引用本文: 劉繞星, 劉志宏, 周莉, 蔣毅. 原發性纖毛運動障礙一例. 中國呼吸與危重監護雜志, 2023, 22(12): 884-885. doi: 10.7507/1671-6205.202206031 復制
版權信息: ?四川大學華西醫院華西期刊社《中國呼吸與危重監護雜志》版權所有,未經授權不得轉載、改編
臨床資料 患者女,33歲。主因“間斷性咳嗽咳痰30余年,加重半個月”于2021年3月18日入院。患者自幼間斷性出現咳嗽咳痰癥狀,咳黃色黏痰,不易咳出。偶有快走時出現胸悶、氣促。自行口服或靜點消炎藥后癥狀可緩解,未正規治療。冬季上述癥狀明顯。2013年1月患者出現咳嗽加重,咳黃綠痰,咳嗽時伴前胸部針刺樣疼痛,以勞累、情緒激動后加重,伴鼻塞,偶有胸悶癥狀,無發熱、心慌、腹痛、腹瀉、皮疹等癥狀,就診于我院,診斷為“Kartagener綜合征”,給予抗感染、止咳祛痰治療后好轉出院。期間一直院外規律復查,近半個月自覺上述癥狀加重,3天前就診于我院門診,胸部CT(2021-03-15)示雙肺支氣管擴張伴多發感染,鏡面右位心,上腹部內臟反位。遂就診我科。自發病以來,食欲、精神尚可,睡眠欠佳,二便正常,體重未見明顯變化。既往3年前因鼻竇炎、鼻中隔偏曲引起“頭痛”行鼻中隔偏曲矯正手術。否認食物及藥物過敏史。20歲結婚,育0子1女,配偶女兒均健康。無煙酒不良嗜好,月經史正常。
入院查體:體溫36.1℃、脈搏100次/min、呼吸20次/min、血壓113/70mm Hg(1 mm Hg=0.133 kPa)。神志清楚,全身皮膚黏膜無黃染,口唇無發紺,雙眼球結膜無水腫,雙肺呼吸音粗,可聞及少許濕啰音,右位心,心律齊,各瓣膜聽診區未聞及病理性雜音。腹軟,全腹無壓痛、反跳痛,肝脾肋下未觸及,雙下肢無水腫。
輔助檢查:入院后積極完善相關化驗檢查,血白細胞計數8.4×109/L,中性粒細胞絕對值5.97×109/L、中性粒細胞百分比71.5%,血紅蛋白130 g/L,血小板計數247×109/L。血氣分析(未吸氧情況下):pH 7.407,PCO2 33. 9 mm Hg,PO2 78.1 mm Hg。白蛋白38.7 g/L。凝血+血漿D-二聚體、谷丙轉氨酶、谷草轉氨酶、肌紅蛋白、肌鈣蛋白、腦鈉肽、降鈣素原等未見明顯異常。胸部CT(2021-03-15本院):(1)雙肺支氣管擴張伴多發感染;(2)鏡面右位心;(3)掃描范圍內:上腹部內臟反位(圖1)。肺功能(2021-03-15本院):(1)彌散功能輕度下降;(2)肺總量、殘氣量正常,殘總比增高。呼吸總阻抗、總氣道阻力、中心氣道阻力正常,周邊彈性阻力增高,共振頻率增高。中重度混合性肺通功能障礙,支管舒張試驗陽性。
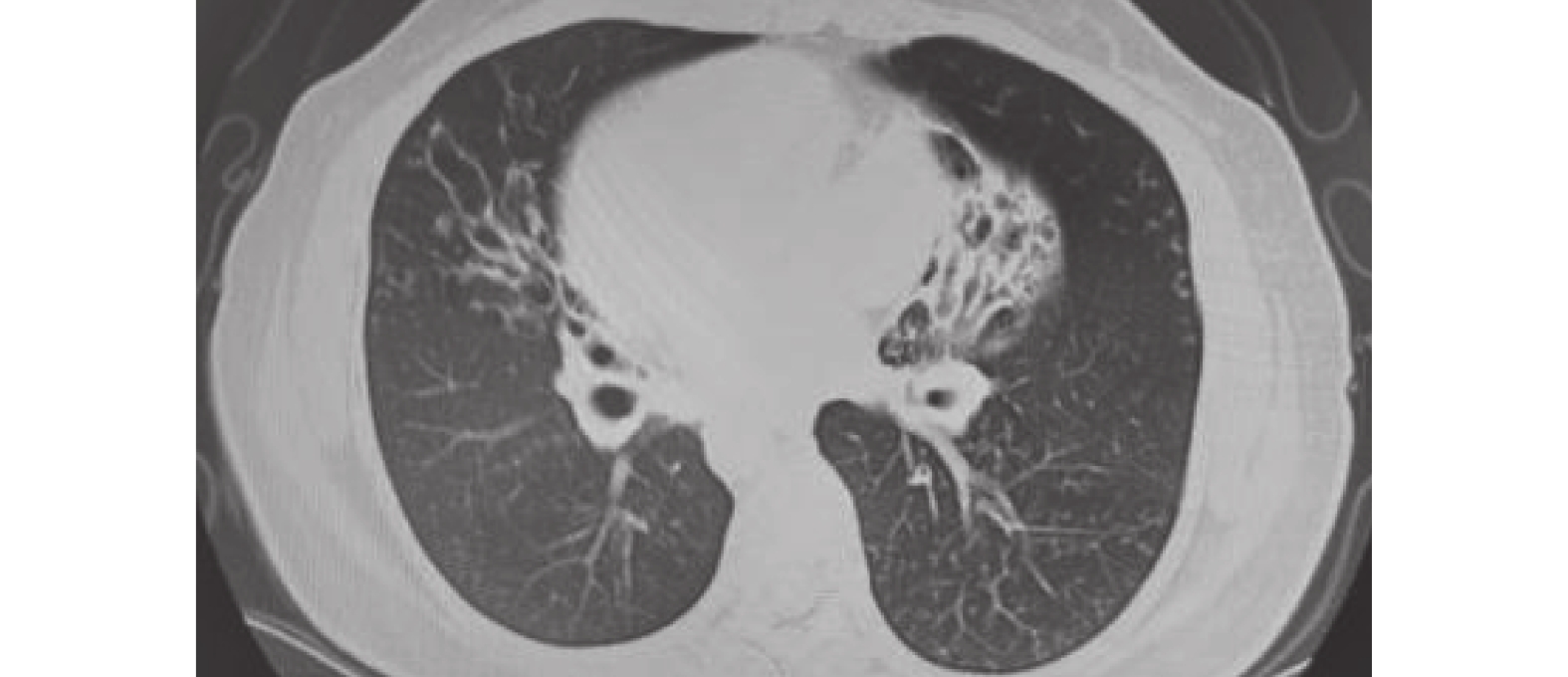
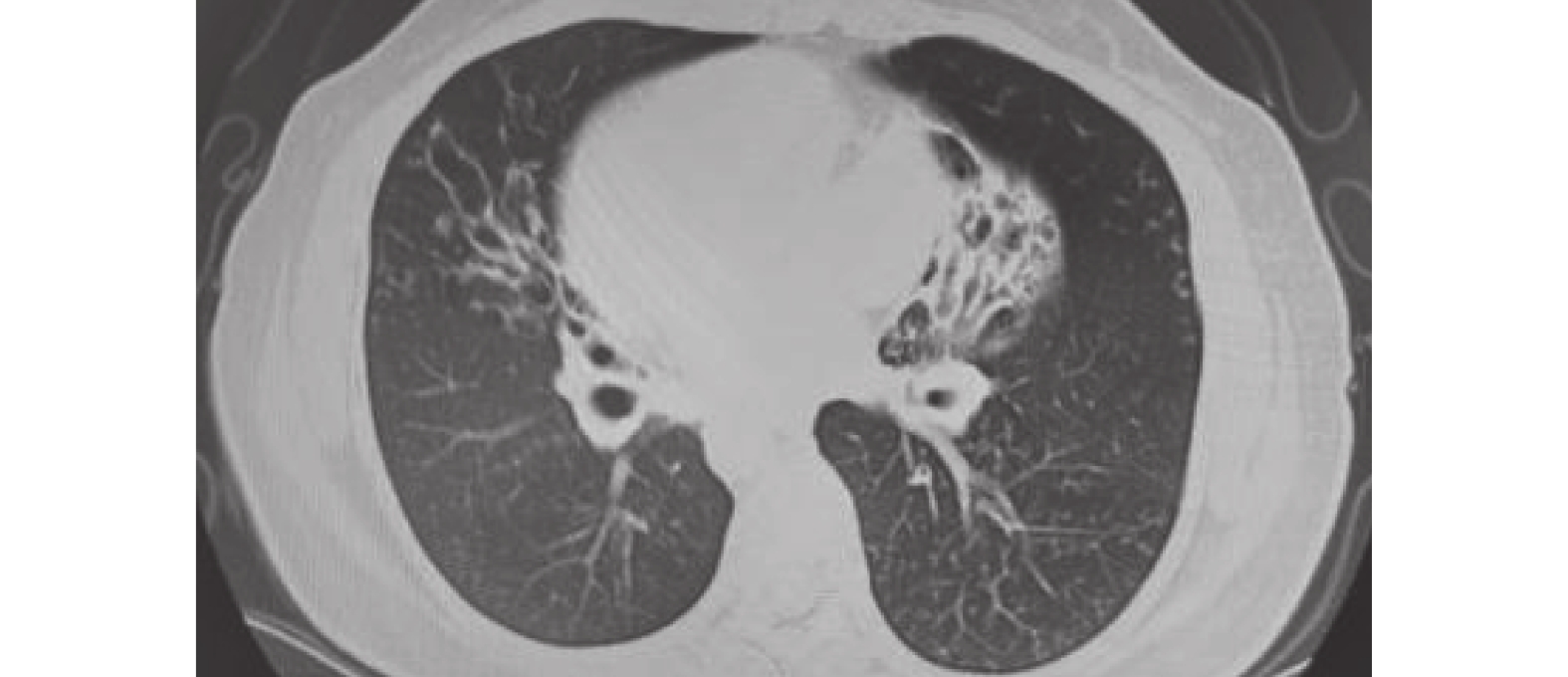 圖1
2021-03-15胸部CT檢查像
圖1
2021-03-15胸部CT檢查像
雙肺支氣管擴張伴多發感染,鏡面右位心。
患者自幼出現咳嗽咳痰癥狀,疾病受季節影響明顯,患者多年受鼻竇炎影響,胸部CT示支氣管擴張、內臟逆位。既往有鼻竇炎,目前支氣管擴張、內臟逆位,故在2013年就診時診斷為Kartagener綜合征。后一直門診規律復查,但在2013年未就診之前患者未正規治療,只是病情加重時自行口服抗生素治療,具體抗生素名稱及劑量患者都不記得。目前患者癥狀加重及復查胸部CT示感染加重入我科治療。
引起支氣管擴張的病因有許多,主要有免疫功能缺陷、感染、遺傳因素、風濕性疾病等。考慮到患者自幼就有咳嗽咳痰疾病,同時追問患者家族史,患者述父母屬近親結婚,患者弟弟因為肺源性心臟病已經去世。所以患者目前遺傳因素導致的支氣管擴張可能性極大,先天性疾病比如α1-抗胰蛋白酶缺乏、纖毛功能異常、軟骨缺陷等都會引起支氣管擴張。結合患者病史有內臟轉位、鼻竇炎、支氣管擴張,因此我們從原發性纖毛運動障礙(primaryciliarydyskinesia,PCD)疾病入手。患者住院后期行全外顯子組測序檢測報告示CFTR、DNAH5、DNAAF1、TAP1變異。歐洲呼吸協會2016年提出PCD診斷參考指南,出現以下1項或多項癥狀時應考慮PCD:長期反復的慢性鼻炎,長期反復慢性咳嗽,內臟異位,先天性心臟病,伴或不伴聽力障礙的慢性中耳疾病,呼吸窘迫的新生兒,兄弟姐妹中有患PCD,女性出現反復異位妊娠,男性精子無法游動等。故診斷PCD。
入院后積極留取痰培養,痰培養示副流感嗜血桿菌1次。同時經驗性給予抗感染、化痰等治療。患者抗感染9天后復查胸部CT平掃+冠狀面重建(2021-03-27):(1)雙肺支氣管擴張伴多發感染,較前炎癥范圍減小;(2)鏡面右位心;(3)所掃范圍內:上腹部內臟反位(圖2)。考慮患者癥狀及影像學較前好轉,建議院外治療,建議患者接種疫苗、增強免疫力,冬春季節可適當口服增強免疫力藥物。在原發性感染的情況下,通常建議根除銅綠假單胞菌,故繼續預防性使用抗感染治療1周后停藥,口服乙酰半胱氨酸泡騰片促進痰液排出。患者目前隨訪過程中病情未再進一步發展。
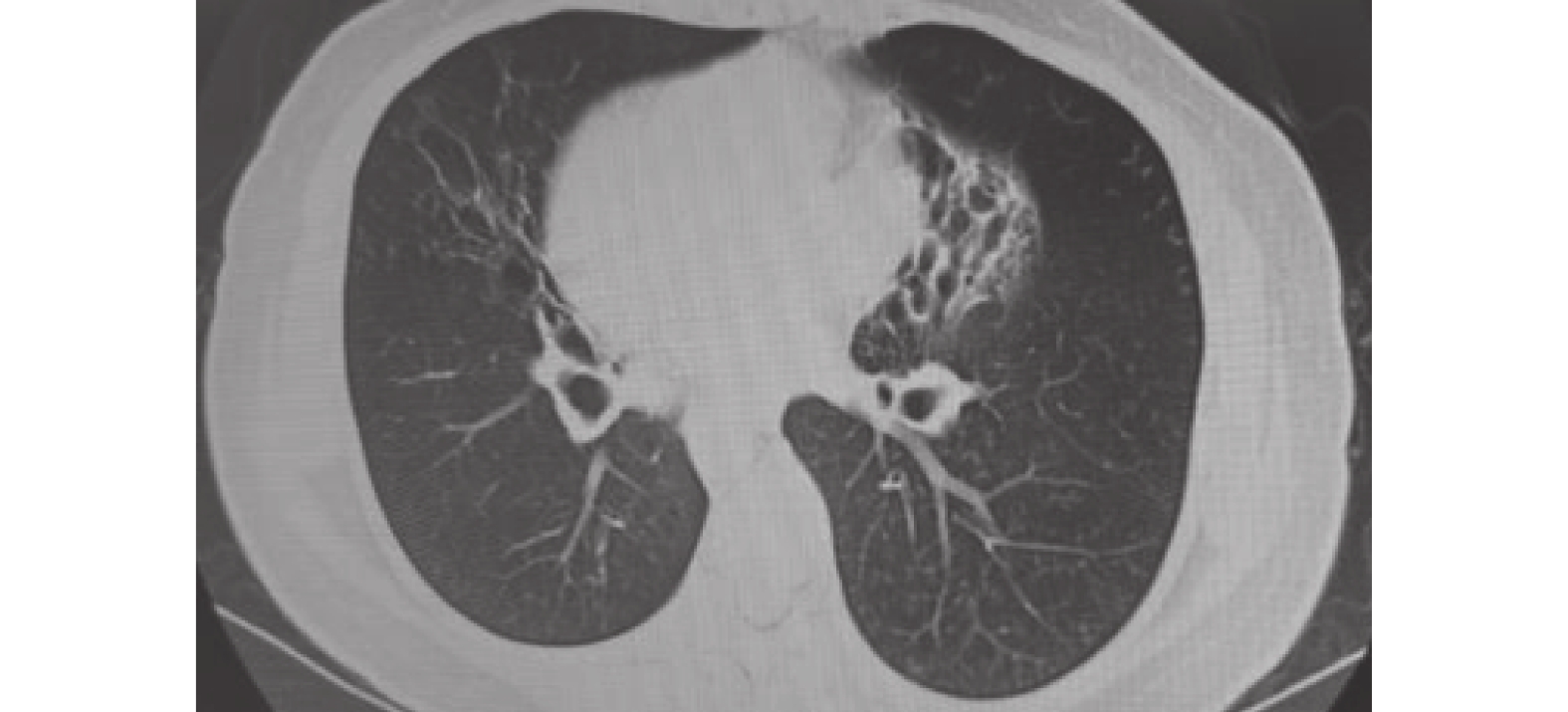
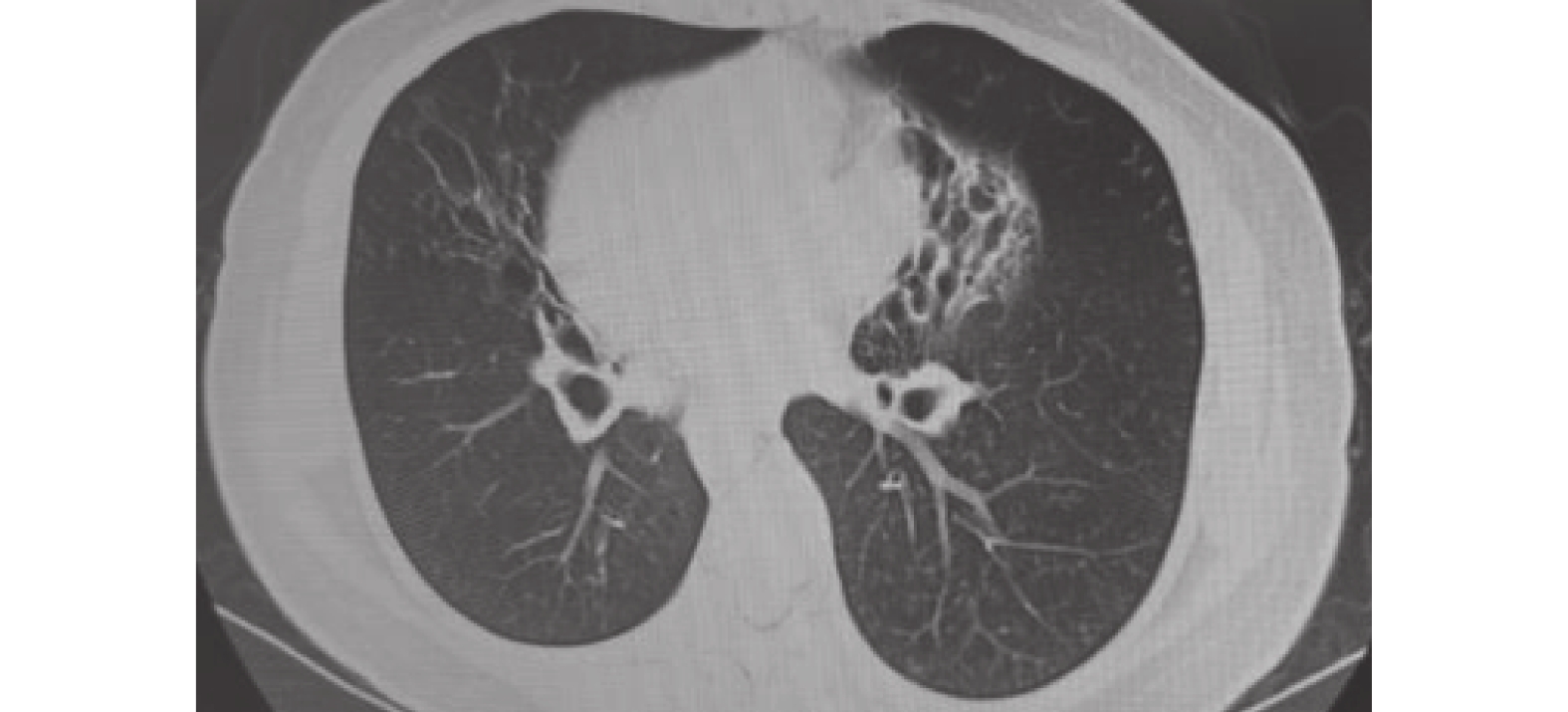 圖2
2021-03-27胸部CT檢查像
圖2
2021-03-27胸部CT檢查像
雙肺支氣管擴張伴多發感染,較前炎癥范圍減小。
討論 PCD是一種少見的遺傳性疾病,通常以常染色體隱性遺傳和X染色體連鎖方式遺傳[1],引起運動纖毛的結構或功能障礙。這種罕見的人類疾病影響1萬~2萬人中的1人[2],臨床表現復雜多樣并且較少見,更容易引起臨床醫師的誤診、漏診。在PCD患者中反復發生肺炎和支氣管炎是常見的,在學齡前80%的患者就會發生復發性下呼吸道感染[3]。PCD患者的檢查方法一般包括測量鼻部一氧化氮、視頻顯微鏡、透射電子顯微鏡、免疫熒光檢測、基因檢查等。鼻一氧化氮測量是一種簡單、無創的方法,鼻一氧化氮具有97%的敏感性和96%的特異性[4]。宏基因組二代測序增加了基因的發現,據報道,超過50個基因的突變會導致PCD[2]。由于大部分基層醫院條件差,視頻顯微鏡、透射電子顯微鏡、免疫熒光檢測等有些檢查無法實施。患者住院后期行全外顯子組測序檢測報告示CFTR、DNAH5、DNAAF1、TAP1變異。
DNAH5突變導致約50%的外動力蛋白臂缺損[5],DNAH5突變患者的呼吸上皮細胞纖毛有時是不動的,有時表現出抽搐運動。DNAAF1突變導致4%~5%的PCD的纖毛功能障礙[6]。一些數據表明,CCDC39、CCDC40、DNAAF1和LRRC6基因突變的患者極有可能發生男性不育[7]。所有報道的DNAAF1突變的女性患者均未自然生育后代[8]。但本例患者自然生育后代。DNAAF1突變通常會引起ODA和IDA的錯位,從而誘發嚴重的臨床癥狀。有研究報道了2例DNAAF1突變患者,其中1例死于嚴重肺部感染和心肺功能低下[8]。有一項研究向斑馬魚DNAAF1突變體注射DNAAF1 mRNA可以逆轉其纖毛缺陷,但外源性DNAAF1是否能促進人類纖毛運動尚不清楚[8]。
本病例的不足之處在于未完善視頻顯微鏡、透射電子顯微鏡、免疫熒光檢測、鼻一氧化氮測量。在今后的隨訪中在條件允許的情況下進一步完善。同時診斷原發性纖毛運動障礙無統一標準,診斷需結合多種檢查結果綜合判斷。
綜上,PCD是一種罕見的疾病,疾病診斷需要多種檢查結合判斷,基因組學的發展正在迅速提高診斷,希望在不久的將來出現基因療法。
利益沖突:本研究不涉及任何利益沖突。
臨床資料 患者女,33歲。主因“間斷性咳嗽咳痰30余年,加重半個月”于2021年3月18日入院。患者自幼間斷性出現咳嗽咳痰癥狀,咳黃色黏痰,不易咳出。偶有快走時出現胸悶、氣促。自行口服或靜點消炎藥后癥狀可緩解,未正規治療。冬季上述癥狀明顯。2013年1月患者出現咳嗽加重,咳黃綠痰,咳嗽時伴前胸部針刺樣疼痛,以勞累、情緒激動后加重,伴鼻塞,偶有胸悶癥狀,無發熱、心慌、腹痛、腹瀉、皮疹等癥狀,就診于我院,診斷為“Kartagener綜合征”,給予抗感染、止咳祛痰治療后好轉出院。期間一直院外規律復查,近半個月自覺上述癥狀加重,3天前就診于我院門診,胸部CT(2021-03-15)示雙肺支氣管擴張伴多發感染,鏡面右位心,上腹部內臟反位。遂就診我科。自發病以來,食欲、精神尚可,睡眠欠佳,二便正常,體重未見明顯變化。既往3年前因鼻竇炎、鼻中隔偏曲引起“頭痛”行鼻中隔偏曲矯正手術。否認食物及藥物過敏史。20歲結婚,育0子1女,配偶女兒均健康。無煙酒不良嗜好,月經史正常。
入院查體:體溫36.1℃、脈搏100次/min、呼吸20次/min、血壓113/70mm Hg(1 mm Hg=0.133 kPa)。神志清楚,全身皮膚黏膜無黃染,口唇無發紺,雙眼球結膜無水腫,雙肺呼吸音粗,可聞及少許濕啰音,右位心,心律齊,各瓣膜聽診區未聞及病理性雜音。腹軟,全腹無壓痛、反跳痛,肝脾肋下未觸及,雙下肢無水腫。
輔助檢查:入院后積極完善相關化驗檢查,血白細胞計數8.4×109/L,中性粒細胞絕對值5.97×109/L、中性粒細胞百分比71.5%,血紅蛋白130 g/L,血小板計數247×109/L。血氣分析(未吸氧情況下):pH 7.407,PCO2 33. 9 mm Hg,PO2 78.1 mm Hg。白蛋白38.7 g/L。凝血+血漿D-二聚體、谷丙轉氨酶、谷草轉氨酶、肌紅蛋白、肌鈣蛋白、腦鈉肽、降鈣素原等未見明顯異常。胸部CT(2021-03-15本院):(1)雙肺支氣管擴張伴多發感染;(2)鏡面右位心;(3)掃描范圍內:上腹部內臟反位(圖1)。肺功能(2021-03-15本院):(1)彌散功能輕度下降;(2)肺總量、殘氣量正常,殘總比增高。呼吸總阻抗、總氣道阻力、中心氣道阻力正常,周邊彈性阻力增高,共振頻率增高。中重度混合性肺通功能障礙,支管舒張試驗陽性。
圖1
2021-03-15胸部CT檢查像
雙肺支氣管擴張伴多發感染,鏡面右位心。
患者自幼出現咳嗽咳痰癥狀,疾病受季節影響明顯,患者多年受鼻竇炎影響,胸部CT示支氣管擴張、內臟逆位。既往有鼻竇炎,目前支氣管擴張、內臟逆位,故在2013年就診時診斷為Kartagener綜合征。后一直門診規律復查,但在2013年未就診之前患者未正規治療,只是病情加重時自行口服抗生素治療,具體抗生素名稱及劑量患者都不記得。目前患者癥狀加重及復查胸部CT示感染加重入我科治療。
引起支氣管擴張的病因有許多,主要有免疫功能缺陷、感染、遺傳因素、風濕性疾病等。考慮到患者自幼就有咳嗽咳痰疾病,同時追問患者家族史,患者述父母屬近親結婚,患者弟弟因為肺源性心臟病已經去世。所以患者目前遺傳因素導致的支氣管擴張可能性極大,先天性疾病比如α1-抗胰蛋白酶缺乏、纖毛功能異常、軟骨缺陷等都會引起支氣管擴張。結合患者病史有內臟轉位、鼻竇炎、支氣管擴張,因此我們從原發性纖毛運動障礙(primaryciliarydyskinesia,PCD)疾病入手。患者住院后期行全外顯子組測序檢測報告示CFTR、DNAH5、DNAAF1、TAP1變異。歐洲呼吸協會2016年提出PCD診斷參考指南,出現以下1項或多項癥狀時應考慮PCD:長期反復的慢性鼻炎,長期反復慢性咳嗽,內臟異位,先天性心臟病,伴或不伴聽力障礙的慢性中耳疾病,呼吸窘迫的新生兒,兄弟姐妹中有患PCD,女性出現反復異位妊娠,男性精子無法游動等。故診斷PCD。
入院后積極留取痰培養,痰培養示副流感嗜血桿菌1次。同時經驗性給予抗感染、化痰等治療。患者抗感染9天后復查胸部CT平掃+冠狀面重建(2021-03-27):(1)雙肺支氣管擴張伴多發感染,較前炎癥范圍減小;(2)鏡面右位心;(3)所掃范圍內:上腹部內臟反位(圖2)。考慮患者癥狀及影像學較前好轉,建議院外治療,建議患者接種疫苗、增強免疫力,冬春季節可適當口服增強免疫力藥物。在原發性感染的情況下,通常建議根除銅綠假單胞菌,故繼續預防性使用抗感染治療1周后停藥,口服乙酰半胱氨酸泡騰片促進痰液排出。患者目前隨訪過程中病情未再進一步發展。
圖2
2021-03-27胸部CT檢查像
雙肺支氣管擴張伴多發感染,較前炎癥范圍減小。
討論 PCD是一種少見的遺傳性疾病,通常以常染色體隱性遺傳和X染色體連鎖方式遺傳[1],引起運動纖毛的結構或功能障礙。這種罕見的人類疾病影響1萬~2萬人中的1人[2],臨床表現復雜多樣并且較少見,更容易引起臨床醫師的誤診、漏診。在PCD患者中反復發生肺炎和支氣管炎是常見的,在學齡前80%的患者就會發生復發性下呼吸道感染[3]。PCD患者的檢查方法一般包括測量鼻部一氧化氮、視頻顯微鏡、透射電子顯微鏡、免疫熒光檢測、基因檢查等。鼻一氧化氮測量是一種簡單、無創的方法,鼻一氧化氮具有97%的敏感性和96%的特異性[4]。宏基因組二代測序增加了基因的發現,據報道,超過50個基因的突變會導致PCD[2]。由于大部分基層醫院條件差,視頻顯微鏡、透射電子顯微鏡、免疫熒光檢測等有些檢查無法實施。患者住院后期行全外顯子組測序檢測報告示CFTR、DNAH5、DNAAF1、TAP1變異。
DNAH5突變導致約50%的外動力蛋白臂缺損[5],DNAH5突變患者的呼吸上皮細胞纖毛有時是不動的,有時表現出抽搐運動。DNAAF1突變導致4%~5%的PCD的纖毛功能障礙[6]。一些數據表明,CCDC39、CCDC40、DNAAF1和LRRC6基因突變的患者極有可能發生男性不育[7]。所有報道的DNAAF1突變的女性患者均未自然生育后代[8]。但本例患者自然生育后代。DNAAF1突變通常會引起ODA和IDA的錯位,從而誘發嚴重的臨床癥狀。有研究報道了2例DNAAF1突變患者,其中1例死于嚴重肺部感染和心肺功能低下[8]。有一項研究向斑馬魚DNAAF1突變體注射DNAAF1 mRNA可以逆轉其纖毛缺陷,但外源性DNAAF1是否能促進人類纖毛運動尚不清楚[8]。
本病例的不足之處在于未完善視頻顯微鏡、透射電子顯微鏡、免疫熒光檢測、鼻一氧化氮測量。在今后的隨訪中在條件允許的情況下進一步完善。同時診斷原發性纖毛運動障礙無統一標準,診斷需結合多種檢查結果綜合判斷。
綜上,PCD是一種罕見的疾病,疾病診斷需要多種檢查結合判斷,基因組學的發展正在迅速提高診斷,希望在不久的將來出現基因療法。
利益沖突:本研究不涉及任何利益沖突。