引用本文: 潘天宇, 鄧圣潔, 林婷婷, 萬慧敏, 單夢田, 陸晶晶. 骨髓間充質干細胞通過前列腺素E2調控NLRP3抑制肺泡巨噬細胞相關炎癥反應. 中國呼吸與危重監護雜志, 2024, 23(7): 495-503. doi: 10.7507/1671-6205.202403037 復制
版權信息: ?四川大學華西醫院華西期刊社《中國呼吸與危重監護雜志》版權所有,未經授權不得轉載、改編
急性肺損傷(acute lung injury,ALI)和急性呼吸窘迫綜合征(acute respiratory distress syndrome,ARDS)發病機制復雜,病死率和致殘率高,其主要的機制是病原體刺激中性粒細胞或者巨噬細胞引起“炎癥因子風暴”,使肺泡-毛細血管屏障的破壞、肺水腫而導致的氣體交換障礙等[1]。大量研究表明肺部炎癥反應的失平衡是ALI/ARDS發病的關鍵環節,因此抑制和減輕肺部炎癥反應是目前ALI/ARDS治療的研究熱點。核苷酸結合寡聚化結構域樣受體3(nucleotide-bound oligomerized domain-like receptor 3,NLRP3)炎癥小體是新的模式識別受體,其活化既可促進多種促炎癥細胞因子的成熟和分泌,還可以調節細胞焦亡,在脂多糖(lipopolysaccharide,LPS)誘導的ALI/ARDS發生發展中起著重要作用。近年來,骨髓間充質干細胞(bone marrow mesenchymal stem cells,BMSC)在抗炎癥、免疫調節及促進血管新生等方面均展現出良好的臨床應用前景,研究表明BMSC在炎癥刺激下可以大量合成并釋放前列腺素E2(prostaglandin E2,PGE2)減輕炎癥反應[2-4]。本研究旨在探究BMSC對ALI中巨噬細胞的作用,機制是否與調控PGE2與NLRP3炎癥小體的活化有關等,以期為開發新的ALI治療策略提供理論依據。
1 材料與方法
1.1 材料
實驗動物:選擇10只SPF級(6周齡,18~22 g)健康成年雄性BALB/c小鼠。購自常州卡文斯,飼養在同濟大學附屬東方醫院動物房內,環境溫度24~26℃,環境濕度55%~60%,光/暗周期為12 h/12 h,小鼠自由飲水和進食。
實驗材料:LPS(Sigma),10%水合氯醛(上海生工),DMEM高糖/低糖培養基(Hyclone),FBS(Hyclone),DIL-ac-LDL(上海生工),Trizol(Invitrogen),DEPC處理水(CTCC),氯仿/異丙醇/無水乙醇(上海國藥),SYBRGreen PCR試劑盒(Thermo F-415XL),逆轉錄試劑盒(Thermo K1622),CD90抗體(Invitrogen 61-0902-80),CD29抗體 (Invitrogen 12-0291-81),CD45抗體(Invitrogen 67-0451-82),CD34抗體(Invitrogen 11-0341-81),BCA蛋白定量試劑盒(凱基KGPBCA),RIPA強效裂解液(碧云天P0013B),丙烯酰胺/電泳緩沖液/過硫酸銨(上海國藥),蛋白預染Marker(Thermo 26616),NLRP3(ABclonal A5652),pro-Caspase-1(ABclonal A0964),Caspase-1(ABclonal A0964),pro-IL-1β(CST 12242S),GAPDH(proteintech 60004-1-Ig),辣根酶標記山羊抗兔IgG(中杉金橋 ZB2301),辣根酶標記山羊抗小鼠IgG(中杉金橋 ZB2305),腫瘤壞死因子α(tumor necrosis factor-α,TNF-α)(Elabscience E-EL-M0049c),白細胞介素1β(interleukin-1β, IL-1β)(Elabscience E-EL-M0037c),IL-10(Elabscience E-EL-M0046c),IL-18(Elabscience E-EL-M0730c),PGE2(酶聯生物)。
實驗儀器:細胞培養箱(Thermo Scientific 8000),熒光倒置顯微鏡(OLYMPUS,IX71),光學顯微鏡(XDS-1A),離心機(Eppendorf),流式細胞儀(BD-FACSVerse),低溫冷凍離心機 (Sigma 3K15), Real-time檢測儀(ABI-7500),電泳儀(BIORAD),酶標儀(ThermoMK3)等。
1.2 方法
1.2.1 支氣管肺泡灌洗獲取巨噬細胞并檢測純度
取健康SPF級成年雄性BALB/c小鼠,麻醉后暴露氣管并氣管插管,用預冷的磷酸鹽緩沖液(phosphate buffered saline,PBS)進行雙肺灌洗,回收獲得支氣管肺泡灌洗液(bronchoalveolar lavage fluid,BALF)。BALF離心重懸后獲得肺泡巨噬細胞,在巨噬細胞培養液中加入DIL-ac-LDL,避光孵育并拍照。
1.2.2 分離及流式細胞術鑒定BMSC
用頸椎脫位法處死小鼠后,用手術剪打開骨髓腔,沖洗、離心、重懸并培養出BMSC。將分離的BMSC收集培養,每孔分別加入200 μL的染色緩沖液,以及CD90、CD29、CD45和CD34抗體。避光孵育30 min后洗滌重懸。流式細胞術定量檢測并分析。
1.2.3 建立穩定的慢病毒轉染BMSC細胞株
(1)觀察慢病毒MOI:商業合成ptges(Gene ID:64292)合成3條干擾慢病毒,1條過表達慢病毒。將病毒按照MOI為10、50、100感染BMSC,48 h后熒光顯微鏡下觀察病毒感染效率。(2)篩選出最佳敲降慢病毒:細胞內總RNA的抽提采用Trizol提取試劑盒(Invitrogen),采用第一鏈cDNA合成試劑盒(天根),SYBRGreen PCR試劑盒(Thermo F-415XL)以及逆轉錄試劑盒(Thermo K1622)等完成實時聚合酶鏈反應(real-time polymerase chain reaction,RT-PCR)。逆轉錄PCR所使用的引物由上海生工公司Primer Premier 5.0軟件設計、合成。根據RT-PCR反應體系配制反應液。在PCR反應管(20 μL)中分別加入ddH2O 7 μL、SybrGreen qPCR Master Mix 10 μL、Forward primer 1 μL、Reverse primer 1 μL、cDNA模板1 μL,充分混勻。94℃ 10 min,以及94℃ 20 s,55℃ 20s,72℃ 20 s共40個循環。PCR擴增后,以管家基因GAPDH為內參,采用2–△△CT法分析目的基因在對照組和各實驗組之間的表達差異。篩選出最佳效率的敲降慢病毒序列。(3)采用慢病毒轉染ptges及ptges shRNA至BMSC,建立穩定的ptges過表達BMSC細胞株[BMSC-PGE2(+)]及ptges沉默的BMSC細胞株[BMSC-PGE2(–)]。調整細胞數為1×106/mL接種于6孔板培養中,吸取上清,PBS沖洗3次,加入含有慢病毒的無血清培養基3 mL后繼續培養、傳代并繼續下一步實驗。
1.2.4 Western blot檢測巨噬細胞中的NLRP3、pro-Caspase-1、Caspase-1及pro-IL-1β蛋白表達水平
(1)樣品準備:肺泡巨噬細胞以2×106/mL接種在Transwell上室中,隨機分為5組,即對照組、LPS組、LPS+BMSC組、LPS+BMSC-PGE2(+)組、LPS+BMSC-PGE2(–)組。共培養的3組中將BMSC/BMSC-PGE2(+)/BMSC-PGE2(–)以1×106/mL接種在Transwell下室。根據分組將肺泡巨噬細胞與BMSC/BMSC-PGE2(+)/BMSC-PGE2(–)置入Transwell共培養體系,LPS組及共培養體系組予以終濃度為10 ng/mL LPS的無血清培養基共培養5 h,然后以終濃度為5 mmol/L的ATP的無血清培養基共培養1 h。(2)收集細胞,向細胞樣本中加入預冷的已加PMSF的RIPA裂解液,混勻,冰上充分裂解離心后收集蛋白。(3)進行SDS-PAGE凝膠電泳。上樣,電泳,切膠,“三明治”法轉膜,漂洗并封閉。加入NLRP3(1∶1 000)、pro-Caspase-1(1∶1 000)、Caspase-1(1∶1 000)、pro-IL-1β(1∶1 000)及GAPDH(1∶5 000)抗體,4℃孵育,漂洗后加入兔二抗或鼠二抗(1∶5 000)。加入ECL發光液顯色拍照。
1.2.5 RT-PCR檢測巨噬細胞中的NLRP3、Caspase-1的mRNA表達水平
收集各組細胞,加入Trizol試劑,提取細胞中的總RNA。隨后按照第一鏈cDNA合成試劑盒(天根)說明書合成cDNA,按照SYBRGreen PCR試劑盒(Thermo F-415XL)說明書配制RT-PCR反應體系,反應程序為:94℃ 10 min,以及94℃ 20s,55℃ 20 s,72℃ 20 s共40個循環。以管家基因GAPDH為內參,采用2–△△CT法計算NLRP3、Caspase-1的mRNA表達水平。
1.2.6 ELISA法檢測細胞上清液中TNF-α、IL-1β、IL-10、IL-18及PGE2水平
采用TNF-α、IL-1β、IL-10、IL-18及PGE2 ELISA試劑盒說明書。
1.3 統計學方法
采用SPSS19.0統計軟件分析,呈正態分布的計量數據以均數±標準差(x±s)表示,多組比較采用單因素方差分析,兩兩比較采用LSD法,方差不齊時采用非參數檢驗。P<0.05為差異有統計學意義。
2 結果
2.1 成功獲取并培養肺泡巨噬細胞
采用原位支氣管肺泡灌洗法收集的BALF中90%的是肺泡巨噬細胞,紅細胞、中性粒細胞及淋巴細胞等經純化培養被去除。獲得的肺泡巨噬細胞呈圓形,偽足不明顯,貼壁后,肺泡巨噬細胞鋪展開,逐漸伸出偽足(圖1a)。熒光顯微鏡下觀察肺泡巨噬細胞內有大量DIL-ac-LDL,提示巨噬細胞吞噬活性良好(圖1b)。
 圖1
小鼠巨噬細胞特征
圖1
小鼠巨噬細胞特征
a. 鏡下特征(×100,×200);b. 熒光顯微鏡下細胞特征(×500)。
2.2 成功提取、培養及鑒定BMSC
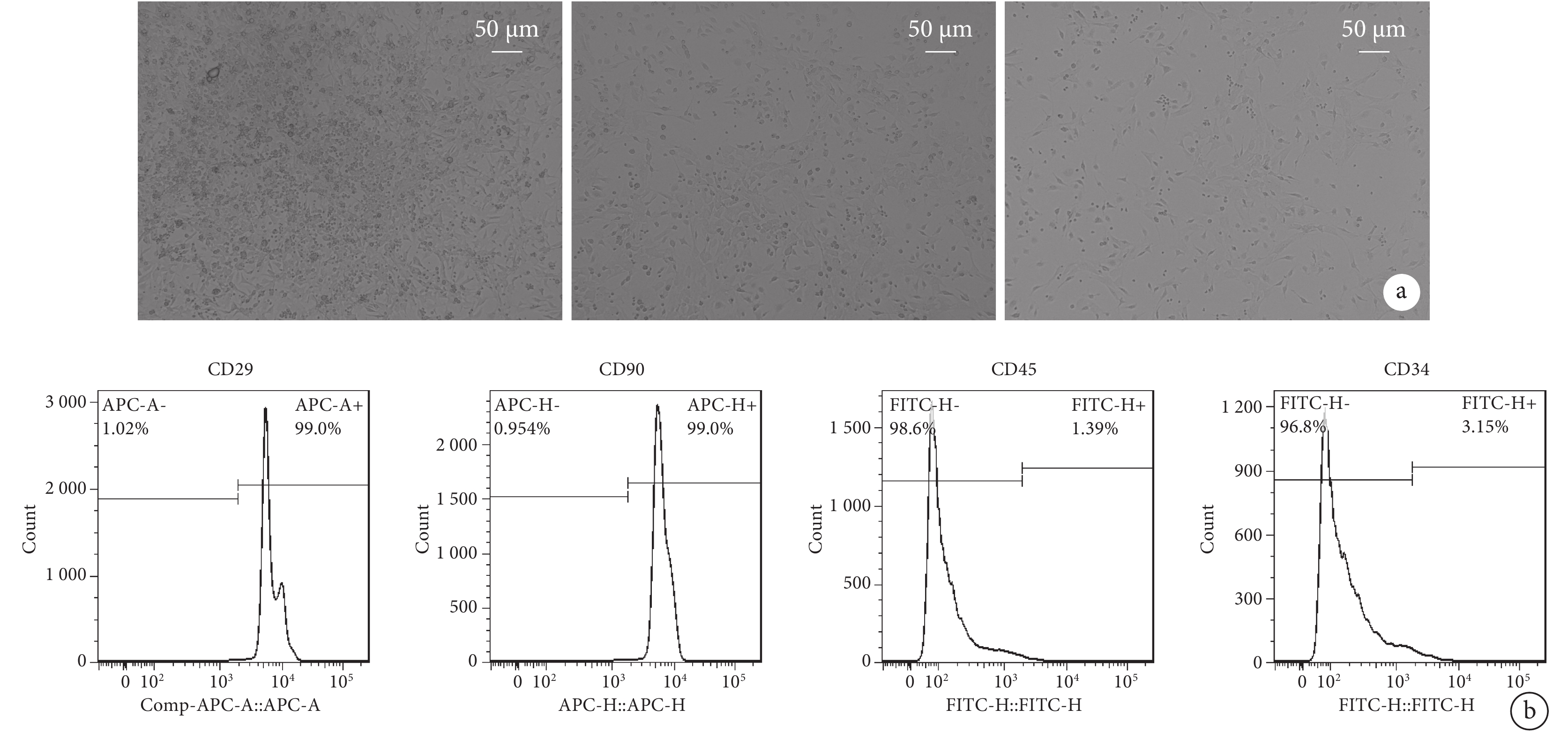
BMSC原代細胞在接種24 h后逐漸貼壁,細胞胞體小,大多為梭形,核居中,偶有寬大扁平的多邊形,細胞折光性強。通過多次換液傳代,可使BMSC更為均一,成梭形成纖維樣(圖2a)。利用流式細胞儀對第3代BMSC進行鑒定,其結果顯示細胞CD29、CD90抗體陽性表達率均為99%,CD45、CD34抗體陽性表達率分別為1.39%和3.15%,提示CD29、CD90為陽性表達,CD45、CD34為陰性表達(圖2b)。符合公認的BMSC免疫表型,提示提取BMSC培養并鑒定成功。
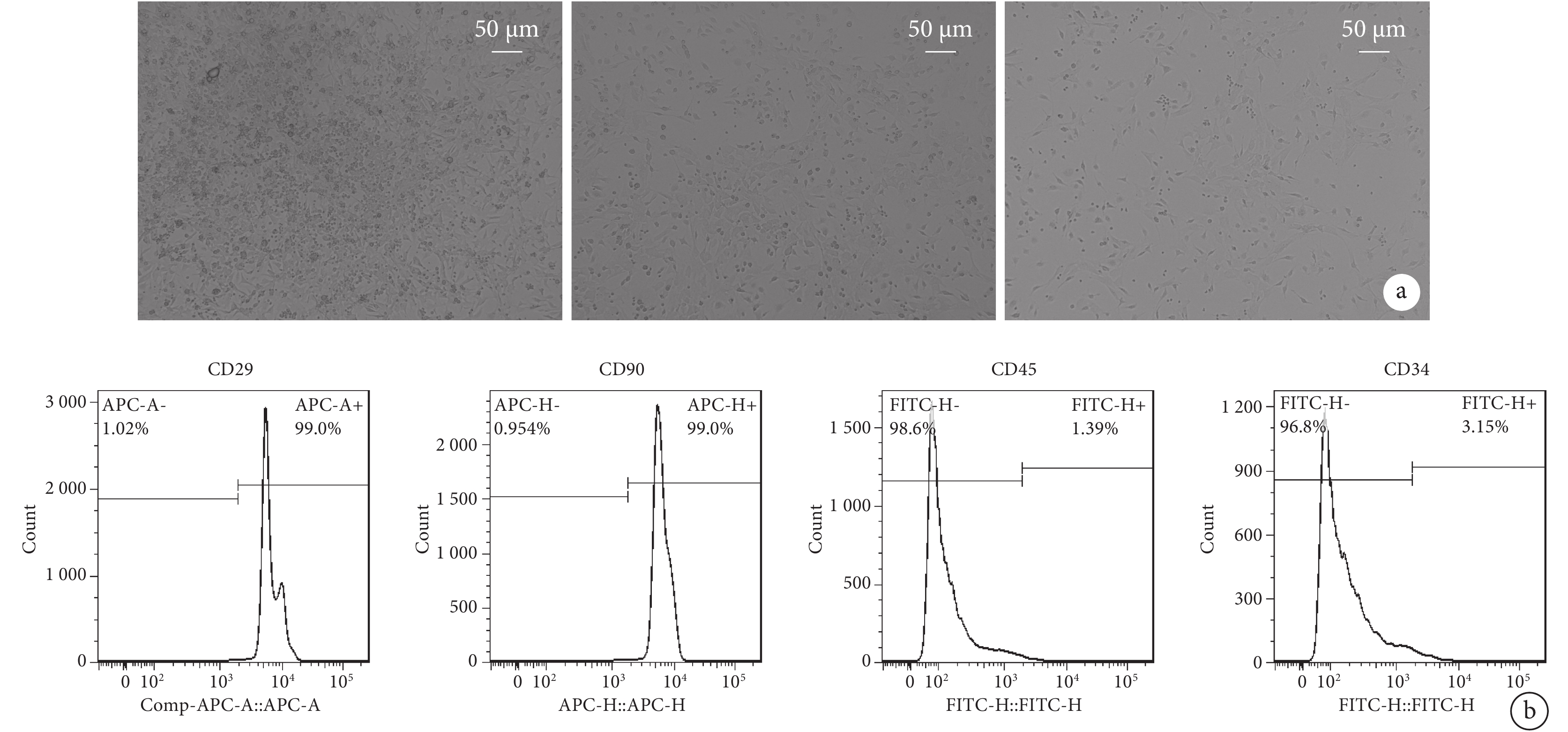 圖2
小鼠骨髓間充質細胞特征
圖2
小鼠骨髓間充質細胞特征
a. 小鼠BMSC的鏡下特征(×200);b. 流式細胞儀鑒定結果:CD29陽性,CD90陽性,CD45陰性、CD34陰性。
2.3 確定慢病毒MOI值及感染效率最佳的序列
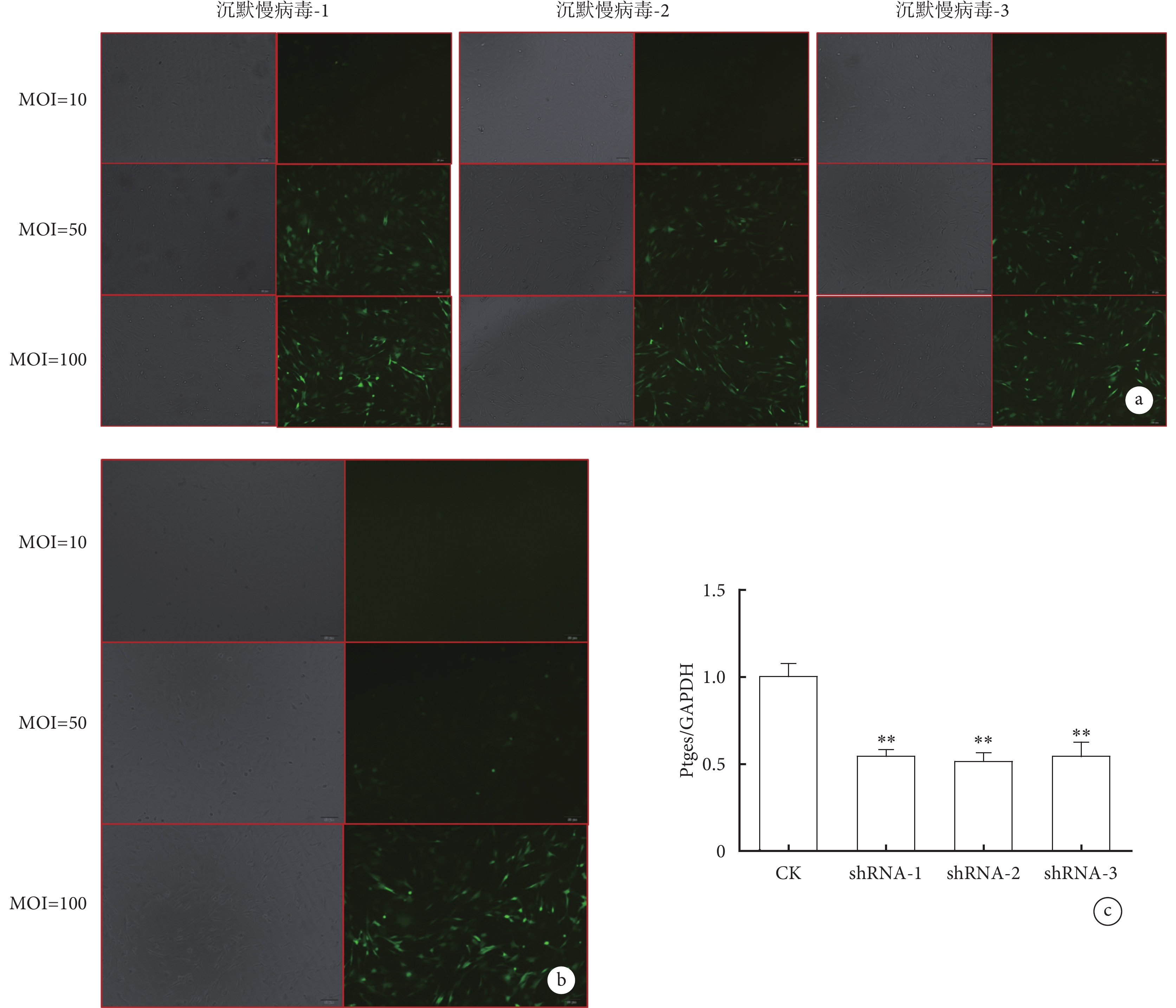
熒光顯微鏡下觀察可見,沉默慢病毒當MOI值為50時,感染效率已達80%以上,故后續選擇MOI值為50進行實驗(圖3a),而過表達慢病毒選擇MOI為100進行后續實驗(圖3b)。RT-PCR實驗結果顯示,相比對照組,當分別感染沉默慢病毒后,目的基因表達水平均有不同程度的降低,其中慢病毒2效果最好(圖3c),所以后期選擇該靶向序列用于實驗。
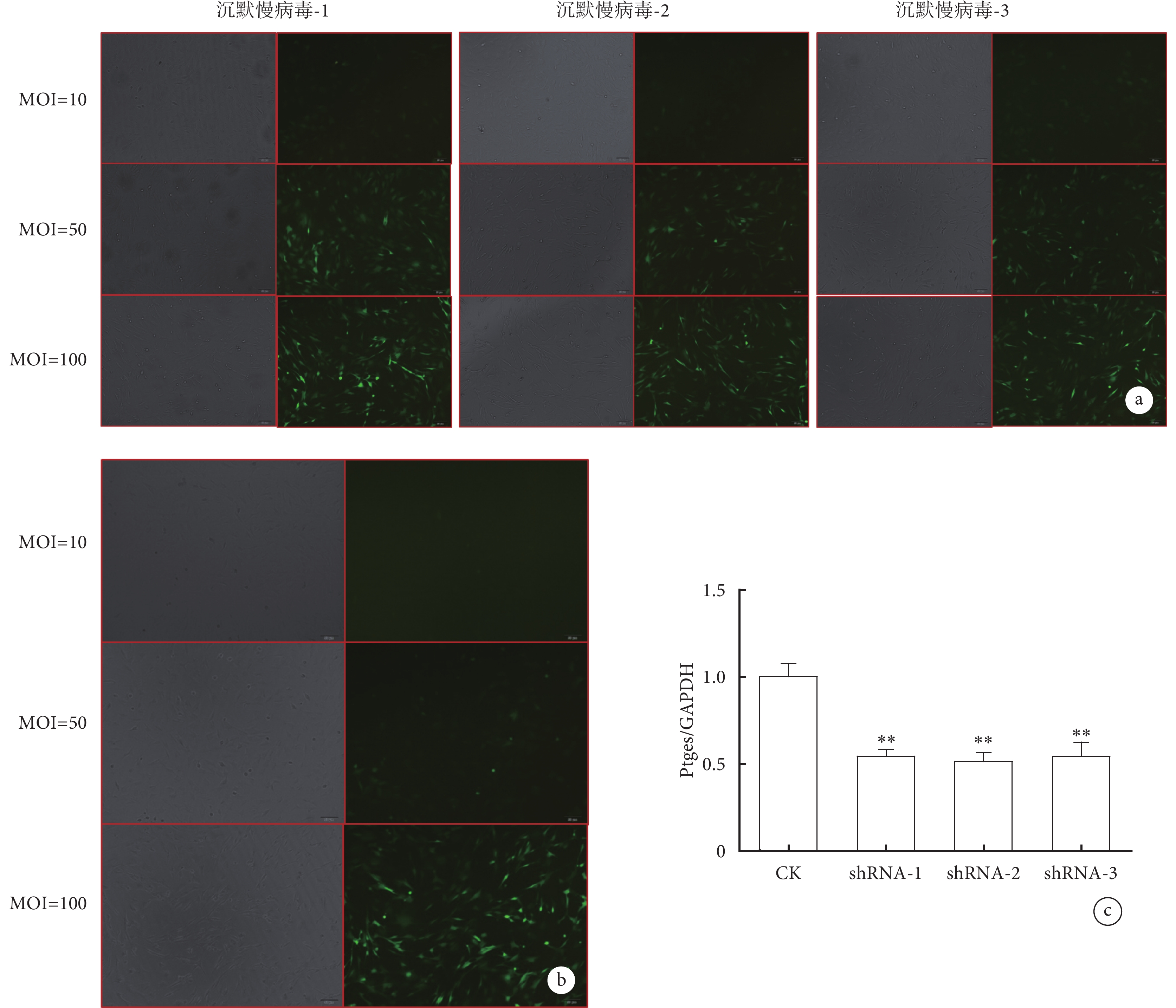 圖3
BMSC細胞株轉染慢病毒熒光顯微鏡下像和RT-PCR結果
圖3
BMSC細胞株轉染慢病毒熒光顯微鏡下像和RT-PCR結果
a. 沉默慢病毒的鏡下特征(×50);b. 過表達慢病毒的鏡下特征(×50);c. RT-PCR顯示shRNA2沉默效率最高。與對照組比較,**
2.4 巨噬細胞中的NLRP3、pro-Caspase-1、Caspase-1、pro-IL-1β蛋白表達水平變化
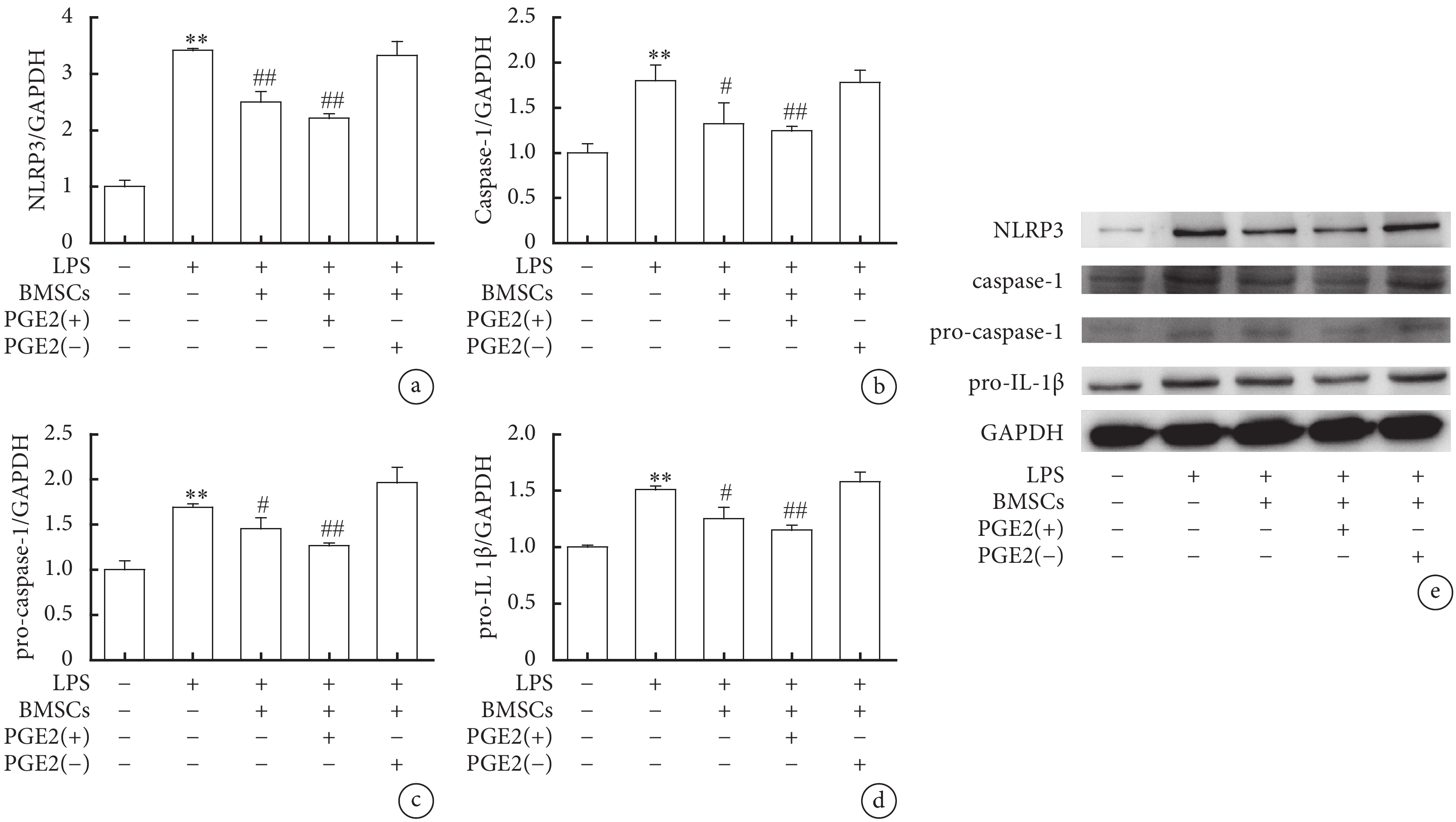
Western blot結果顯示,LPS組NLRP3、pro-Caspase-1、Caspase-1及pro-IL-1β蛋白表達量較對照組明顯升高,差異有統計學意義(P<0.01);LPS+BMSC組巨噬細胞與BMSC共培養后,NLRP3、pro-Caspase-1、Caspase-1及pro-IL-1β蛋白表達較LPS組明顯減少,差異有統計學意義(P<0.05,P<0.01);LPS+BMSC-PGE2(+)組中巨噬細胞與BMSC-PGE2(+)共培養后,NLRP3、pro-Caspase-1、Caspase-1及pro-IL-1β蛋白表達進一步下降,差異有統計學意義(P<0.01);而LPS+BMSC-PGE2(–)組與LPS組無明顯變化,差異無統計學意義(圖4)。表明LPS刺激后的巨噬細胞中,BMSC通過分泌PGE2,進一步抑制下游NLRP3炎癥小體的活化,減輕炎癥反應。
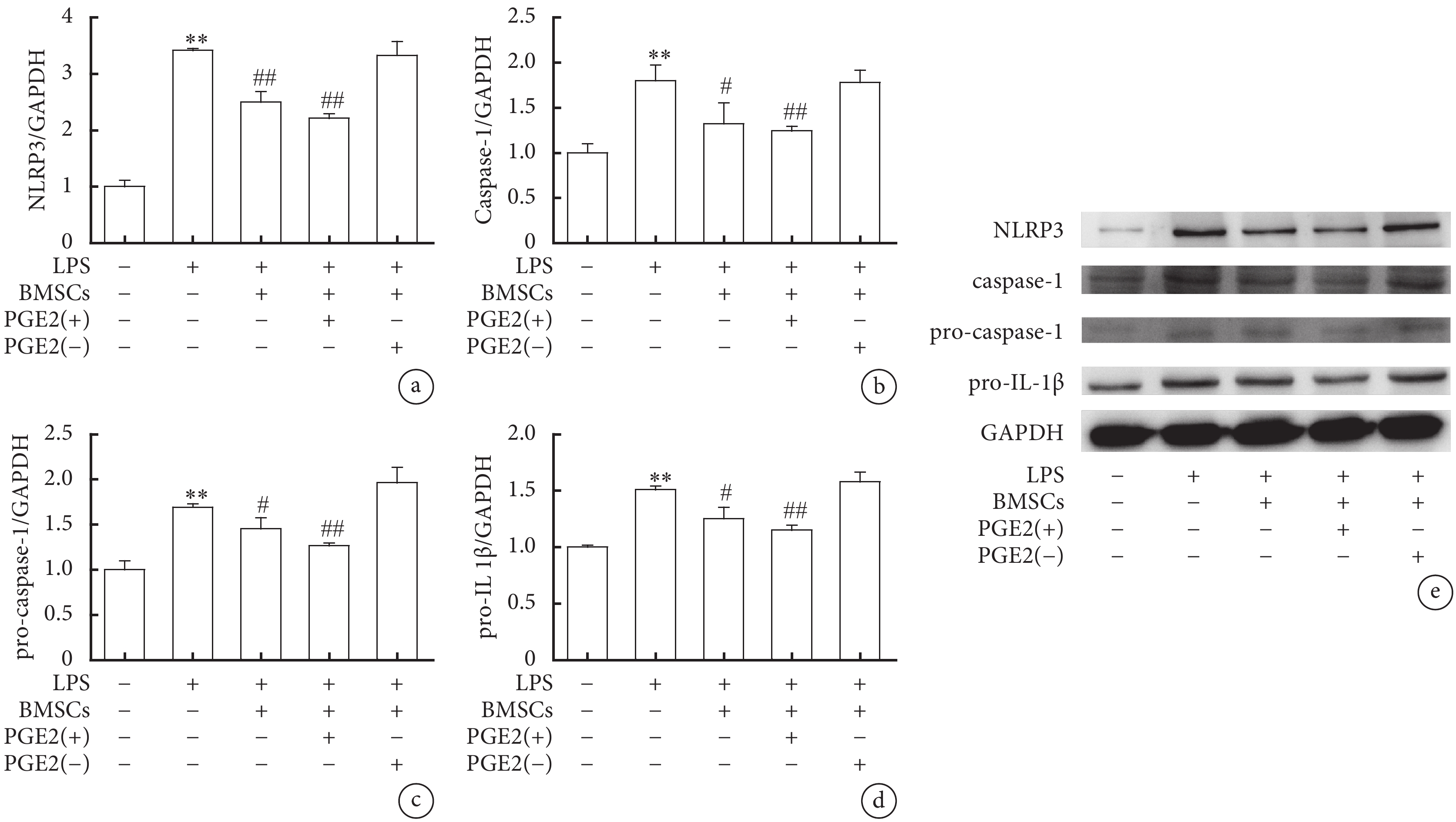 圖4
NLRP3、pro-Caspase-1、Caspase-1、pro-IL-1β蛋白表達Western blot結果
圖4
NLRP3、pro-Caspase-1、Caspase-1、pro-IL-1β蛋白表達Western blot結果
a. NLRP3的表達水平;b. Caspase-1的表達水平;c. pro-Caspase-1的表達水平;d. pro-IL-1β的表達水平;e. NLRP3、pro-Caspase-1、Caspase-1及pro-IL-1β蛋白表達水平的代表性圖像。與對照組比較,**
2.5 巨噬細胞中的NLRP3、Caspase-1的mRNA表達水平變化
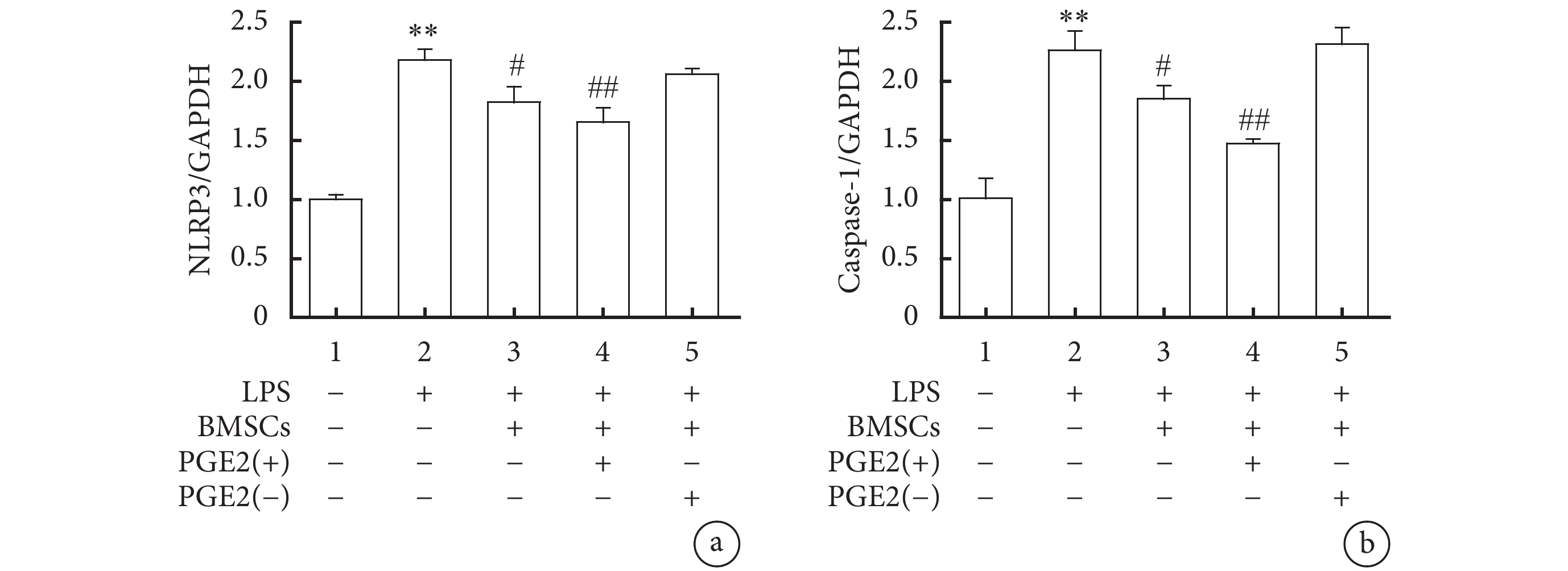
RT-PCR實驗結果顯示,LPS組NLRP3、Caspase-1的mRNA表達量較對照組明顯升高,差異顯著,具有統計學意義(P<0.01);LPS+BMSC組巨噬細胞與BMSC共培養后,NLRP3、Caspase-1的mRNA表達量較LPS組明顯減少,差異有統計學意義(P<0.05);LPS+BMSC-PGE2(+)組中,巨噬細胞與BMSC-PGE2(+)共培養后,NLRP3、Caspase-1的mRNA表達量進一步下降,差異有統計學意義(P<0.01);而LPS+BMSC-PGE2(–)組與LPS組無明顯變化,差異無統計學意義(圖5)。
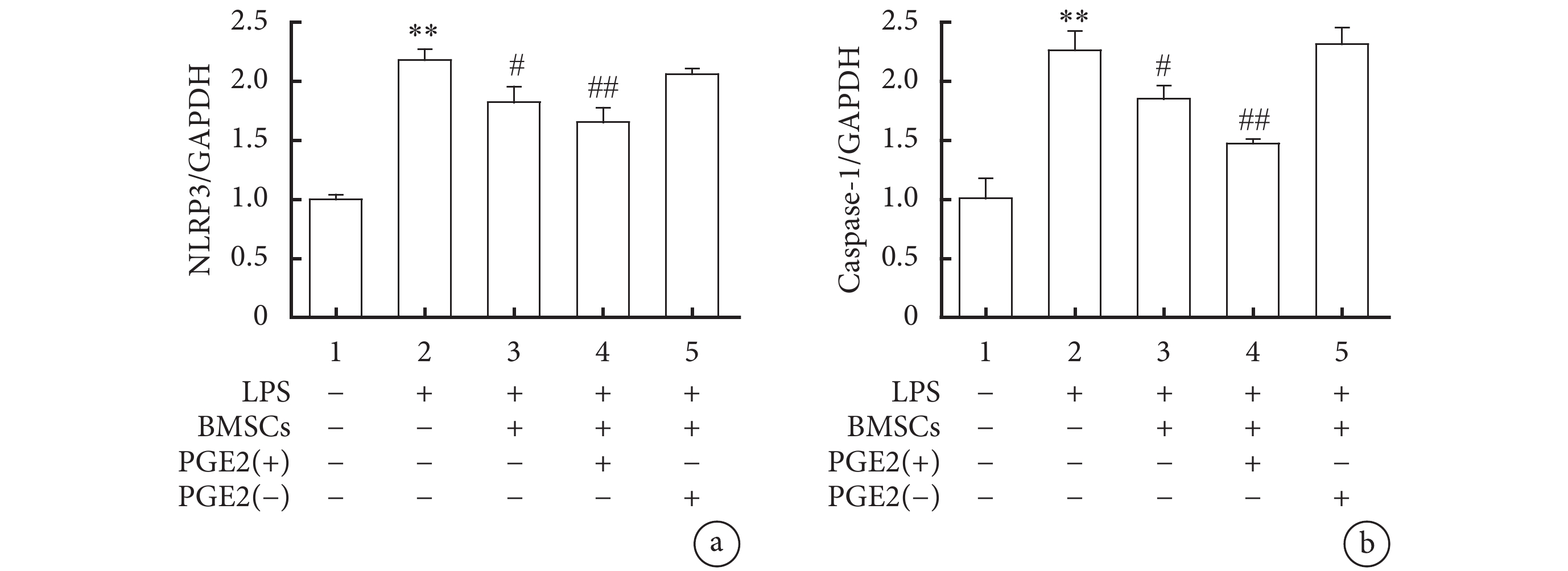 圖5
各組NLRP3的表達水平(a)以及Caspase-1的表達水平(b)
圖5
各組NLRP3的表達水平(a)以及Caspase-1的表達水平(b)
與對照組比較,**
2.6 細胞上清液中相關炎癥因子的水平變化
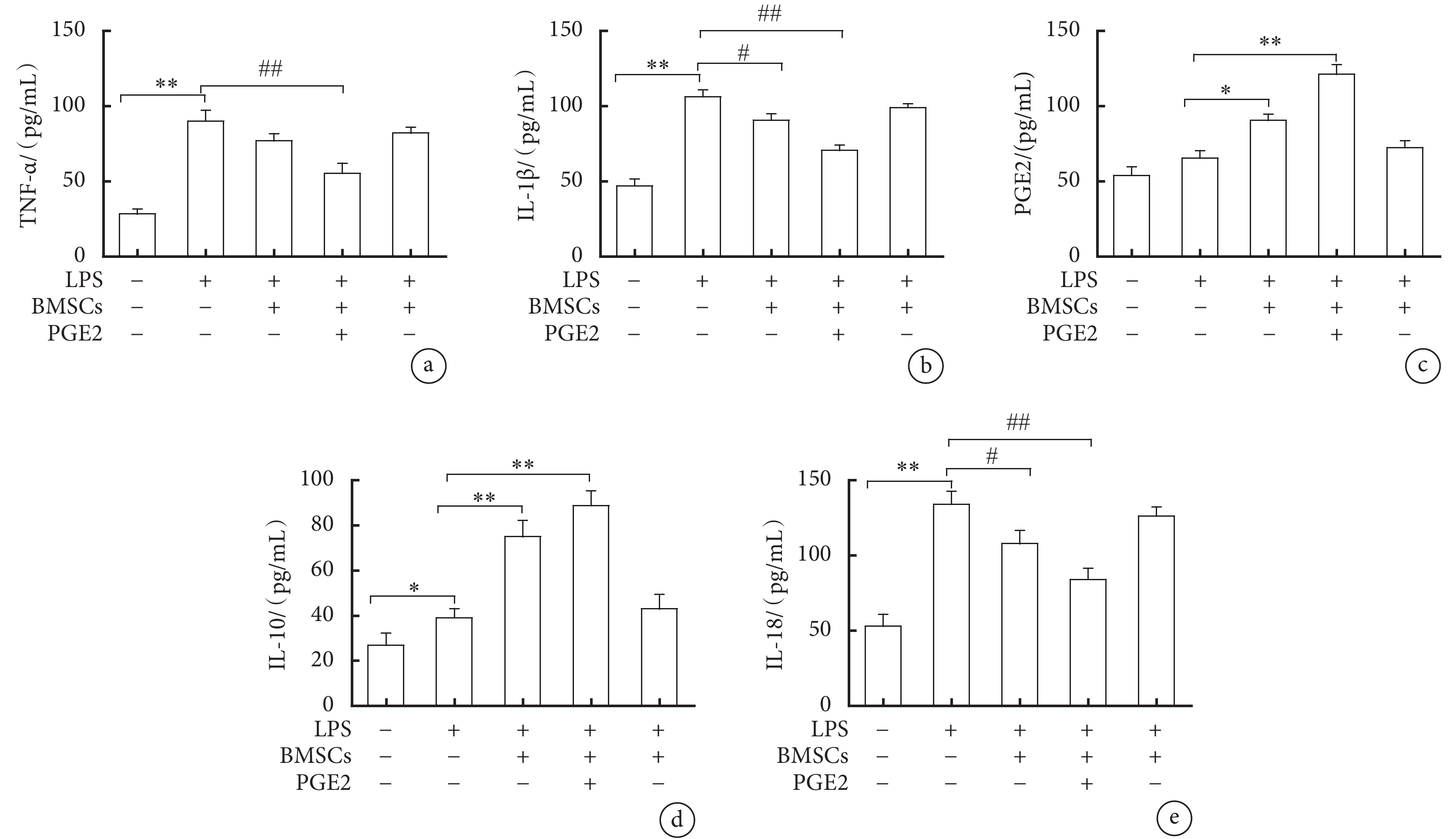
ELISA結果顯示,培養液上清中TNF-α、IL-1β及IL-18含量在LPS組中最高,顯著高于對照組、LPS+BMSC組和LPS+BMSC-PGE2(+)組,差異有統計學意義(P<0.05,P<0.01)。培養液上清中IL-10和PGE2含量,LPS組較對照組有升高趨勢,LPS+BMSC組和LPS+BMSC-PGE2(+)組進一步增加,與LPS組比較差異有統計學意義(P<0.05,P<0.01),LPS+BMSC-PGE2(–)組與LPS組沒有顯著差異(圖6)。
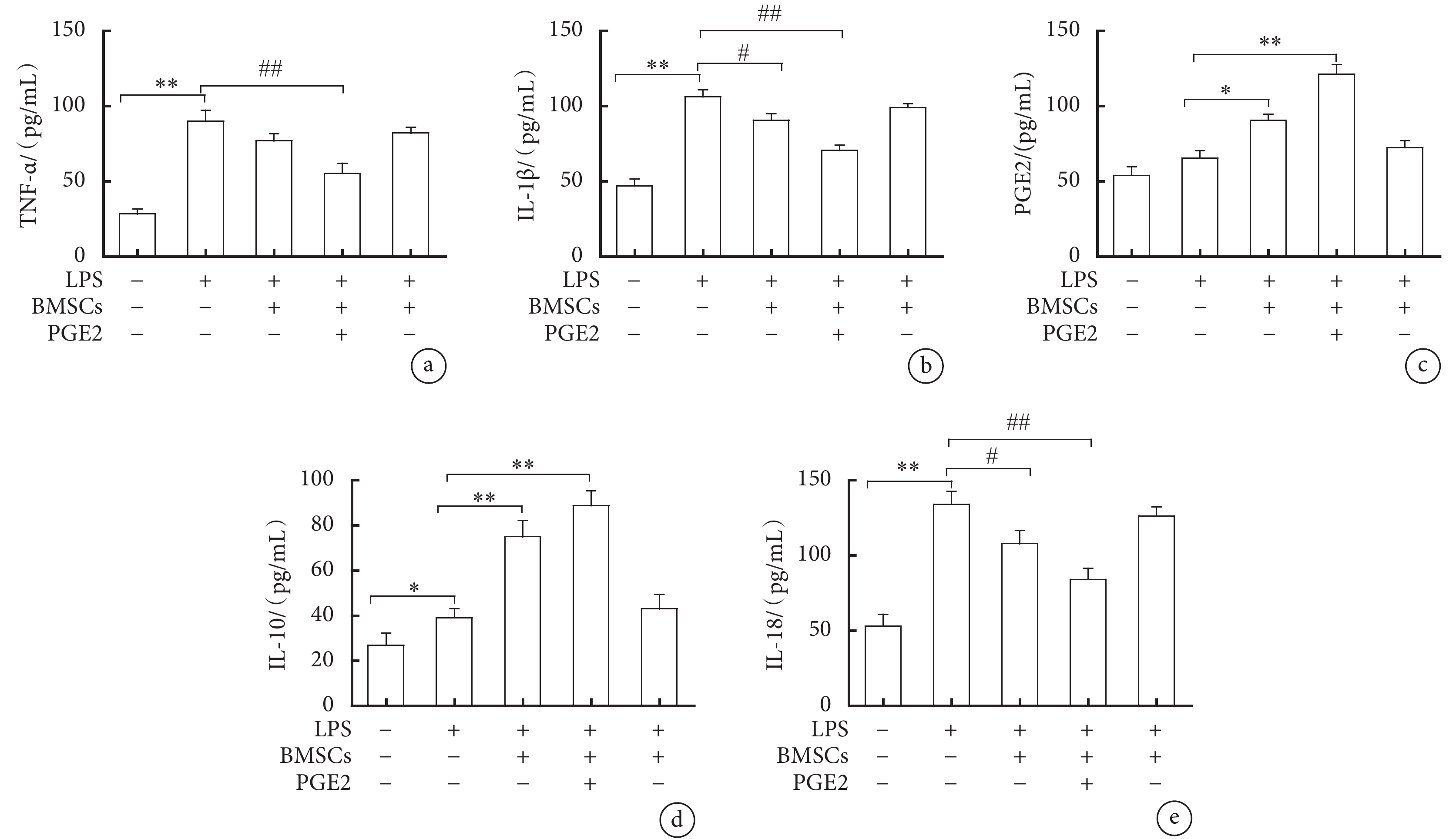 圖6
細胞上清液中相關炎癥因子表達的ELISA結果
圖6
細胞上清液中相關炎癥因子表達的ELISA結果
a. TNF-α的表達水平;b. IL-1β的表達水平;c. PGE2的表達水平;d. IL-10的表達水平;e. IL-18的表達水平。與對照組比較,*
3 討論
ALI和ARDS起病急,病情非常兇險,病死率高,是嚴重威脅人類健康的呼吸系統急危重癥[5]。其本質是由多種炎性介質和效應細胞共同參與,呈瀑布樣級聯放大的炎癥反應,發病過程中涉及炎癥、凋亡、氧化應激等一系列信號通路。肺泡巨噬細胞的激活是ALI/ARDS進程中的始動環節[6],在受到內外致病因素的刺激下,一方面分泌TNF-α、IL-1β、IL-6和巨噬細胞炎性蛋白2(macrophage inflammatory protein 2,MIP-2)等促炎因子,后者募集大量中性粒細胞,啟動和維持肺部炎癥反應,造成肺泡上皮細胞和血管內皮細胞損傷;另一方面,肺泡巨噬細胞還可以分泌IL-4、IL-10等抗炎因子,后者促進免疫細胞吞噬壞死組織細胞和凋亡的中性粒細胞,促進炎癥消散和組織修復。因此,肺泡巨噬細胞介導的促炎和抗炎失衡是ALI/ARDS發生發展的關鍵性因素。
近年來許多研究證實了BMSC可調節肺部炎癥反應,恢復細胞因子網絡之間的平衡,減少促炎因子的分泌,增加抗炎標志物和細胞因子的釋放[7]。在我們的研究中,通過流式細胞儀得到CD29、CD90為陽性表達,而CD45、CD34為陰性表達的較為純化的BMSC,可以用于進一步實驗。在LPS誘導的ALI模型中,肺泡巨噬細胞相關炎性因子TNF-α、IL-1β、IL-18的表達明顯增加,而BMSC能夠明顯逆轉這些促炎因子的表達并增強抗炎因子IL-10 的表達水平。同時,與BMSC相比,過表達PGE2的BMSC可進一步抑制炎癥反應,表明過表達PGE2可增強BMSC對LPS誘導的肺泡巨噬細胞損傷的保護作用。
NLRP3是一種模式識別受體(pattern-recognition receptors,PRRs)[8],主要在中性粒細胞和巨噬細胞的細胞質中表達,其組成部分包括先天免疫受體蛋白NLRP3、炎性蛋白酶Caspase-1與銜接蛋白ASC。作為機體固有免疫的重要組成部分[9-10],NLRP3蛋白可被病原體相關分子模式(pathogen-associated molecular patterns,PAMPs)識別并激活,如NLRP3炎癥小體可以被細菌LPS、活性氧、K+外流、Ca2+信號、毒素、顆粒物質、病毒RNA和溶酶體破壞等。NLRP3炎癥小體激活機制有三種不同信號通路:經典NLRP3炎性體激活、非經典NLRP3炎性體激活和替代NLRP3 炎性體激活(也稱為一步NLRP3 炎性體激活)[11-12]。NLRP3炎性體的激活將誘導pro-Caspase-1的自我切割和激活,從而促進包括IL-1β和IL-18在內的多種促炎性細胞因子的剪切和分泌[13],另一方面活化的Caspase-1還將切割消皮素D(gasdermin D,GSDMD)并釋放其N端結構域,這一結構域將轉移并在細胞膜處形成孔,從而釋放細胞內容物,進而調節程序性細胞焦亡(pyroptosis),誘導細胞在炎性和應激的病理條件下死亡[3,14-17]。研究表明在小鼠ALI模型中,NLRP3炎癥小體可被激活,而抑制NLRP3炎癥小體可以顯著抑制TNF-α、IL-1β的合成和分泌,改善膿毒癥所致的肺泡上皮細胞通透性的增加[18]。因此,NLRP3炎癥小體的活化在ALI的發生發展中起到重要作用。
其中,包括PGE2在內的許多因子都與NLRP3炎癥小體通路激活密切相關[4,19]。PGE合酶(PGE synthase,PTGES)作為PGE2合成中的終末限速酶,參與PGE2的合成[20]。PGE2通過前列腺素E受體(prostaglandin E receptor,EP)家族的4種不同受體發出信號:EP1、EP2、EP3和EP4[21]。研究發現NLRP3炎癥體的快速激活被蛋白激酶A(protein kinase A,PKA)直接抑制,PKA直接磷酸化胞漿受體NLRP3,減弱其ATP酶功能。而PKA是由PGE2通過PGE2受體EP4信號傳導誘導的[1]。還有研究發現PGE2通過促進抗炎因子IL-10的分泌,能夠抑制中性粒細胞的黏附和內皮細胞激活,對NLRP3炎性小體有拮抗作用[22-23]。阻斷巨噬細胞IL-10受體可促進NLRP3炎性體的激活[24],NLRP3炎癥小體的活化也可以抑制單核細胞中IL-10的分泌[25]。IL-10基因敲除的小鼠在LPS刺激后,NLRP3、pro-Caspase-1和pro-IL-1β水平均明顯高于野生型[26],因此通過IL-10來抑制NLRP3炎癥小體的活化可能是PGE2的作用機制之一。本研究中,BMSC可明顯抑制NLRP3、pro-Caspase-1、Caspase-1及pro-IL-1β的表達,降低TNF-α、IL-1β表達水平,增加IL-10的表達水平,當給予過表達PGE2的BMSC后,PGE2可抑制NLRP3活化,與BMSC相比,對ALI的抑制作用進一步增強,從而發揮對ALI的治療作用。
綜上所述,研究結果從細胞層面表明,在LPS誘導的ALI發病過程中,BMSC通過攜帶并分泌PGE2來促進肺泡巨噬細胞中抗炎因子的釋放,從而抑制肺泡巨噬細胞中NLRP3炎癥小體的活化,減輕肺部炎性損害。本研究為運用BMSC來調控ALI時的炎癥反應提供理論基礎,為臨床救治ALI患者提供新的治療策略和重要的理論依據。
利益沖突:本研究不涉及任何利益沖突。
急性肺損傷(acute lung injury,ALI)和急性呼吸窘迫綜合征(acute respiratory distress syndrome,ARDS)發病機制復雜,病死率和致殘率高,其主要的機制是病原體刺激中性粒細胞或者巨噬細胞引起“炎癥因子風暴”,使肺泡-毛細血管屏障的破壞、肺水腫而導致的氣體交換障礙等[1]。大量研究表明肺部炎癥反應的失平衡是ALI/ARDS發病的關鍵環節,因此抑制和減輕肺部炎癥反應是目前ALI/ARDS治療的研究熱點。核苷酸結合寡聚化結構域樣受體3(nucleotide-bound oligomerized domain-like receptor 3,NLRP3)炎癥小體是新的模式識別受體,其活化既可促進多種促炎癥細胞因子的成熟和分泌,還可以調節細胞焦亡,在脂多糖(lipopolysaccharide,LPS)誘導的ALI/ARDS發生發展中起著重要作用。近年來,骨髓間充質干細胞(bone marrow mesenchymal stem cells,BMSC)在抗炎癥、免疫調節及促進血管新生等方面均展現出良好的臨床應用前景,研究表明BMSC在炎癥刺激下可以大量合成并釋放前列腺素E2(prostaglandin E2,PGE2)減輕炎癥反應[2-4]。本研究旨在探究BMSC對ALI中巨噬細胞的作用,機制是否與調控PGE2與NLRP3炎癥小體的活化有關等,以期為開發新的ALI治療策略提供理論依據。
1 材料與方法
1.1 材料
實驗動物:選擇10只SPF級(6周齡,18~22 g)健康成年雄性BALB/c小鼠。購自常州卡文斯,飼養在同濟大學附屬東方醫院動物房內,環境溫度24~26℃,環境濕度55%~60%,光/暗周期為12 h/12 h,小鼠自由飲水和進食。
實驗材料:LPS(Sigma),10%水合氯醛(上海生工),DMEM高糖/低糖培養基(Hyclone),FBS(Hyclone),DIL-ac-LDL(上海生工),Trizol(Invitrogen),DEPC處理水(CTCC),氯仿/異丙醇/無水乙醇(上海國藥),SYBRGreen PCR試劑盒(Thermo F-415XL),逆轉錄試劑盒(Thermo K1622),CD90抗體(Invitrogen 61-0902-80),CD29抗體 (Invitrogen 12-0291-81),CD45抗體(Invitrogen 67-0451-82),CD34抗體(Invitrogen 11-0341-81),BCA蛋白定量試劑盒(凱基KGPBCA),RIPA強效裂解液(碧云天P0013B),丙烯酰胺/電泳緩沖液/過硫酸銨(上海國藥),蛋白預染Marker(Thermo 26616),NLRP3(ABclonal A5652),pro-Caspase-1(ABclonal A0964),Caspase-1(ABclonal A0964),pro-IL-1β(CST 12242S),GAPDH(proteintech 60004-1-Ig),辣根酶標記山羊抗兔IgG(中杉金橋 ZB2301),辣根酶標記山羊抗小鼠IgG(中杉金橋 ZB2305),腫瘤壞死因子α(tumor necrosis factor-α,TNF-α)(Elabscience E-EL-M0049c),白細胞介素1β(interleukin-1β, IL-1β)(Elabscience E-EL-M0037c),IL-10(Elabscience E-EL-M0046c),IL-18(Elabscience E-EL-M0730c),PGE2(酶聯生物)。
實驗儀器:細胞培養箱(Thermo Scientific 8000),熒光倒置顯微鏡(OLYMPUS,IX71),光學顯微鏡(XDS-1A),離心機(Eppendorf),流式細胞儀(BD-FACSVerse),低溫冷凍離心機 (Sigma 3K15), Real-time檢測儀(ABI-7500),電泳儀(BIORAD),酶標儀(ThermoMK3)等。
1.2 方法
1.2.1 支氣管肺泡灌洗獲取巨噬細胞并檢測純度
取健康SPF級成年雄性BALB/c小鼠,麻醉后暴露氣管并氣管插管,用預冷的磷酸鹽緩沖液(phosphate buffered saline,PBS)進行雙肺灌洗,回收獲得支氣管肺泡灌洗液(bronchoalveolar lavage fluid,BALF)。BALF離心重懸后獲得肺泡巨噬細胞,在巨噬細胞培養液中加入DIL-ac-LDL,避光孵育并拍照。
1.2.2 分離及流式細胞術鑒定BMSC
用頸椎脫位法處死小鼠后,用手術剪打開骨髓腔,沖洗、離心、重懸并培養出BMSC。將分離的BMSC收集培養,每孔分別加入200 μL的染色緩沖液,以及CD90、CD29、CD45和CD34抗體。避光孵育30 min后洗滌重懸。流式細胞術定量檢測并分析。
1.2.3 建立穩定的慢病毒轉染BMSC細胞株
(1)觀察慢病毒MOI:商業合成ptges(Gene ID:64292)合成3條干擾慢病毒,1條過表達慢病毒。將病毒按照MOI為10、50、100感染BMSC,48 h后熒光顯微鏡下觀察病毒感染效率。(2)篩選出最佳敲降慢病毒:細胞內總RNA的抽提采用Trizol提取試劑盒(Invitrogen),采用第一鏈cDNA合成試劑盒(天根),SYBRGreen PCR試劑盒(Thermo F-415XL)以及逆轉錄試劑盒(Thermo K1622)等完成實時聚合酶鏈反應(real-time polymerase chain reaction,RT-PCR)。逆轉錄PCR所使用的引物由上海生工公司Primer Premier 5.0軟件設計、合成。根據RT-PCR反應體系配制反應液。在PCR反應管(20 μL)中分別加入ddH2O 7 μL、SybrGreen qPCR Master Mix 10 μL、Forward primer 1 μL、Reverse primer 1 μL、cDNA模板1 μL,充分混勻。94℃ 10 min,以及94℃ 20 s,55℃ 20s,72℃ 20 s共40個循環。PCR擴增后,以管家基因GAPDH為內參,采用2–△△CT法分析目的基因在對照組和各實驗組之間的表達差異。篩選出最佳效率的敲降慢病毒序列。(3)采用慢病毒轉染ptges及ptges shRNA至BMSC,建立穩定的ptges過表達BMSC細胞株[BMSC-PGE2(+)]及ptges沉默的BMSC細胞株[BMSC-PGE2(–)]。調整細胞數為1×106/mL接種于6孔板培養中,吸取上清,PBS沖洗3次,加入含有慢病毒的無血清培養基3 mL后繼續培養、傳代并繼續下一步實驗。
1.2.4 Western blot檢測巨噬細胞中的NLRP3、pro-Caspase-1、Caspase-1及pro-IL-1β蛋白表達水平
(1)樣品準備:肺泡巨噬細胞以2×106/mL接種在Transwell上室中,隨機分為5組,即對照組、LPS組、LPS+BMSC組、LPS+BMSC-PGE2(+)組、LPS+BMSC-PGE2(–)組。共培養的3組中將BMSC/BMSC-PGE2(+)/BMSC-PGE2(–)以1×106/mL接種在Transwell下室。根據分組將肺泡巨噬細胞與BMSC/BMSC-PGE2(+)/BMSC-PGE2(–)置入Transwell共培養體系,LPS組及共培養體系組予以終濃度為10 ng/mL LPS的無血清培養基共培養5 h,然后以終濃度為5 mmol/L的ATP的無血清培養基共培養1 h。(2)收集細胞,向細胞樣本中加入預冷的已加PMSF的RIPA裂解液,混勻,冰上充分裂解離心后收集蛋白。(3)進行SDS-PAGE凝膠電泳。上樣,電泳,切膠,“三明治”法轉膜,漂洗并封閉。加入NLRP3(1∶1 000)、pro-Caspase-1(1∶1 000)、Caspase-1(1∶1 000)、pro-IL-1β(1∶1 000)及GAPDH(1∶5 000)抗體,4℃孵育,漂洗后加入兔二抗或鼠二抗(1∶5 000)。加入ECL發光液顯色拍照。
1.2.5 RT-PCR檢測巨噬細胞中的NLRP3、Caspase-1的mRNA表達水平
收集各組細胞,加入Trizol試劑,提取細胞中的總RNA。隨后按照第一鏈cDNA合成試劑盒(天根)說明書合成cDNA,按照SYBRGreen PCR試劑盒(Thermo F-415XL)說明書配制RT-PCR反應體系,反應程序為:94℃ 10 min,以及94℃ 20s,55℃ 20 s,72℃ 20 s共40個循環。以管家基因GAPDH為內參,采用2–△△CT法計算NLRP3、Caspase-1的mRNA表達水平。
1.2.6 ELISA法檢測細胞上清液中TNF-α、IL-1β、IL-10、IL-18及PGE2水平
采用TNF-α、IL-1β、IL-10、IL-18及PGE2 ELISA試劑盒說明書。
1.3 統計學方法
采用SPSS19.0統計軟件分析,呈正態分布的計量數據以均數±標準差(x±s)表示,多組比較采用單因素方差分析,兩兩比較采用LSD法,方差不齊時采用非參數檢驗。P<0.05為差異有統計學意義。
2 結果
2.1 成功獲取并培養肺泡巨噬細胞
采用原位支氣管肺泡灌洗法收集的BALF中90%的是肺泡巨噬細胞,紅細胞、中性粒細胞及淋巴細胞等經純化培養被去除。獲得的肺泡巨噬細胞呈圓形,偽足不明顯,貼壁后,肺泡巨噬細胞鋪展開,逐漸伸出偽足(圖1a)。熒光顯微鏡下觀察肺泡巨噬細胞內有大量DIL-ac-LDL,提示巨噬細胞吞噬活性良好(圖1b)。
圖1
小鼠巨噬細胞特征
a. 鏡下特征(×100,×200);b. 熒光顯微鏡下細胞特征(×500)。
2.2 成功提取、培養及鑒定BMSC
BMSC原代細胞在接種24 h后逐漸貼壁,細胞胞體小,大多為梭形,核居中,偶有寬大扁平的多邊形,細胞折光性強。通過多次換液傳代,可使BMSC更為均一,成梭形成纖維樣(圖2a)。利用流式細胞儀對第3代BMSC進行鑒定,其結果顯示細胞CD29、CD90抗體陽性表達率均為99%,CD45、CD34抗體陽性表達率分別為1.39%和3.15%,提示CD29、CD90為陽性表達,CD45、CD34為陰性表達(圖2b)。符合公認的BMSC免疫表型,提示提取BMSC培養并鑒定成功。
圖2
小鼠骨髓間充質細胞特征
a. 小鼠BMSC的鏡下特征(×200);b. 流式細胞儀鑒定結果:CD29陽性,CD90陽性,CD45陰性、CD34陰性。
2.3 確定慢病毒MOI值及感染效率最佳的序列
熒光顯微鏡下觀察可見,沉默慢病毒當MOI值為50時,感染效率已達80%以上,故后續選擇MOI值為50進行實驗(圖3a),而過表達慢病毒選擇MOI為100進行后續實驗(圖3b)。RT-PCR實驗結果顯示,相比對照組,當分別感染沉默慢病毒后,目的基因表達水平均有不同程度的降低,其中慢病毒2效果最好(圖3c),所以后期選擇該靶向序列用于實驗。
圖3
BMSC細胞株轉染慢病毒熒光顯微鏡下像和RT-PCR結果
a. 沉默慢病毒的鏡下特征(×50);b. 過表達慢病毒的鏡下特征(×50);c. RT-PCR顯示shRNA2沉默效率最高。與對照組比較,**
2.4 巨噬細胞中的NLRP3、pro-Caspase-1、Caspase-1、pro-IL-1β蛋白表達水平變化
Western blot結果顯示,LPS組NLRP3、pro-Caspase-1、Caspase-1及pro-IL-1β蛋白表達量較對照組明顯升高,差異有統計學意義(P<0.01);LPS+BMSC組巨噬細胞與BMSC共培養后,NLRP3、pro-Caspase-1、Caspase-1及pro-IL-1β蛋白表達較LPS組明顯減少,差異有統計學意義(P<0.05,P<0.01);LPS+BMSC-PGE2(+)組中巨噬細胞與BMSC-PGE2(+)共培養后,NLRP3、pro-Caspase-1、Caspase-1及pro-IL-1β蛋白表達進一步下降,差異有統計學意義(P<0.01);而LPS+BMSC-PGE2(–)組與LPS組無明顯變化,差異無統計學意義(圖4)。表明LPS刺激后的巨噬細胞中,BMSC通過分泌PGE2,進一步抑制下游NLRP3炎癥小體的活化,減輕炎癥反應。
圖4
NLRP3、pro-Caspase-1、Caspase-1、pro-IL-1β蛋白表達Western blot結果
a. NLRP3的表達水平;b. Caspase-1的表達水平;c. pro-Caspase-1的表達水平;d. pro-IL-1β的表達水平;e. NLRP3、pro-Caspase-1、Caspase-1及pro-IL-1β蛋白表達水平的代表性圖像。與對照組比較,**
2.5 巨噬細胞中的NLRP3、Caspase-1的mRNA表達水平變化
RT-PCR實驗結果顯示,LPS組NLRP3、Caspase-1的mRNA表達量較對照組明顯升高,差異顯著,具有統計學意義(P<0.01);LPS+BMSC組巨噬細胞與BMSC共培養后,NLRP3、Caspase-1的mRNA表達量較LPS組明顯減少,差異有統計學意義(P<0.05);LPS+BMSC-PGE2(+)組中,巨噬細胞與BMSC-PGE2(+)共培養后,NLRP3、Caspase-1的mRNA表達量進一步下降,差異有統計學意義(P<0.01);而LPS+BMSC-PGE2(–)組與LPS組無明顯變化,差異無統計學意義(圖5)。
圖5
各組NLRP3的表達水平(a)以及Caspase-1的表達水平(b)
與對照組比較,**
2.6 細胞上清液中相關炎癥因子的水平變化
ELISA結果顯示,培養液上清中TNF-α、IL-1β及IL-18含量在LPS組中最高,顯著高于對照組、LPS+BMSC組和LPS+BMSC-PGE2(+)組,差異有統計學意義(P<0.05,P<0.01)。培養液上清中IL-10和PGE2含量,LPS組較對照組有升高趨勢,LPS+BMSC組和LPS+BMSC-PGE2(+)組進一步增加,與LPS組比較差異有統計學意義(P<0.05,P<0.01),LPS+BMSC-PGE2(–)組與LPS組沒有顯著差異(圖6)。
圖6
細胞上清液中相關炎癥因子表達的ELISA結果
a. TNF-α的表達水平;b. IL-1β的表達水平;c. PGE2的表達水平;d. IL-10的表達水平;e. IL-18的表達水平。與對照組比較,*
3 討論
ALI和ARDS起病急,病情非常兇險,病死率高,是嚴重威脅人類健康的呼吸系統急危重癥[5]。其本質是由多種炎性介質和效應細胞共同參與,呈瀑布樣級聯放大的炎癥反應,發病過程中涉及炎癥、凋亡、氧化應激等一系列信號通路。肺泡巨噬細胞的激活是ALI/ARDS進程中的始動環節[6],在受到內外致病因素的刺激下,一方面分泌TNF-α、IL-1β、IL-6和巨噬細胞炎性蛋白2(macrophage inflammatory protein 2,MIP-2)等促炎因子,后者募集大量中性粒細胞,啟動和維持肺部炎癥反應,造成肺泡上皮細胞和血管內皮細胞損傷;另一方面,肺泡巨噬細胞還可以分泌IL-4、IL-10等抗炎因子,后者促進免疫細胞吞噬壞死組織細胞和凋亡的中性粒細胞,促進炎癥消散和組織修復。因此,肺泡巨噬細胞介導的促炎和抗炎失衡是ALI/ARDS發生發展的關鍵性因素。
近年來許多研究證實了BMSC可調節肺部炎癥反應,恢復細胞因子網絡之間的平衡,減少促炎因子的分泌,增加抗炎標志物和細胞因子的釋放[7]。在我們的研究中,通過流式細胞儀得到CD29、CD90為陽性表達,而CD45、CD34為陰性表達的較為純化的BMSC,可以用于進一步實驗。在LPS誘導的ALI模型中,肺泡巨噬細胞相關炎性因子TNF-α、IL-1β、IL-18的表達明顯增加,而BMSC能夠明顯逆轉這些促炎因子的表達并增強抗炎因子IL-10 的表達水平。同時,與BMSC相比,過表達PGE2的BMSC可進一步抑制炎癥反應,表明過表達PGE2可增強BMSC對LPS誘導的肺泡巨噬細胞損傷的保護作用。
NLRP3是一種模式識別受體(pattern-recognition receptors,PRRs)[8],主要在中性粒細胞和巨噬細胞的細胞質中表達,其組成部分包括先天免疫受體蛋白NLRP3、炎性蛋白酶Caspase-1與銜接蛋白ASC。作為機體固有免疫的重要組成部分[9-10],NLRP3蛋白可被病原體相關分子模式(pathogen-associated molecular patterns,PAMPs)識別并激活,如NLRP3炎癥小體可以被細菌LPS、活性氧、K+外流、Ca2+信號、毒素、顆粒物質、病毒RNA和溶酶體破壞等。NLRP3炎癥小體激活機制有三種不同信號通路:經典NLRP3炎性體激活、非經典NLRP3炎性體激活和替代NLRP3 炎性體激活(也稱為一步NLRP3 炎性體激活)[11-12]。NLRP3炎性體的激活將誘導pro-Caspase-1的自我切割和激活,從而促進包括IL-1β和IL-18在內的多種促炎性細胞因子的剪切和分泌[13],另一方面活化的Caspase-1還將切割消皮素D(gasdermin D,GSDMD)并釋放其N端結構域,這一結構域將轉移并在細胞膜處形成孔,從而釋放細胞內容物,進而調節程序性細胞焦亡(pyroptosis),誘導細胞在炎性和應激的病理條件下死亡[3,14-17]。研究表明在小鼠ALI模型中,NLRP3炎癥小體可被激活,而抑制NLRP3炎癥小體可以顯著抑制TNF-α、IL-1β的合成和分泌,改善膿毒癥所致的肺泡上皮細胞通透性的增加[18]。因此,NLRP3炎癥小體的活化在ALI的發生發展中起到重要作用。
其中,包括PGE2在內的許多因子都與NLRP3炎癥小體通路激活密切相關[4,19]。PGE合酶(PGE synthase,PTGES)作為PGE2合成中的終末限速酶,參與PGE2的合成[20]。PGE2通過前列腺素E受體(prostaglandin E receptor,EP)家族的4種不同受體發出信號:EP1、EP2、EP3和EP4[21]。研究發現NLRP3炎癥體的快速激活被蛋白激酶A(protein kinase A,PKA)直接抑制,PKA直接磷酸化胞漿受體NLRP3,減弱其ATP酶功能。而PKA是由PGE2通過PGE2受體EP4信號傳導誘導的[1]。還有研究發現PGE2通過促進抗炎因子IL-10的分泌,能夠抑制中性粒細胞的黏附和內皮細胞激活,對NLRP3炎性小體有拮抗作用[22-23]。阻斷巨噬細胞IL-10受體可促進NLRP3炎性體的激活[24],NLRP3炎癥小體的活化也可以抑制單核細胞中IL-10的分泌[25]。IL-10基因敲除的小鼠在LPS刺激后,NLRP3、pro-Caspase-1和pro-IL-1β水平均明顯高于野生型[26],因此通過IL-10來抑制NLRP3炎癥小體的活化可能是PGE2的作用機制之一。本研究中,BMSC可明顯抑制NLRP3、pro-Caspase-1、Caspase-1及pro-IL-1β的表達,降低TNF-α、IL-1β表達水平,增加IL-10的表達水平,當給予過表達PGE2的BMSC后,PGE2可抑制NLRP3活化,與BMSC相比,對ALI的抑制作用進一步增強,從而發揮對ALI的治療作用。
綜上所述,研究結果從細胞層面表明,在LPS誘導的ALI發病過程中,BMSC通過攜帶并分泌PGE2來促進肺泡巨噬細胞中抗炎因子的釋放,從而抑制肺泡巨噬細胞中NLRP3炎癥小體的活化,減輕肺部炎性損害。本研究為運用BMSC來調控ALI時的炎癥反應提供理論基礎,為臨床救治ALI患者提供新的治療策略和重要的理論依據。
利益沖突:本研究不涉及任何利益沖突。