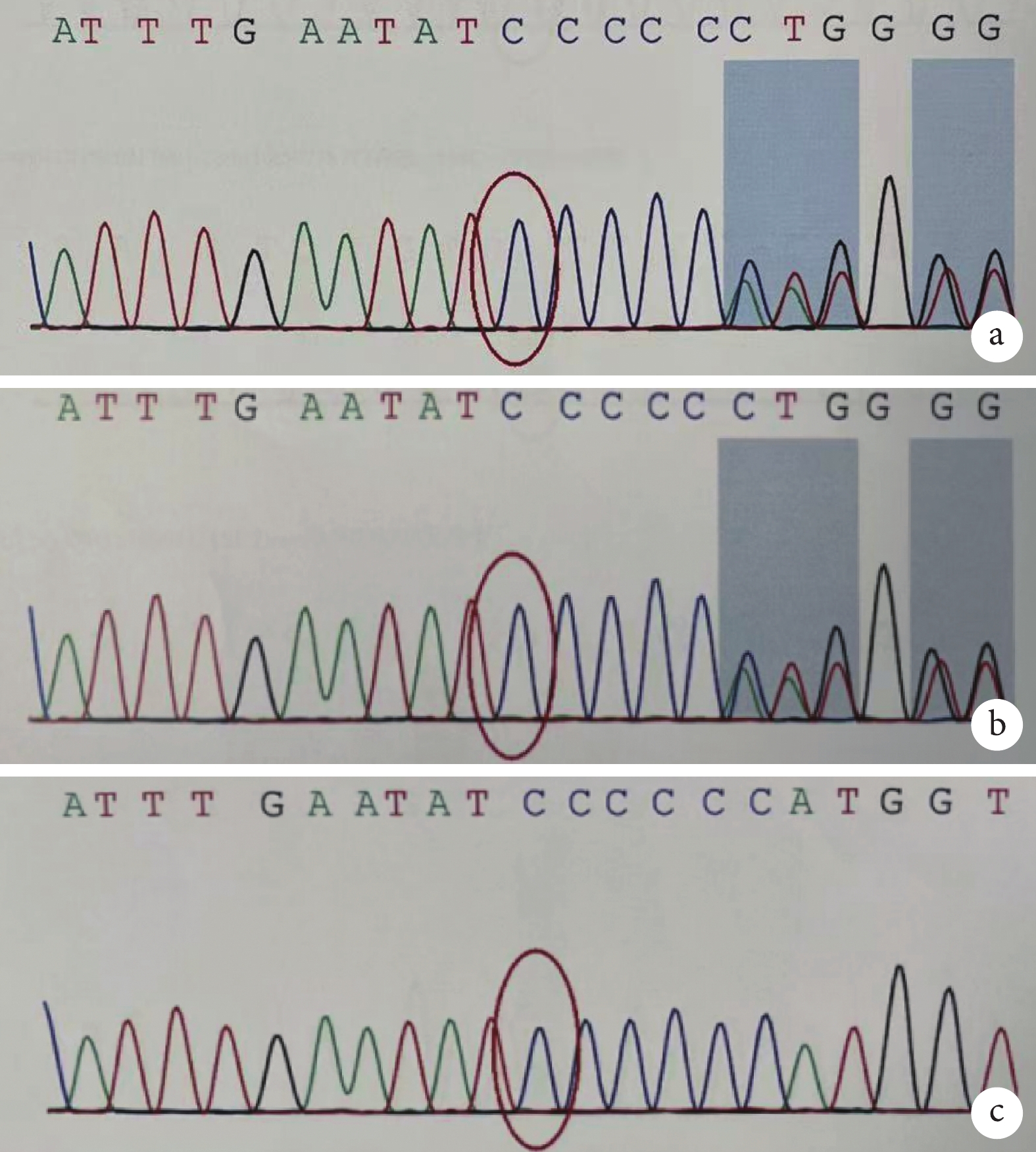 圖2
患兒及雙親 ERCC6 基因 c.2144delG 的 Sanger 測序結果
圖2
患兒及雙親 ERCC6 基因 c.2144delG 的 Sanger 測序結果
a. 患兒 chr10:50690758 存在 c.2144delG 的雜合突變;b. 患兒父親 chr10:50690758 存在 c.2144delG 的雜合突變;c. 患兒母親 chr10:50690758 無突變
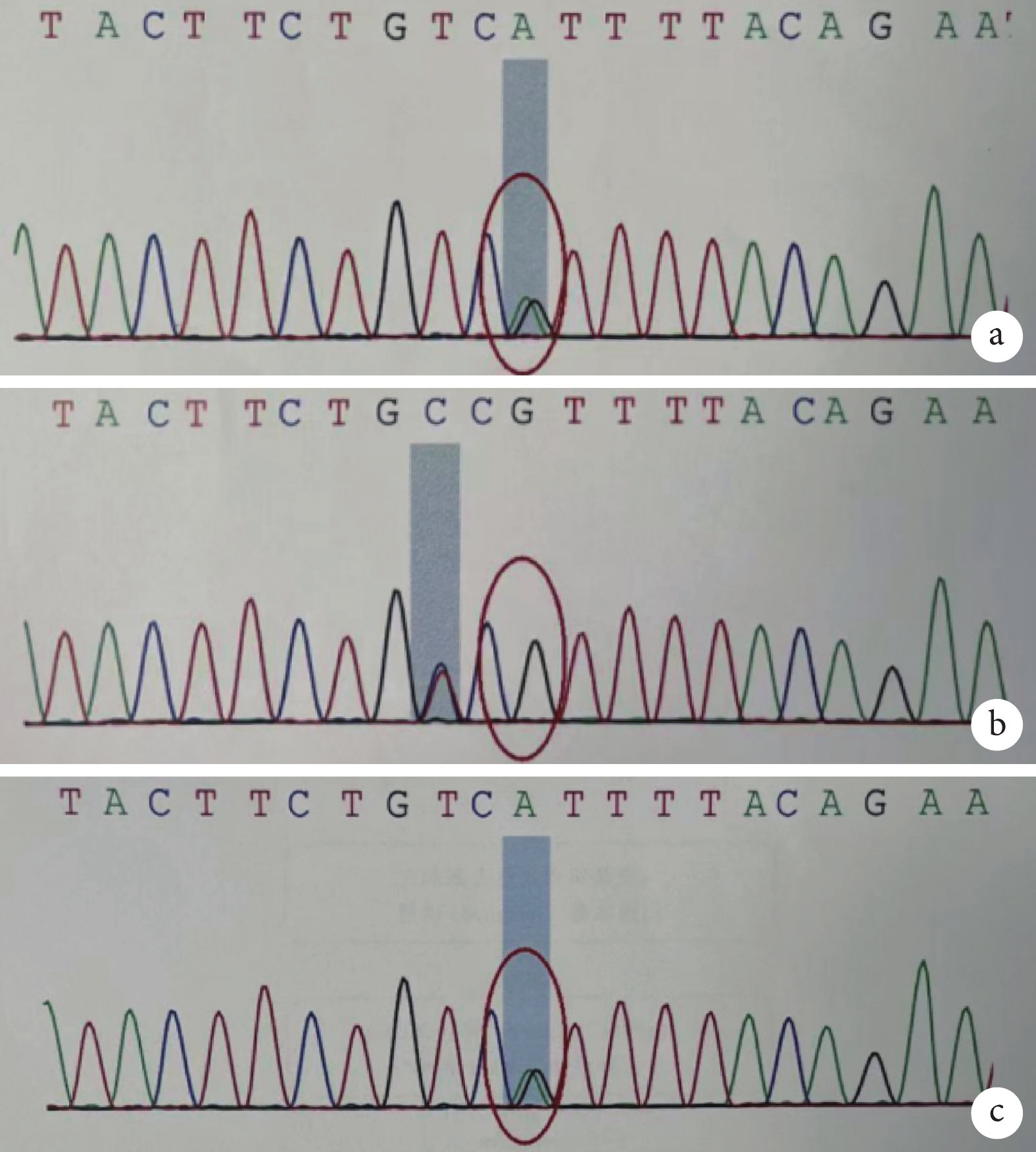 圖3
患兒及雙親 ERCC6 基因 c.526C>T 的 Sanger 測序結果
圖3
患兒及雙親 ERCC6 基因 c.526C>T 的 Sanger 測序結果
a. 患兒 chr10:50738783 存在 c.526C>T 的雜合突變;b. 患兒父親 chr10:50738783 無突變;c. 患兒母親 chr10:50738783 存在 c.526C>T 的雜合突變
綜合臨床表現、影像學資料及基因檢查結果,患兒診斷為 Cockayne 綜合征(Cockayne syndrome, CS)Ⅰ型。
討論 CS 又稱 Neill-Dingwall 綜合征、侏儒-視網膜萎縮-耳聾綜合征,是一種罕見的具有多種臨床特征的常染色體隱性遺傳疾病,于 1936 年由 Cockayne[1]首次報道,目前在世界范圍內有 200 多例報道。CS 全球范圍發病率極低,歐洲報道發病率約為 2.7/100 萬[2],與美國、日本的發病率相近[3-5],我國尚無其發病率統計,僅有散發病例。CS 主要臨床特征包括惡病質侏儒癥、小頭畸形、認知障礙,還可出現色素性視網膜病變、白內障、感音性神經性耳聾、行走困難和進食困難等癥狀。該病是一種進行性疾病,臨床癥狀隨著時間的推移而進行性加重。
目前已知與 CS 發病相關的基因有 5 個,分別是 ERCC6、ERCC8、XPB、XPD 和 XPG[6],其中 90% 的患者與 ERCC6(CSB)和 ERCC8(CSA)基因突變相關,其中 ERCC6 基因突變占 65%[7]。CSB 是由
CS 包括了廣泛的臨床表型,從最嚴重的產前亞型到最輕的晚發亞型,目前根據發病年齡、臨床表現嚴重程度和疾病進展速度將其主要分為 3 種類型,包括:CSⅠ型,為經典類型,胎兒在孕期發育正常,通常在 1 歲前表現出臨床癥狀,包括神經功能缺損、皮膚光敏、眼窩凹陷和發育遲緩,隨著年齡增長而癥狀惡化,大多數患兒在 20 歲之前死亡;CSⅡ型,為嚴重型,出生后主要表現為生長發育障礙,神經系統發育嚴重受損,大多數患者在 6~7 歲前死亡;CSⅢ型,為遲發性,程度輕型,癥狀在兒童期和成年期逐漸出現[11-13]。CS 患者中病情較輕的是紫外線敏感綜合征,該類型的臨床特征是皮膚的光敏性,患者皮膚暴露在陽光下可出現雀斑和色素異常,但不涉及癌變傾向或神經損害[14-15]。與此同時,還有許多 CS 其他亞型,其中大腦眼面部骨骼綜合征是最嚴重的 CS 類型,被認為是一種產前形式的 CS。
CS 通常出現在嬰兒期或幼兒期,大多數在新生兒期發育良好,后逐漸落后于同齡人。受累兒童常表現為反應欠佳,眼窩凹陷,鼻梁高聳,齲齒,易曬傷,進行性共濟失調、痙攣和脫髓鞘神經病變影響行走,患者的運動能力、語言和認知發育都相當遲緩,聽力和視力可受到損害[16]。在疾病過程中,患者的神經系統、視網膜、內耳、血管、骨和脂肪組織受到的影響尤為嚴重。CS 患者可出現多個系統損害,神經系統可出現破壞性改變,可出現腦白質萎縮、小頭畸形等情況,導致認知功能障礙和發育里程碑推遲。進行性的小頭畸形被認為是 CS 的特征性改變[17],患者出生時頭圍通常正常,隨著年齡增長,頭圍增長越來越慢,通常在 1~2 歲完全停止,幾乎所有 2 歲以上的 CS 患者都有小頭畸形。少部分 CS 患者可出現痙攣、震顫、癲癇、肌肉萎縮等情況,患者的周圍神經病變一般出現在 2 歲之后,肌電圖檢查可出現感覺、運動神經傳導速度均減慢[18-19]。因小腦受累,在合并視覺損害和周圍神經病變時可出現明顯共濟失調表現[2]。在聽力方面,CS 患者的感音神經性高音調聽力損失是主要特征,部分患者在 2~3 歲后出現聽力喪失,極個別嚴重個體可在新生兒期出現,其他 CS 患者存在輕度和晚期的聽力障礙,有研究表明,聽力損傷的嚴重程度可能與疾病整體的嚴重程度相關[3]。在眼部病變中,具有特征性表現的是視網膜營養不良,可與白內障并存,還包括報道的其他視覺異常,如角膜渾濁、角膜潰瘍、小角膜、遠視、斜視和眼球震顫等[3]。在皮膚方面,CS 患者可在早期即出現光敏性皮炎,日光曬后出現皮膚紅腫,并留有色素沉著。在外形上,CS 患者存在體格生長發育受限,可出現營養不良、脫水等影響,皮下脂肪尤其是眼周皮下脂肪的減少,導致眼眶凹陷明顯。在其他系統上,CS 患者可出現動脈粥樣硬化、鈣化性血管病變、腎功能衰竭、喂養困難和齲齒等癥狀[2]。值得注意的是,據 Nance 等[11]報道,CS 患者中約 10% 出現腎臟并發癥,對于腎衰竭引起的高血壓和貧血,合理的管理很重要。
在影像學方面,腦成像在 CS 的診斷中起著至關重要的作用。CS 患者可出現嚴重的腦白質萎縮,腦白質損傷可能是 CS 患者較早的神經影像學表現。CT 上顯示的基底節區鈣化和腦萎縮(主要發生在后顱窩)是本病的主要特征,出現顱內鈣化的年齡大多在 3 歲后[2]。同時有研究表明 CS 是一種低髓鞘疾病,在腦影像學上也發現該病亞型之間的顯著差異,較嚴重的類型(CSⅡ型和大腦眼面部骨骼綜合征)以早期腦萎縮、嚴重的脫髓鞘改變和鈣化為特征,相比之下,CSⅢ型腦萎縮程度輕,脫髓鞘概率發生較低[20]。有研究報道了 CS 患者口服甲硝唑后出現急性肝功能衰竭,有患者甚至因此死亡[3, 21],所以要警惕在 CS 兒童患者中使用甲硝唑的危險性。
Nance 等[11]在 1992 年提出 CS 的診斷標準,Laugel[17]在此基礎上進行修訂,診斷標準主要包括 3 項主要標準和 5 項次要標準,3 項主要標準有發育遲緩、進行性生長不足和進行性小頭畸形,5 個次要標準包括皮膚光敏性、色素性視網膜病變和/或白內障、進行性感音神經性聽力損失、牙釉質發育不良和眼球凹陷。滿足 3 項主要標準及至少 3 項次要標準即可診斷 CS;在疾病的早期,在滿足所有主要標準的情況下,至少需滿足 2 項次要標準,這將提高臨床篩查的敏感性。在 CS 的早期,患者可能只滿足部分診斷標準,而隨著病情進展,會逐漸出現更多癥狀,因此,對于疑似 CS 的患者,應進行詳細的臨床評估和基因檢測以明確診斷。
本例患者存在典型臨床表現和影像學特征性改變,且基因檢測提示 ERCC6 突變,根據起病年齡和臨床表現嚴重程度,本例患兒可確診為 CS,并根據臨床表現、影像學資料及基因檢查結果綜合考慮為 CSⅠ型。目前對于 CS 患者,尚無特殊治療手段,大多數患者預后不良,目前治療手段以對癥治療為主,包括積極改善患者營養狀態,避免日光照曬等紫外線損傷,每年評估聽力、視力等并發癥,定期監測血壓、肝功能和腎功能。
綜上所述,CS 是一種罕見疾病,在國內僅有散發報道。本文回顧分析了 1 例 CS 兒童患者的臨床特征、影像學表現及基因突變結果,希望通過對本例 CS 患兒的報道,提高對該病的認識,以盡早識別和診斷。其更好的治療策略有待進一步探索。
利益沖突:所有作者聲明不存在利益沖突。
病例介紹 患兒,男,8 歲,因“精神運動發育落后 7 年”于 2023 年 7 月前往四川大學華西第二醫院兒童神經科門診就診。該男性患兒系孕 1 產 1,父母體健,非近親婚配,母親孕期無異常,足月順產,產時無窒息、搶救史。患兒 1 歲 7 個月時開始獨立行走,后逐漸出現走路姿勢異常,易摔跤,至今不可蹲、跑;1 歲會說話,現僅可說短句,后面容逐漸改變,出現鼻梁高聳、眼窩凹陷和大耳;2 歲后曬后面部皮膚出現紅腫,后出現蛻皮,伴隨少許色素沉著;3 歲時有“熱性驚厥”病史,后未再抽搐。患兒家族中無類似患者。體格檢查:神志清楚,精神可;身高 121 cm(<P3),體重 22 kg(<P10),頭圍 46 cm(<P3),體質量指數 15 kg/m2;皮膚干燥、粗糙,面部皮膚散在少許色素沉著,大耳,雙側眼窩凹陷,鼻梁高聳,小下頜(圖1a、1b);心肺腹未見異常;脊柱未見異常;雙手掌紋正常,雙手精細動作差,四肢肌力正常,雙下肢肌張力稍增高,雙側膝反射亢進,雙踝關節背屈受限,病理征陰性;肢體協調性欠佳,易摔倒,可理解并說出簡單短句。輔助檢查:2018 年患兒頭顱 CT 顯示雙側基底節區對稱性分布高密度影,雙側額葉少許小點片狀高密度影(圖1c、1d);同年頭顱 MRI 有髓鞘化成熟障礙征象(圖1e、1f);2023 年頭顱 CT 提示雙側額頂葉皮髓質交界區、豆狀核、齒狀核見對稱分布鈣化灶。聽誘發電位檢查顯示雙側Ⅲ波潛伏期可疑延長,雙側Ⅳ、Ⅴ波潛伏期延長,雙側Ⅰ-Ⅴ波間潛伏期延長,聽閾顯示雙側Ⅴ波均未引出。視覺誘發電位顯示雙側 P100 波潛伏期可疑延長。肝功能檢查顯示丙氨酸轉氨酶及天冬氨酸轉氨酶有一過性升高,后復查水平正常。下肢周圍神經功能檢測、視頻腦電圖、血尿代謝篩查、血氨、乳酸、銅藍蛋白及腎功能檢查均未見異常。患兒就診時行韋氏兒童智力量表測試,顯示其處于智力低下水平(總智商分數<41 分)。
圖1
患兒面部特征及頭顱影像學檢查圖像
a、b. 患兒目前外貌;c、d. 2018 年患兒頭顱 CT 檢查圖像,顯示雙側基底節區對稱性分布高密度影,雙側額葉少許小點片狀高密度影(白箭);e、f. 2018 年患兒頭顱 MRI 檢查圖像,顯示髓鞘化成熟障礙
全外顯子基因測序發現,患兒 ERCC6 基因 NM_000124 轉錄本外顯子存在 2 處突變,分別來自于患兒父親和母親,為復雜雜合突變,其中 c.2144delG 為缺失突變(圖2),導致氨基酸改變 p.G715Dfs*24(移碼突變-24 位后終止);另一突變位點 c.526C>T(胞嘧啶>胸腺嘧啶)(圖3),導致氨基酸改變 p.R176*(精氨酸>終止)。2 處突變均導致蛋白翻譯提前終止,對蛋白功能的影響可能較大,可以解釋患者表型的致病性變異。
圖2
患兒及雙親 ERCC6 基因 c.2144delG 的 Sanger 測序結果
a. 患兒 chr10:50690758 存在 c.2144delG 的雜合突變;b. 患兒父親 chr10:50690758 存在 c.2144delG 的雜合突變;c. 患兒母親 chr10:50690758 無突變
圖3
患兒及雙親 ERCC6 基因 c.526C>T 的 Sanger 測序結果
a. 患兒 chr10:50738783 存在 c.526C>T 的雜合突變;b. 患兒父親 chr10:50738783 無突變;c. 患兒母親 chr10:50738783 存在 c.526C>T 的雜合突變
綜合臨床表現、影像學資料及基因檢查結果,患兒診斷為 Cockayne 綜合征(Cockayne syndrome, CS)Ⅰ型。
討論 CS 又稱 Neill-Dingwall 綜合征、侏儒-視網膜萎縮-耳聾綜合征,是一種罕見的具有多種臨床特征的常染色體隱性遺傳疾病,于 1936 年由 Cockayne[1]首次報道,目前在世界范圍內有 200 多例報道。CS 全球范圍發病率極低,歐洲報道發病率約為 2.7/100 萬[2],與美國、日本的發病率相近[3-5],我國尚無其發病率統計,僅有散發病例。CS 主要臨床特征包括惡病質侏儒癥、小頭畸形、認知障礙,還可出現色素性視網膜病變、白內障、感音性神經性耳聾、行走困難和進食困難等癥狀。該病是一種進行性疾病,臨床癥狀隨著時間的推移而進行性加重。
目前已知與 CS 發病相關的基因有 5 個,分別是 ERCC6、ERCC8、XPB、XPD 和 XPG[6],其中 90% 的患者與 ERCC6(CSB)和 ERCC8(CSA)基因突變相關,其中 ERCC6 基因突變占 65%[7]。CSB 是由
CS 包括了廣泛的臨床表型,從最嚴重的產前亞型到最輕的晚發亞型,目前根據發病年齡、臨床表現嚴重程度和疾病進展速度將其主要分為 3 種類型,包括:CSⅠ型,為經典類型,胎兒在孕期發育正常,通常在 1 歲前表現出臨床癥狀,包括神經功能缺損、皮膚光敏、眼窩凹陷和發育遲緩,隨著年齡增長而癥狀惡化,大多數患兒在 20 歲之前死亡;CSⅡ型,為嚴重型,出生后主要表現為生長發育障礙,神經系統發育嚴重受損,大多數患者在 6~7 歲前死亡;CSⅢ型,為遲發性,程度輕型,癥狀在兒童期和成年期逐漸出現[11-13]。CS 患者中病情較輕的是紫外線敏感綜合征,該類型的臨床特征是皮膚的光敏性,患者皮膚暴露在陽光下可出現雀斑和色素異常,但不涉及癌變傾向或神經損害[14-15]。與此同時,還有許多 CS 其他亞型,其中大腦眼面部骨骼綜合征是最嚴重的 CS 類型,被認為是一種產前形式的 CS。
CS 通常出現在嬰兒期或幼兒期,大多數在新生兒期發育良好,后逐漸落后于同齡人。受累兒童常表現為反應欠佳,眼窩凹陷,鼻梁高聳,齲齒,易曬傷,進行性共濟失調、痙攣和脫髓鞘神經病變影響行走,患者的運動能力、語言和認知發育都相當遲緩,聽力和視力可受到損害[16]。在疾病過程中,患者的神經系統、視網膜、內耳、血管、骨和脂肪組織受到的影響尤為嚴重。CS 患者可出現多個系統損害,神經系統可出現破壞性改變,可出現腦白質萎縮、小頭畸形等情況,導致認知功能障礙和發育里程碑推遲。進行性的小頭畸形被認為是 CS 的特征性改變[17],患者出生時頭圍通常正常,隨著年齡增長,頭圍增長越來越慢,通常在 1~2 歲完全停止,幾乎所有 2 歲以上的 CS 患者都有小頭畸形。少部分 CS 患者可出現痙攣、震顫、癲癇、肌肉萎縮等情況,患者的周圍神經病變一般出現在 2 歲之后,肌電圖檢查可出現感覺、運動神經傳導速度均減慢[18-19]。因小腦受累,在合并視覺損害和周圍神經病變時可出現明顯共濟失調表現[2]。在聽力方面,CS 患者的感音神經性高音調聽力損失是主要特征,部分患者在 2~3 歲后出現聽力喪失,極個別嚴重個體可在新生兒期出現,其他 CS 患者存在輕度和晚期的聽力障礙,有研究表明,聽力損傷的嚴重程度可能與疾病整體的嚴重程度相關[3]。在眼部病變中,具有特征性表現的是視網膜營養不良,可與白內障并存,還包括報道的其他視覺異常,如角膜渾濁、角膜潰瘍、小角膜、遠視、斜視和眼球震顫等[3]。在皮膚方面,CS 患者可在早期即出現光敏性皮炎,日光曬后出現皮膚紅腫,并留有色素沉著。在外形上,CS 患者存在體格生長發育受限,可出現營養不良、脫水等影響,皮下脂肪尤其是眼周皮下脂肪的減少,導致眼眶凹陷明顯。在其他系統上,CS 患者可出現動脈粥樣硬化、鈣化性血管病變、腎功能衰竭、喂養困難和齲齒等癥狀[2]。值得注意的是,據 Nance 等[11]報道,CS 患者中約 10% 出現腎臟并發癥,對于腎衰竭引起的高血壓和貧血,合理的管理很重要。
在影像學方面,腦成像在 CS 的診斷中起著至關重要的作用。CS 患者可出現嚴重的腦白質萎縮,腦白質損傷可能是 CS 患者較早的神經影像學表現。CT 上顯示的基底節區鈣化和腦萎縮(主要發生在后顱窩)是本病的主要特征,出現顱內鈣化的年齡大多在 3 歲后[2]。同時有研究表明 CS 是一種低髓鞘疾病,在腦影像學上也發現該病亞型之間的顯著差異,較嚴重的類型(CSⅡ型和大腦眼面部骨骼綜合征)以早期腦萎縮、嚴重的脫髓鞘改變和鈣化為特征,相比之下,CSⅢ型腦萎縮程度輕,脫髓鞘概率發生較低[20]。有研究報道了 CS 患者口服甲硝唑后出現急性肝功能衰竭,有患者甚至因此死亡[3, 21],所以要警惕在 CS 兒童患者中使用甲硝唑的危險性。
Nance 等[11]在 1992 年提出 CS 的診斷標準,Laugel[17]在此基礎上進行修訂,診斷標準主要包括 3 項主要標準和 5 項次要標準,3 項主要標準有發育遲緩、進行性生長不足和進行性小頭畸形,5 個次要標準包括皮膚光敏性、色素性視網膜病變和/或白內障、進行性感音神經性聽力損失、牙釉質發育不良和眼球凹陷。滿足 3 項主要標準及至少 3 項次要標準即可診斷 CS;在疾病的早期,在滿足所有主要標準的情況下,至少需滿足 2 項次要標準,這將提高臨床篩查的敏感性。在 CS 的早期,患者可能只滿足部分診斷標準,而隨著病情進展,會逐漸出現更多癥狀,因此,對于疑似 CS 的患者,應進行詳細的臨床評估和基因檢測以明確診斷。
本例患者存在典型臨床表現和影像學特征性改變,且基因檢測提示 ERCC6 突變,根據起病年齡和臨床表現嚴重程度,本例患兒可確診為 CS,并根據臨床表現、影像學資料及基因檢查結果綜合考慮為 CSⅠ型。目前對于 CS 患者,尚無特殊治療手段,大多數患者預后不良,目前治療手段以對癥治療為主,包括積極改善患者營養狀態,避免日光照曬等紫外線損傷,每年評估聽力、視力等并發癥,定期監測血壓、肝功能和腎功能。
綜上所述,CS 是一種罕見疾病,在國內僅有散發報道。本文回顧分析了 1 例 CS 兒童患者的臨床特征、影像學表現及基因突變結果,希望通過對本例 CS 患兒的報道,提高對該病的認識,以盡早識別和診斷。其更好的治療策略有待進一步探索。
利益沖突:所有作者聲明不存在利益沖突。