引用本文: 楊迷玲, 王立峰, 徐小艷, 姜黃. 單形性親上皮性腸道T細胞淋巴瘤: 附3例患者臨床病理分析. 中國普外基礎與臨床雜志, 2024, 31(9): 1125-1129. doi: 10.7507/1007-9424.202404086 復制
版權信息: ?四川大學華西醫院華西期刊社《中國普外基礎與臨床雜志》版權所有,未經授權不得轉載、改編
原發性結外非霍奇金淋巴瘤好發于胃腸道,且大多數為B細胞淋巴瘤,而起源于胃腸道的外周T細胞淋巴瘤較為罕見,其中腸道外周T細胞淋巴瘤可以伴或不伴腸病。2008年世界衛生組織基于修訂后的歐美淋巴腫瘤分類將腸病相關T細胞淋巴瘤分為2型:經典型和單形型[1]。2016年世界衛生組織將單形型命名為獨立的疾病體,即“單形性親上皮性腸道T細胞淋巴瘤(monomorphic epitheliotropic intestinal T-cell lymphoma,MEITL)” [2]。MEITL作為一種新的疾病實體,與腸病無關,不伴有乳糜瀉,在2022年《第5版世界衛生組織血液淋巴腫瘤分類:淋巴腫瘤》分類系統中繼續沿用該分類及命名[3]。MEITL缺乏特異性的臨床表現,侵襲性強,預后差,給臨床診斷和治療帶來一定的困難,對病理醫生也是一個挑戰。現回顧性分析鄭州人民醫院收治的3例MEITL患者的臨床資料、診療經過、組織病理學、免疫表型等特點,同時復習國內外文獻,以提高臨床醫生和病理醫生對該病的認識,降低漏診率。
1 臨床資料
回顧性分析2014年4月至2023年4月期間鄭州人民醫院收治的3例MEITL患者的臨床病理資料。MEITL的病理診斷參照2022年《第5版世界衛生組織血液淋巴腫瘤分類:淋巴腫瘤》分類系統中MEITL的診斷標準[3]。3例患者的具體資料見表1。其中男2例、女1例,年齡49~57歲。2例患者既往體健。3例患者的腫瘤均來源于小腸,其中病例1和病例3明確提及腫瘤位于空腸;病例2在CT檢查和術中檢查均未提及腫瘤明確的部位,而僅提及腫瘤位于小腸。3例患者術前均行CT檢查,術后大體標本均可見明確的腫瘤,腫瘤長徑6~10 cm,均位于黏膜下,2例可見穿孔。均采用手術治療。3例患者的手術切除標本經10%中性甲醛緩沖液固定,常規取材、脫水、石蠟包埋,行蘇木精-伊紅(hematoxylin-eosin,HE)染色和免疫組織化學EnVision二步法檢測細胞角蛋白、CD3、CD7、CD8、CD56、T 細胞胞漿內抗原(T-cell intracytoplasmic antigen,TIA-1)、顆粒酶B、穿孔素、CD20、CD79α、CD5、Pax-5、CD30、末端脫氧核苷酸轉移酶(terminal deoxynucleotidyl transferase,TDT)、Ki67,操作流程嚴格按照說明書進行。3例患者的手術切除標本均進行了Epstein-Barr病毒編碼的小RNA原位雜交檢測(所有抗體及檢測試劑盒均購自福州邁新生物技術開發有限公司)。
2 結果
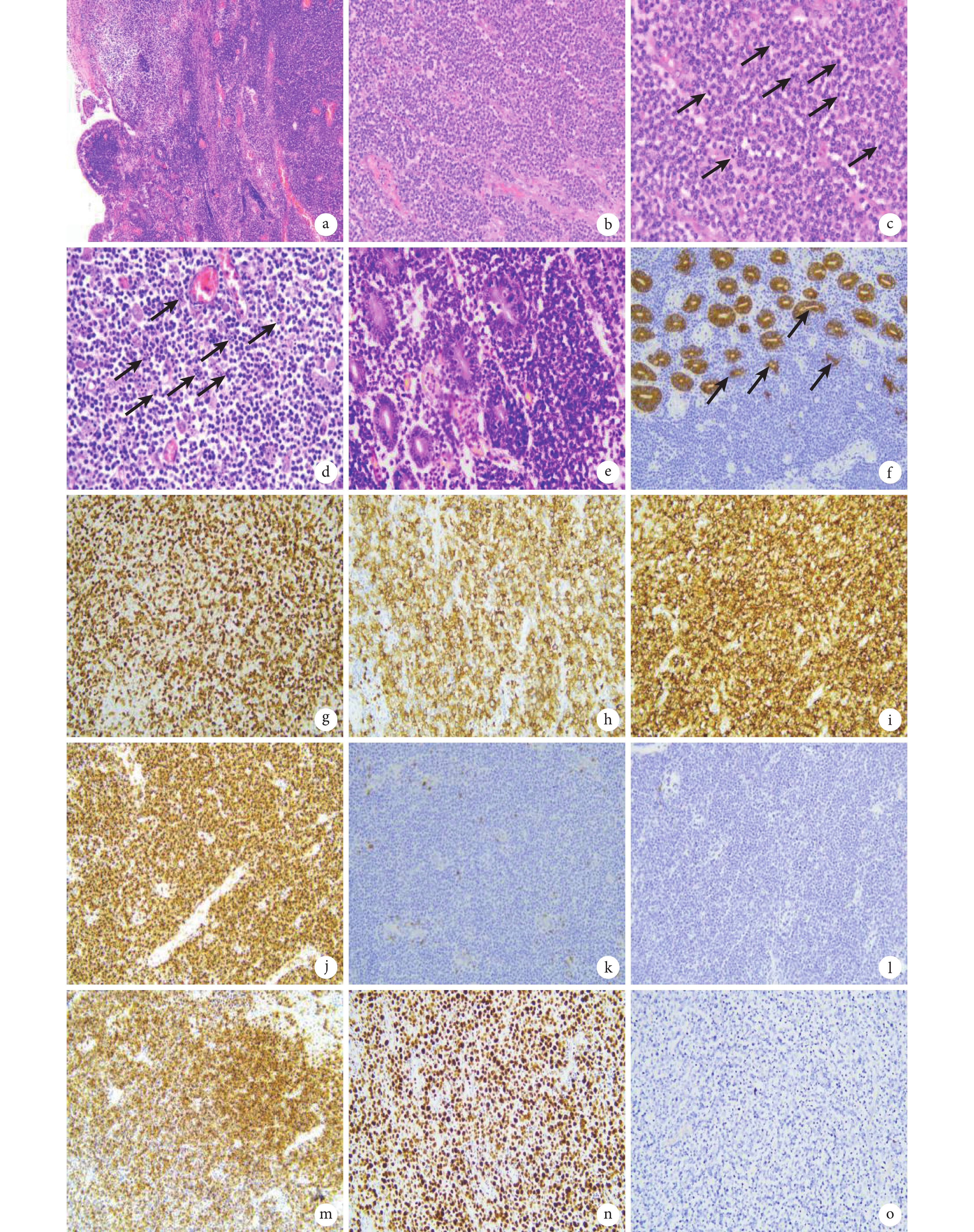
3例患者的腫瘤均來源于小腸,其中病例1和病例3明確提示位于空腸。3例患者均可見明確的腫瘤,腫瘤長徑6~10 cm,均位于黏膜下,2例可見腸穿孔。病例3腫瘤約8.0 cm×5.5 cm×5.0 cm大小,切面灰白灰紅,局部灰黃色,灶區呈囊性,內可見灰白灰黃色壞死物,黏膜面見潰瘍。3例患者術后半年內有2例死亡,另1例至今仍存活。3例患者的組織病理學HE染色結果:見腫瘤細胞周圍小腸絨毛結構破壞、絨毛萎縮或消失(圖1a);3例患者的腫瘤細胞鏡下形態類似,腫瘤細胞形態單一,缺乏炎性腸病的炎癥背景,小至中等大小(圖1b),細胞漿少,部分可以出現淡染的胞漿,核圓形或略不規則,染色質致密,核仁不明顯,有的可見小核仁;3例腫瘤細胞均彌漫浸潤腸壁全層,均可見腫瘤性壞死及較多核分裂象(圖1c),病例3可見較多核碎裂(圖1d),病例2腸周淋巴結見腫瘤累及。黏膜表面可見潰瘍,隱窩上皮內可見腫瘤性淋巴細胞浸潤形成“親上皮現象” (圖1e、1f)。免疫表型結果:3例患者組織樣本中的腫瘤細胞CD3(圖1g)、CD7、CD8(圖1h)、CD56(圖1i)、TIA-1(圖1j)均呈陽性表達,CD79α、CD5(圖1k)、CD20(圖1l)、Pax-5、CD30、TDT均為陰性表達,2例腫瘤細胞顆粒酶B(圖1m)和穿孔素部分陽性表達,Ki67增殖指數約70%~90%(圖1n)。原位雜交檢測Epstein-Barr病毒編碼的小RNA在3例患者的組織樣本中均為陰性(圖1o)。
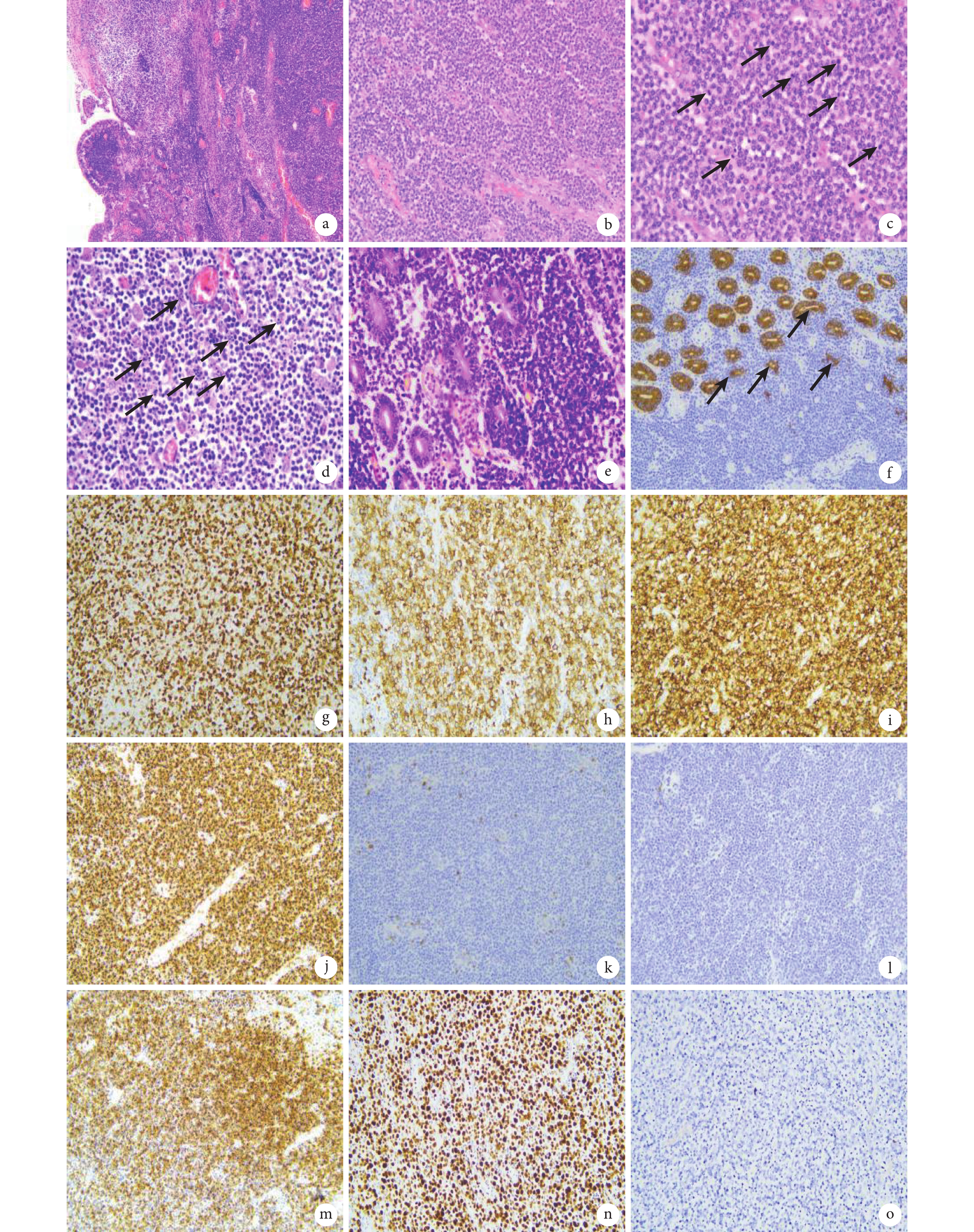 圖1
示MEITL的組織病理形態學特征
圖1
示MEITL的組織病理形態學特征
a:腫瘤細胞周圍小腸絨毛結構破壞、絨毛萎縮或消失(HE ×100);b:腫瘤細胞形態單一,小至中等大小(HE ×200);c:黑箭示較多核分裂象(HE ×400);d:黑箭示腫瘤內大量核碎裂(HE ×200);e:隱窩上皮內可見腫瘤性淋巴細胞浸潤形成“親上皮現象” (HE ×400);f:黑箭所指細胞角蛋白“親上皮現象” (EnVision ×200);g~j:分別為腫瘤細胞CD3、CD8、CD56及TIA-1均呈彌漫陽性表達(EnVision ×200);k、l:分別為腫瘤細胞CD5和CD20表達陰性(EnVision ×200);m:腫瘤細胞顆粒酶B部分陽性表達(EnVision ×200);n:腫瘤細胞Ki67增殖指數70%~90%(EnVision ×200);o:腫瘤細胞Epstein-Barr病毒編碼的小RNA陰性(原位雜交 ×200)
3 討論
3.1 MEITL的流行病學特征及臨床表現
MEITL發病率男性多于女性[3];本組患者中男∶女為2∶1。MEITL的原發部位主要位于小腸(尤其是空腸),本組病例中病例1和病例3均發生于空腸,病例2未明確報道其具體部位而僅提及發生于小腸;也有發生于其他部位的病例報道,如胃[4]、闌尾[5]、食管[6]、卵巢[7]等腸外臟器;Suzuki等[8]報道1例原發于小腸的MEITL同時累及肺和腦。MEITL臨床表現無特異性,患者常出現腹痛、腹瀉、腹脹等常見消化道疾病癥狀。有病例以急腹癥就診,易合并腸梗阻及腸穿孔[9];有病例以消化道出血就診,甚至反復出現消化道出血[10];也有類似克羅恩病癥狀的報道,發生部位在結腸[11]。本病缺乏典型的臨床癥狀,且腸鏡不易到達病變部位,因此容易漏診和誤診;本病容易發生腸梗阻和腸穿孔,后者可導致彌漫性腹膜炎等并發癥,常需進行手術治療。本組3例病例均以腹痛就診,病例1和病例3出現急腹癥,病例3行腹部CT檢查發現左側下腹部團塊狀軟組織影,與鄰近腸管分界不清,周圍見滲出性改變,腹膜后、腸系膜區見多發淋巴結腫大,考慮腸癌;病例1和病例2經檢查發現合并腸梗阻和腸穿孔。3例患者均經手術切除后標本病理檢查明確診斷,術前均未行腸鏡檢查。
3.2 MEITL的組織病理學特征
MEITL具有較為獨特的病理學特征,腫瘤細胞形態單一,小至中等大小,細胞漿少,核圓形,染色質致密,核仁不明顯,小腸絨毛結構破壞形成潰瘍,病變周圍小腸絨毛消失或萎縮,隱窩上皮內可見腫瘤性淋巴細胞浸潤即“親上皮現象” [9, 12]。本組3例患者的組織形態學與文獻報道類似,腫瘤細胞大小一致,彌漫浸潤腸壁全層,病例3可見較多核碎裂。也有文獻[12]報道腫瘤細胞形態多形性,核異型性明顯,同時伴有淋巴結累及的病例報道,推測可能與細胞的多形性有關。本組病例中病例2累及腸周淋巴結,細胞形態比較單一,可見片狀腫瘤性壞死及大量核分裂象,這2個特征是侵襲性強的特征。3例患者的免疫組織化學染色CD3、CD7、CD8、CD56、TIA-1均呈陽性表達,2例患者的腫瘤細胞顆粒酶B和穿孔素部分陽性表達,CD20、CD79α、CD5、Pax-5、CD30、TDT均為陰性表達,Ki67增殖指數較高(70%~90%)。有研究者[13-14]報道有少部分病例可出現CD20和CD79α的異常表達。
3.3 鑒別診斷
雖然MEITL具有較為獨特的病理學特征,但臨床表現不特異,很容易誤診,需與其他腸道疾病或腫瘤進行鑒別。
3.3.1 炎性腸病
需與克羅恩病[11]鑒別。克羅恩病為多發性跳躍性病變,有較深的裂隙狀潰瘍,也易發生腸穿孔,鏡下特點為腸壁全層炎細胞浸潤及無干酪樣壞死的肉芽腫,可見大量成熟的淋巴細胞浸潤。雖然部分MEITL的黏膜也可表現為粗鵝卵石樣增厚,但克羅恩病的鏡下淋巴細胞無異型性。
3.3.2 胃腸道惰性T細胞淋巴瘤
該類疾病屬于交界性病變,可發生于消化道任何部位,病程進展緩慢或反復性,生物學行為惰性,侵襲性弱;在形態學上表現為相對一致的小淋巴細胞、圓形或輕度不規則,核分裂象少見或罕見,累及范圍通常不超過黏膜下層,一般不見“親上皮現象” ;免疫組織化學染色及分子學檢查方面表達T系標記,CD4+CD8– 的病例較多見,CD56陰性,TIA-1陽性,顆粒酶B陰性,Ki67 <10%,Epstein-Barr病毒編碼的小RNA陰性、 T細胞受體重排陽性。
3.3.3 胃腸道惰性自然殺傷細胞淋巴組織增殖性疾病
2022年《第5版世界衛生組織血液淋巴腫瘤分類:淋巴腫瘤》分類系統中將該疾病劃分為交界性病變[15]。該病臨床表現通常無癥狀,可以自發消退,也可以持續形成新的病變。形態學上局限于黏膜層,浸潤黏膜下層有限,腫瘤細胞中等至大細胞,胞質透亮,常見嗜酸性顆粒,染色質細膩,核仁不明顯,無壞死,缺乏血管中心性生長及血管壁浸潤,免疫表型與MEITL相似,Ki67為10%~90%,但T細胞受體重排陰性,EBER陰性。
3.3.4 結外自然殺傷細胞/T細胞淋巴瘤
該病腹部癥狀明顯,可以有腸穿孔或梗阻,影像學表現為消化道明顯的腫塊,呈侵襲性進展。腫瘤細胞形態多樣,顯著的細胞異型性,典型特征是血管中心性病變,破壞血管壁,壞死顯著,大部分病例T細胞受體重排陰性,所有病例Epstein-Barr病毒編碼的小RNA陽性。
3.3.5 腸病相關T細胞淋巴瘤
該病好發于歐美國家,與乳糜瀉有關;腹部癥狀明顯,可以表現為腸穿孔或梗阻,呈侵襲性進展。腫瘤細胞通常表現為多形性或中等大的腫瘤性淋巴細胞,背景炎癥細胞明顯,免疫組織化學染色CD4、CD8和CD56均陰性。
3.3.6 腸道T細胞淋巴瘤-非特指型
該病腹部癥狀明顯,可以有腸穿孔或梗阻;影像學表現為消化道明顯的腫塊,呈侵襲性進展。腫瘤細胞形態多樣,背景通常混有很多反應性小淋巴細胞、嗜酸性粒細胞、組織細胞和漿細胞,腫瘤細胞CD4和CD5均陽性,CD8和CD56多數呈陰性,Epstein-Barr病毒編碼的小RNA陰性,T細胞受體重排陽性。該疾病診斷需要排除消化道其他自然殺傷細胞/T淋巴組織增殖性疾病或者其他部位外周T細胞淋巴瘤累及消化道。
3.4 治療
MEITL目前尚無統一有效的標準治療方法。腫瘤細胞增殖指數高,侵襲性強,易合并腸穿孔和腸梗阻,通常采用手術治療。MEITL患者的預后差,中位生存時間7個月[16]。手術聯合化療的效果優于單純的手術治療,目前常用的化療方案是CHOP方案,但也有文獻[17]報道它在治療MEITL中的效果并不理想。有研究者[18]認為,應用大劑量化療聯合自體造血干細胞移植的療效更好。本組病例中病例1術后采用改良CHOP方案化療4個周期后出現高熱、腹瀉、粒細胞缺乏,術后4個月時因感染死亡;病例2手術治療后轉外院化療,具體化療方案不詳,術后6個月也因感染死亡;病例3手術聯合化療,化療采用GDPT方案,但術后5個月余時發現腹部腫瘤,CT提示小腸壁增厚,考慮淋巴瘤復發,未進一步做病理檢查證實,目前術后7個月仍存活,無其他不適。
總之,MEITL是一種罕見的高度侵襲性的腸道T細胞淋巴瘤,病程進展快,預后差。MEITL缺乏特異性的臨床表現,明確診斷需結合組織病理學、免疫組織化學及分子檢測結果進行鑒別。早期發現、早期治療可提高患者的生存率,改善患者的預后。目前缺乏有效的標準治療方案,尚需積累病例進一步研究,尋找有效的治療措施以改善患者的預后。
重要聲明
利益沖突聲明:本文全體作者閱讀并理解了《中國普外基礎與臨床雜志》的政策聲明,我們沒有相互競爭的利益。
作者貢獻聲明:楊迷玲撰寫及修改文章;王立峰指導論文;徐小艷審閱及修改文章;姜黃提供病例及查閱文獻。
倫理聲明:本研究通過了鄭州人民醫院科研倫理委員會審批(批文編號:2024011134)。
原發性結外非霍奇金淋巴瘤好發于胃腸道,且大多數為B細胞淋巴瘤,而起源于胃腸道的外周T細胞淋巴瘤較為罕見,其中腸道外周T細胞淋巴瘤可以伴或不伴腸病。2008年世界衛生組織基于修訂后的歐美淋巴腫瘤分類將腸病相關T細胞淋巴瘤分為2型:經典型和單形型[1]。2016年世界衛生組織將單形型命名為獨立的疾病體,即“單形性親上皮性腸道T細胞淋巴瘤(monomorphic epitheliotropic intestinal T-cell lymphoma,MEITL)” [2]。MEITL作為一種新的疾病實體,與腸病無關,不伴有乳糜瀉,在2022年《第5版世界衛生組織血液淋巴腫瘤分類:淋巴腫瘤》分類系統中繼續沿用該分類及命名[3]。MEITL缺乏特異性的臨床表現,侵襲性強,預后差,給臨床診斷和治療帶來一定的困難,對病理醫生也是一個挑戰。現回顧性分析鄭州人民醫院收治的3例MEITL患者的臨床資料、診療經過、組織病理學、免疫表型等特點,同時復習國內外文獻,以提高臨床醫生和病理醫生對該病的認識,降低漏診率。
1 臨床資料
回顧性分析2014年4月至2023年4月期間鄭州人民醫院收治的3例MEITL患者的臨床病理資料。MEITL的病理診斷參照2022年《第5版世界衛生組織血液淋巴腫瘤分類:淋巴腫瘤》分類系統中MEITL的診斷標準[3]。3例患者的具體資料見表1。其中男2例、女1例,年齡49~57歲。2例患者既往體健。3例患者的腫瘤均來源于小腸,其中病例1和病例3明確提及腫瘤位于空腸;病例2在CT檢查和術中檢查均未提及腫瘤明確的部位,而僅提及腫瘤位于小腸。3例患者術前均行CT檢查,術后大體標本均可見明確的腫瘤,腫瘤長徑6~10 cm,均位于黏膜下,2例可見穿孔。均采用手術治療。3例患者的手術切除標本經10%中性甲醛緩沖液固定,常規取材、脫水、石蠟包埋,行蘇木精-伊紅(hematoxylin-eosin,HE)染色和免疫組織化學EnVision二步法檢測細胞角蛋白、CD3、CD7、CD8、CD56、T 細胞胞漿內抗原(T-cell intracytoplasmic antigen,TIA-1)、顆粒酶B、穿孔素、CD20、CD79α、CD5、Pax-5、CD30、末端脫氧核苷酸轉移酶(terminal deoxynucleotidyl transferase,TDT)、Ki67,操作流程嚴格按照說明書進行。3例患者的手術切除標本均進行了Epstein-Barr病毒編碼的小RNA原位雜交檢測(所有抗體及檢測試劑盒均購自福州邁新生物技術開發有限公司)。
2 結果
3例患者的腫瘤均來源于小腸,其中病例1和病例3明確提示位于空腸。3例患者均可見明確的腫瘤,腫瘤長徑6~10 cm,均位于黏膜下,2例可見腸穿孔。病例3腫瘤約8.0 cm×5.5 cm×5.0 cm大小,切面灰白灰紅,局部灰黃色,灶區呈囊性,內可見灰白灰黃色壞死物,黏膜面見潰瘍。3例患者術后半年內有2例死亡,另1例至今仍存活。3例患者的組織病理學HE染色結果:見腫瘤細胞周圍小腸絨毛結構破壞、絨毛萎縮或消失(圖1a);3例患者的腫瘤細胞鏡下形態類似,腫瘤細胞形態單一,缺乏炎性腸病的炎癥背景,小至中等大小(圖1b),細胞漿少,部分可以出現淡染的胞漿,核圓形或略不規則,染色質致密,核仁不明顯,有的可見小核仁;3例腫瘤細胞均彌漫浸潤腸壁全層,均可見腫瘤性壞死及較多核分裂象(圖1c),病例3可見較多核碎裂(圖1d),病例2腸周淋巴結見腫瘤累及。黏膜表面可見潰瘍,隱窩上皮內可見腫瘤性淋巴細胞浸潤形成“親上皮現象” (圖1e、1f)。免疫表型結果:3例患者組織樣本中的腫瘤細胞CD3(圖1g)、CD7、CD8(圖1h)、CD56(圖1i)、TIA-1(圖1j)均呈陽性表達,CD79α、CD5(圖1k)、CD20(圖1l)、Pax-5、CD30、TDT均為陰性表達,2例腫瘤細胞顆粒酶B(圖1m)和穿孔素部分陽性表達,Ki67增殖指數約70%~90%(圖1n)。原位雜交檢測Epstein-Barr病毒編碼的小RNA在3例患者的組織樣本中均為陰性(圖1o)。
圖1
示MEITL的組織病理形態學特征
a:腫瘤細胞周圍小腸絨毛結構破壞、絨毛萎縮或消失(HE ×100);b:腫瘤細胞形態單一,小至中等大小(HE ×200);c:黑箭示較多核分裂象(HE ×400);d:黑箭示腫瘤內大量核碎裂(HE ×200);e:隱窩上皮內可見腫瘤性淋巴細胞浸潤形成“親上皮現象” (HE ×400);f:黑箭所指細胞角蛋白“親上皮現象” (EnVision ×200);g~j:分別為腫瘤細胞CD3、CD8、CD56及TIA-1均呈彌漫陽性表達(EnVision ×200);k、l:分別為腫瘤細胞CD5和CD20表達陰性(EnVision ×200);m:腫瘤細胞顆粒酶B部分陽性表達(EnVision ×200);n:腫瘤細胞Ki67增殖指數70%~90%(EnVision ×200);o:腫瘤細胞Epstein-Barr病毒編碼的小RNA陰性(原位雜交 ×200)
3 討論
3.1 MEITL的流行病學特征及臨床表現
MEITL發病率男性多于女性[3];本組患者中男∶女為2∶1。MEITL的原發部位主要位于小腸(尤其是空腸),本組病例中病例1和病例3均發生于空腸,病例2未明確報道其具體部位而僅提及發生于小腸;也有發生于其他部位的病例報道,如胃[4]、闌尾[5]、食管[6]、卵巢[7]等腸外臟器;Suzuki等[8]報道1例原發于小腸的MEITL同時累及肺和腦。MEITL臨床表現無特異性,患者常出現腹痛、腹瀉、腹脹等常見消化道疾病癥狀。有病例以急腹癥就診,易合并腸梗阻及腸穿孔[9];有病例以消化道出血就診,甚至反復出現消化道出血[10];也有類似克羅恩病癥狀的報道,發生部位在結腸[11]。本病缺乏典型的臨床癥狀,且腸鏡不易到達病變部位,因此容易漏診和誤診;本病容易發生腸梗阻和腸穿孔,后者可導致彌漫性腹膜炎等并發癥,常需進行手術治療。本組3例病例均以腹痛就診,病例1和病例3出現急腹癥,病例3行腹部CT檢查發現左側下腹部團塊狀軟組織影,與鄰近腸管分界不清,周圍見滲出性改變,腹膜后、腸系膜區見多發淋巴結腫大,考慮腸癌;病例1和病例2經檢查發現合并腸梗阻和腸穿孔。3例患者均經手術切除后標本病理檢查明確診斷,術前均未行腸鏡檢查。
3.2 MEITL的組織病理學特征
MEITL具有較為獨特的病理學特征,腫瘤細胞形態單一,小至中等大小,細胞漿少,核圓形,染色質致密,核仁不明顯,小腸絨毛結構破壞形成潰瘍,病變周圍小腸絨毛消失或萎縮,隱窩上皮內可見腫瘤性淋巴細胞浸潤即“親上皮現象” [9, 12]。本組3例患者的組織形態學與文獻報道類似,腫瘤細胞大小一致,彌漫浸潤腸壁全層,病例3可見較多核碎裂。也有文獻[12]報道腫瘤細胞形態多形性,核異型性明顯,同時伴有淋巴結累及的病例報道,推測可能與細胞的多形性有關。本組病例中病例2累及腸周淋巴結,細胞形態比較單一,可見片狀腫瘤性壞死及大量核分裂象,這2個特征是侵襲性強的特征。3例患者的免疫組織化學染色CD3、CD7、CD8、CD56、TIA-1均呈陽性表達,2例患者的腫瘤細胞顆粒酶B和穿孔素部分陽性表達,CD20、CD79α、CD5、Pax-5、CD30、TDT均為陰性表達,Ki67增殖指數較高(70%~90%)。有研究者[13-14]報道有少部分病例可出現CD20和CD79α的異常表達。
3.3 鑒別診斷
雖然MEITL具有較為獨特的病理學特征,但臨床表現不特異,很容易誤診,需與其他腸道疾病或腫瘤進行鑒別。
3.3.1 炎性腸病
需與克羅恩病[11]鑒別。克羅恩病為多發性跳躍性病變,有較深的裂隙狀潰瘍,也易發生腸穿孔,鏡下特點為腸壁全層炎細胞浸潤及無干酪樣壞死的肉芽腫,可見大量成熟的淋巴細胞浸潤。雖然部分MEITL的黏膜也可表現為粗鵝卵石樣增厚,但克羅恩病的鏡下淋巴細胞無異型性。
3.3.2 胃腸道惰性T細胞淋巴瘤
該類疾病屬于交界性病變,可發生于消化道任何部位,病程進展緩慢或反復性,生物學行為惰性,侵襲性弱;在形態學上表現為相對一致的小淋巴細胞、圓形或輕度不規則,核分裂象少見或罕見,累及范圍通常不超過黏膜下層,一般不見“親上皮現象” ;免疫組織化學染色及分子學檢查方面表達T系標記,CD4+CD8– 的病例較多見,CD56陰性,TIA-1陽性,顆粒酶B陰性,Ki67 <10%,Epstein-Barr病毒編碼的小RNA陰性、 T細胞受體重排陽性。
3.3.3 胃腸道惰性自然殺傷細胞淋巴組織增殖性疾病
2022年《第5版世界衛生組織血液淋巴腫瘤分類:淋巴腫瘤》分類系統中將該疾病劃分為交界性病變[15]。該病臨床表現通常無癥狀,可以自發消退,也可以持續形成新的病變。形態學上局限于黏膜層,浸潤黏膜下層有限,腫瘤細胞中等至大細胞,胞質透亮,常見嗜酸性顆粒,染色質細膩,核仁不明顯,無壞死,缺乏血管中心性生長及血管壁浸潤,免疫表型與MEITL相似,Ki67為10%~90%,但T細胞受體重排陰性,EBER陰性。
3.3.4 結外自然殺傷細胞/T細胞淋巴瘤
該病腹部癥狀明顯,可以有腸穿孔或梗阻,影像學表現為消化道明顯的腫塊,呈侵襲性進展。腫瘤細胞形態多樣,顯著的細胞異型性,典型特征是血管中心性病變,破壞血管壁,壞死顯著,大部分病例T細胞受體重排陰性,所有病例Epstein-Barr病毒編碼的小RNA陽性。
3.3.5 腸病相關T細胞淋巴瘤
該病好發于歐美國家,與乳糜瀉有關;腹部癥狀明顯,可以表現為腸穿孔或梗阻,呈侵襲性進展。腫瘤細胞通常表現為多形性或中等大的腫瘤性淋巴細胞,背景炎癥細胞明顯,免疫組織化學染色CD4、CD8和CD56均陰性。
3.3.6 腸道T細胞淋巴瘤-非特指型
該病腹部癥狀明顯,可以有腸穿孔或梗阻;影像學表現為消化道明顯的腫塊,呈侵襲性進展。腫瘤細胞形態多樣,背景通常混有很多反應性小淋巴細胞、嗜酸性粒細胞、組織細胞和漿細胞,腫瘤細胞CD4和CD5均陽性,CD8和CD56多數呈陰性,Epstein-Barr病毒編碼的小RNA陰性,T細胞受體重排陽性。該疾病診斷需要排除消化道其他自然殺傷細胞/T淋巴組織增殖性疾病或者其他部位外周T細胞淋巴瘤累及消化道。
3.4 治療
MEITL目前尚無統一有效的標準治療方法。腫瘤細胞增殖指數高,侵襲性強,易合并腸穿孔和腸梗阻,通常采用手術治療。MEITL患者的預后差,中位生存時間7個月[16]。手術聯合化療的效果優于單純的手術治療,目前常用的化療方案是CHOP方案,但也有文獻[17]報道它在治療MEITL中的效果并不理想。有研究者[18]認為,應用大劑量化療聯合自體造血干細胞移植的療效更好。本組病例中病例1術后采用改良CHOP方案化療4個周期后出現高熱、腹瀉、粒細胞缺乏,術后4個月時因感染死亡;病例2手術治療后轉外院化療,具體化療方案不詳,術后6個月也因感染死亡;病例3手術聯合化療,化療采用GDPT方案,但術后5個月余時發現腹部腫瘤,CT提示小腸壁增厚,考慮淋巴瘤復發,未進一步做病理檢查證實,目前術后7個月仍存活,無其他不適。
總之,MEITL是一種罕見的高度侵襲性的腸道T細胞淋巴瘤,病程進展快,預后差。MEITL缺乏特異性的臨床表現,明確診斷需結合組織病理學、免疫組織化學及分子檢測結果進行鑒別。早期發現、早期治療可提高患者的生存率,改善患者的預后。目前缺乏有效的標準治療方案,尚需積累病例進一步研究,尋找有效的治療措施以改善患者的預后。
重要聲明
利益沖突聲明:本文全體作者閱讀并理解了《中國普外基礎與臨床雜志》的政策聲明,我們沒有相互競爭的利益。
作者貢獻聲明:楊迷玲撰寫及修改文章;王立峰指導論文;徐小艷審閱及修改文章;姜黃提供病例及查閱文獻。
倫理聲明:本研究通過了鄭州人民醫院科研倫理委員會審批(批文編號:2024011134)。