引用本文: 劉翔, 彭聰, 王子欣, 孫翔. 腸道菌群與泌尿道感染的關系:雙樣本孟德爾隨機化研究. 中國循證醫學雜志, 2025, 25(1): 50-56. doi: 10.7507/1672-2531.202403179 復制
版權信息: ?四川大學華西醫院華西期刊社《中國循證醫學雜志》版權所有,未經授權不得轉載、改編
泌尿道感染(UTI)是腎臟、輸尿管、膀胱、尿道等泌尿系統各個部位感染的總稱,是尿路上皮對細菌等病原體侵入的炎癥反應,UTI的癥狀可以從輕微的尿頻和尿急到嚴重的腹痛、發熱和尿液混濁,根據感染的部位和嚴重程度而異[1]。全球范圍內,UTI廣泛分布,每年有約1.3億至1.75億人患病,位居第二大感染性疾病。UTI的復發率極高,大約27%的患者在6個月內會再次發生尿路感染,而3%的患者在同一期間內可能會經歷超過3次感染[2]。這些情況導致了大量抗菌藥物的使用和醫療開支增加,因此需要更多的研究來深入了解UTI的發生和發展,以減輕相關疾病負擔。尿道感染可由來源于陰道菌群和會陰部皮膚的表皮葡萄球菌和白念珠菌等引起,但大多數尿路感染是由來源于腸道菌群的兼性厭氧菌菌群引起的,所以本質上是一種內源性感染[3]。腸道微生物組,是人體腸道內的微生物生態系統,是人體內最多樣化的微生物群落之一,這些微生物與宿主的互動對于多個方面的生理和病理過程至關重要[4]。近年來,許多研究表明腸道微生物組生態失調與尿路感染之間存在著各種各樣的聯系[5,6]。但腸道微生物群與UTI之間的關聯是否為因果關系以及因果關系的方向仍然未知。
本研究基于全基因組關聯研究(GWAS)的數據,采用雙樣本孟德爾隨機化(MR)方法[7],探討腸道菌群與UTI之間的潛在因果關系,并確定特定的致病菌分類群。同時進行反向MR分析檢查UTI風險的遺傳易感性是否會影響腸道微生物群。試圖闡明腸道微生物群在UTI發生發展中的作用,以最終幫助開發新的UTI治療策略。
1 資料與方法
1.1 暴露數據
使用來自國際聯盟MiBioGen的GWAS數據集中與人腸道微生物組組成相關的單核苷酸多態性(SNP)作為工具變量(IV)[8]。MiBioGen的GWAS數據集來自一項多種族大規模GWAS,協調了來自美國、加拿大、以色列、韓國、德國、丹麥、荷蘭、比利時、瑞典、芬蘭和英國的24個隊列的18 340名參與者的16S核糖體RNA基因測序譜和基因分型數據,以探索常染色體人類遺傳變異與腸道微生物組之間的關聯。共包括9門16綱20目35科131屬211個分類群。在本研究中,最低的分類學分類是在屬的水平[9]。研究結果表明,131個屬被鑒定為平均豐度超過1%,其中12個屬無法鑒定[10]。因此,本次分析限于可鑒定的119個細菌屬。
1.2 結局數據
UTI的遺傳匯總統計量來自UK Biobank(數據集:ieu-b-
1.3 IV的選擇
為了滿足MR的3個關鍵假設[11]:① 相關性假設:基因工具與感興趣的暴露顯著相關;② 獨立性假設:遺傳工具與暴露-結果關聯的任何混雜因素均不相關;③ 排除假設:遺傳工具僅通過暴露影響結果[12],確保關于腸道菌群對UTI的因果影響結果的準確性,實施了一系列質量控制程序以選擇與腸道微生物組特征相關的遺傳預測因子。首先,選擇用于分析的IV必須證明與暴露因子顯著相關。在本研究中,為獲取一定數量的SNP以供分析,將基因座范圍顯著性閾值定為P值低于1×10?5[13,14],并以0.01的閾值排除次要等位基因頻率(MAF)。第二,由于強連鎖不平衡可能導致結果偏倚,為確保所有IV的獨立性,我們以r2<0.000 1為閾值,以10 000 kb為聚集窗口進行連鎖不平衡分析,這種方法最小化了連鎖不平衡的影響,避免了違反隨機化等位基因分配。第三,MR風險分析中的重要步驟是確保SNP對暴露的影響對應于與對結果的影響相同的等位基因。為了避免鏈取向或等位基因編碼的失真,在協調暴露和結果的影響后,我們去除了模糊和重復的SNP。并采用SNP的F統計量來確定IV和暴露因素之間聯系的強度和穩定性,只有F>10的SNP才會被用于分析[15]。F=R2/(1?R2)×(N?K?1)/K。其中N是樣本量,K是有效SNP的數量,R2=2×MAF×(1?MAF)×β2/[2×MAF×(1?MAF)×β2+2×MAF×(1?MAF)×N×SE2][16]。MAF通常為0至0.5之間的值。β代表回歸系數,表示遺傳變異和表型特征之間的關聯強度。SE代表標準誤差。它們通常用于衡量回歸系數估計的準確性。
1.4 MR分析
使用6種方法來研究腸道微生物群對UTI的因果關系,包括逆方差加權法(IVW)[17]、MR-Egger[18]、加權中位數法[19]、加權模型法[20]、簡單模型法[21]和最大似然法(ML)[22]。在IVW模型中,所有IV都被假定為有效IV,然后使用元分析技術進行組合,該方法被用作主要分析[19]。當存在異質性時,采用隨機效應的IVW模型;否則,將固定效應的IVW估計作為主要分析。MR-Egger和加權中位數方法在即使存在多效性的情況下也能產生較為穩健的結果。此外,為了減少因有限樣本而引起的偏倚,提高結果的準確性和可信度,還使用了ML方法,ML方法通過最大化給定數據條件下參數的似然函數來估計因果效應。
敏感性分析方面,采用Cochran’s Q檢驗評價IV的異質性,P<0.05的Q統計表明存在異質性[17]。使用MR-Egger回歸,我們可以通過判斷截距是否顯著偏離零點來評估水平多效性的存在。同時采用留一法,系統地每次消除一個納入的SNP,并重新計算其余IV所產生的結果,以檢驗是否有單個離群SNP導致結果產生偏倚[23]。而MR-PRESSO方法可以通過比較IVW模型和MR-Egger模型的殘差來檢測是否存在多效性的同時可以識別并排除可能由于單個SNP導致的異常值,這些異常值可能會扭曲因果估計[24]。
此外,為了驗證不存在多效性和反向因果關系,使用UTI作為暴露,腸道微生物群作為結果進行反向MR分析,并使用MR Steiger方向性檢驗[25]來檢查暴露是否與結局有方向性因果關系。
所有統計分析均使用R 4.3.1版本進行。使用“Two Sample MR”“MR-PRESSO”和“Mendelian Randomization”函數包進行數據分析。使用“ggplot 2”函數包繪制圖。
2 結果
經過IVW方法分析后,三種腸道微生物細菌屬:Eggerthella、Ruminococcaceae(UCG005)和Defluviitaleaceae(UCG011)與UTI之間存在關聯。IVW分析確定了細菌屬Eggerthella和Ruminococcaceae(UCG005)豐度增加與UTI風險升高有關。相反,細菌屬Defluviitaleaceae(UCG011)對UTI有保護作用。其中在對Eggerthella菌屬的所有MR分析中觀察到了一致的方向:IVW[OR=1.08,95%CI(1.01,1.16),P=0.03]、ML[OR=1.08,95%CI(1.00,1.16),P=0.04]、MR-Egger[OR=1.22,95%CI(0.89,1.67),P=0.26]、簡單模型法[OR=1.07,95%CI(0.93,1.23),P=0.39]、加權模型法[OR=1.06,95%CI(0.93,1.23),P=0.42]、加權中位數法[OR=1.07,95%CI(0.98,1.17),P=0.15]。對Defluviitaleaceae(UCG011)菌屬的所有MR分析中同樣觀察到了一致的方向:IVW[OR=0.90,95%CI(0.82,0.99),P=0.02]、ML[OR=0.90,95%CI(0.82,0.99),P=0.03]、MR-Egger[OR=0.79,95%CI(0.59,1.07),P=0.19]、簡單模型法[OR=0.89,95%CI(0.74,1.06),P=0.24]、加權模型法[OR=0.89,95%CI(0.75,1.06),P=0.22]、加權中位數法[OR=0.90,95%CI(0.80,1.01),P=0.08]。對Ruminococcaceae(UCG005)菌屬的MR分析中:IVW[OR=1.10,95%CI(1.01,1.20),P=0.02]、ML[OR=1.11,95%CI(1.02,1.21),P=0.02]、MR-Egger[OR=0.98,95%CI(0.77,1.24),P=0.87]、簡單模型法[OR=1.03,95%CI(0.86,1.22),P=0.64]、加權模型法[OR=1.02,95%CI(0.88,1.19),P=0.71]、加權中位數法[OR=1.04,95%CI(0.93,1.17),P=0.41]。
敏感性分析中,Cochran’s Q檢驗證明了IV間的均勻性,未檢測到明顯異質性(P>0.05)。MR-Egger(P-intecept>0.05)和MR-PRESSO(P-global test>0.05)分析進一步驗證了這一點,這在很大程度上排除了這些IV中的水平多效性效應。MR-PRESSO分析沒有將任何SNP鑒定為離群值,并且在仔細檢查數據圖(圖1和圖2)時,沒有發現明顯的偏差或異常。MR Steiger方向性測試進一步加強了所有評價結果之間的穩健連接。詳細結果見表1。
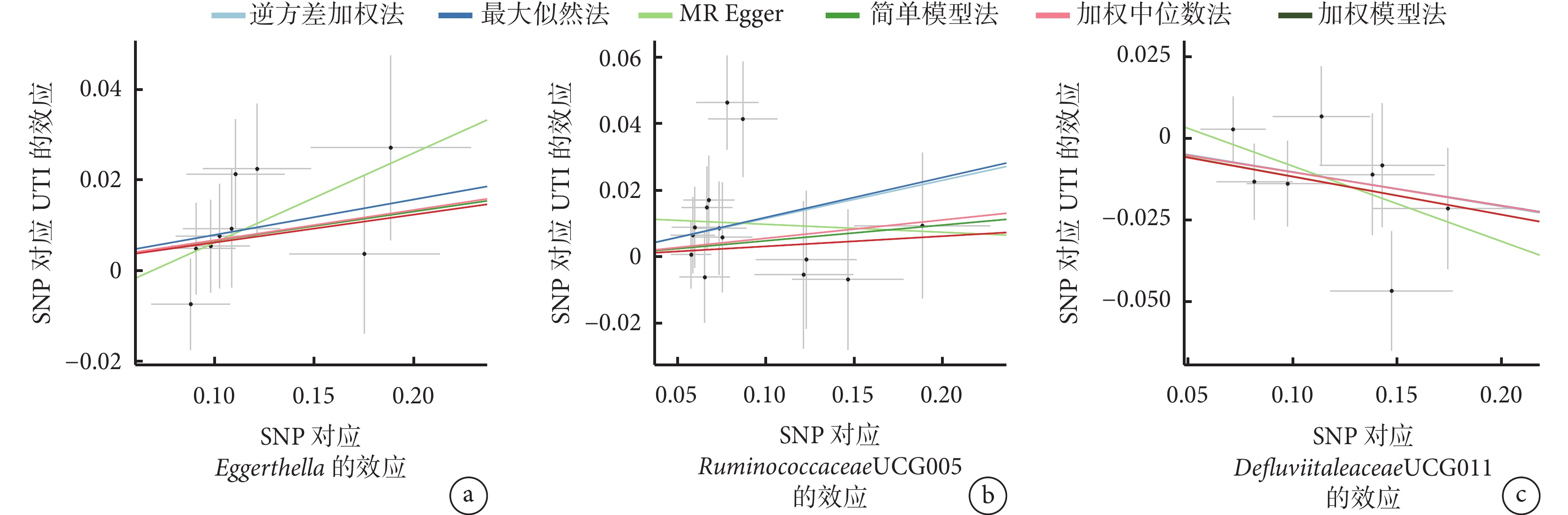
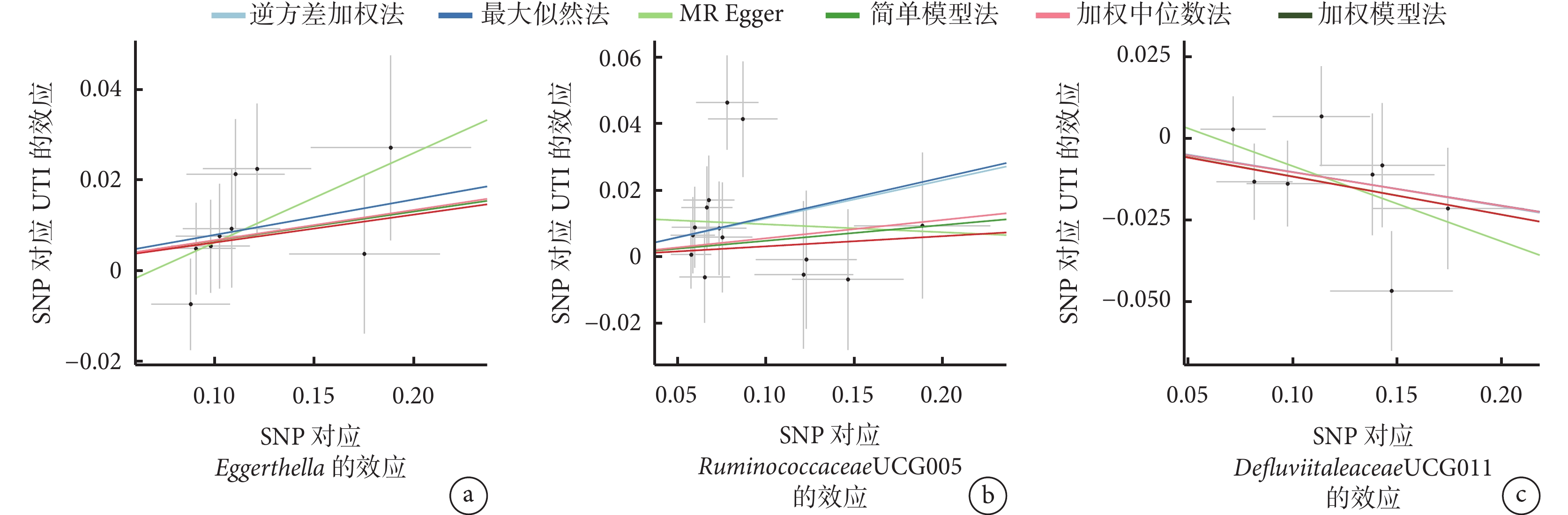 圖1
腸道菌群和泌尿道感染之間因果關系的散點圖
圖1
腸道菌群和泌尿道感染之間因果關系的散點圖
該圖每個SNP對腸道菌群和UTI風險的效應大小和95%可信區間的散點圖。橫軸反映每個SNP對腸道菌群的遺傳效應。縱軸代表每個SNP對UTI風險的遺傳效應。MR:孟德爾隨機化;UTI:泌尿道感染;SNP:單核苷酸多態性。
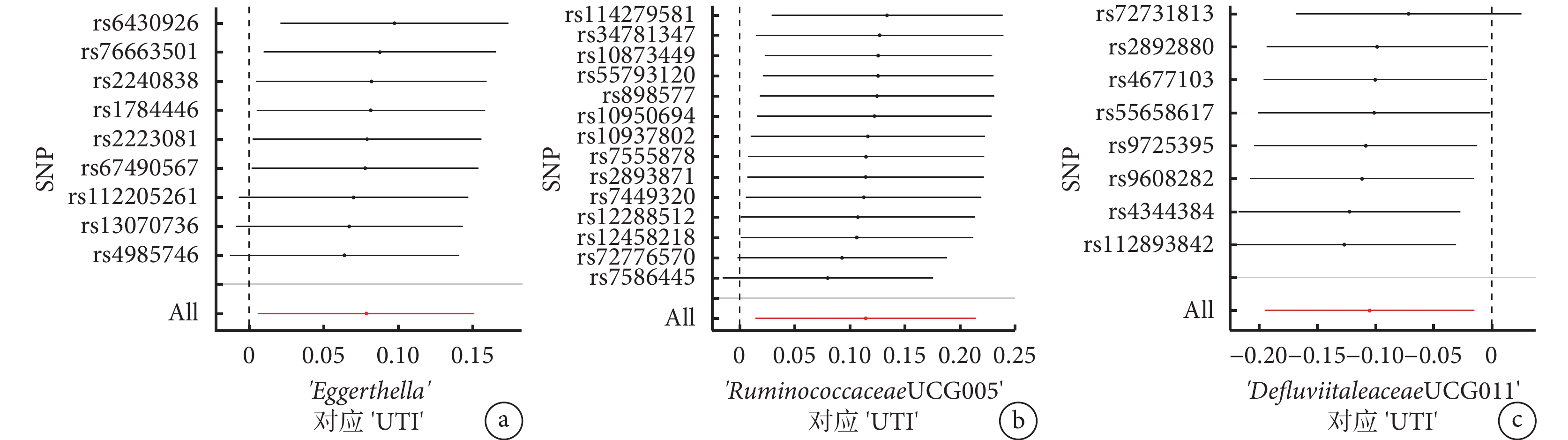
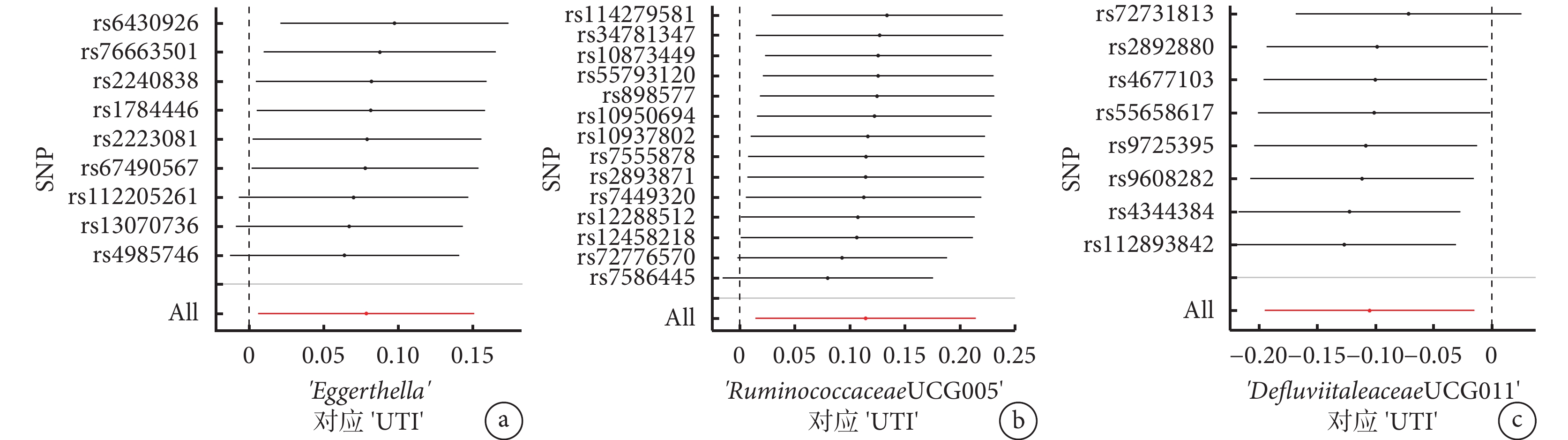 圖2
單個SNP對腸道菌群和泌尿道感染風險之間關聯的影響的留一法分析
圖2
單個SNP對腸道菌群和泌尿道感染風險之間關聯的影響的留一法分析
MR:孟德爾隨機化;SNP:單核苷酸多態性;UTI:泌尿道感染。
反向MR分析評估了三種細菌性狀與UTI的潛在反向關聯。篩選SNP時,我們嘗試使用傳統的P<5×10?8作為篩選門檻,但未篩選到任何SNP,選用P<5×10?7作為篩選門檻時,經和諧后獲取到2個SNP以進行MR分析,SNP數量過少可能導致IV偏倚,同時導致部分敏感性分析無法進行,故我們最終選擇P<5×10?6作為篩選門檻,經和諧匹配后獲取到了8個SNP,使用IVW方法進行分析后,我們沒有發現UTI與這三種腸道細菌屬中的任何一種之間存在統計學關聯。敏感性分析結果均顯示不存在顯著異質性或水平多效性。
3 討論
本研究使用了基于超過50萬個歐洲個體的綜合遺傳數據,并從中鑒定出了強相關的基因變異,在基因預測水平上對腸道菌群和UTI之間的因果關系進行了初步的廣泛的MR研究,我們發現遺傳預測的腸道細菌屬Eggerthella和Ruminococcaceae(UCG005)與患UTI的風險正相關,而腸道細菌屬Defluviitaleaceae(UCG011)與患UTI的風險呈負相關。反向MR分析沒有觀察到任何證據表明UTI對腸道各菌群豐度有因果作用。
腸道微生物群被認為是尿路致病性腸道菌的來源,腸道菌群可定植于尿道周圍空間并上升至膀胱,導致UTI。越來越多的研究發現,本研究中選擇的腸道菌群與UTI之間可能存在其他聯系。如Paalanne等[5]進行的一項比較37名急性UTI住院兒童與69名健康對照兒童的腸道微生物組的病例-對照研究中,發現腸桿菌屬在UTI兒童腸道微生物組中的富集,以及消化鏈球菌科大芬戈爾德菌和梭菌屬在健康受試者中的富集,后二者均為厭氧菌,揭示了較高的厭氧菌相對豐度可能是UTI的保護因素。Magruder等[6]使用V4~V5高變區的16S rRNA基因測序對168名腎移植受者的510份糞便標本進行了腸道微生物組分析,結果顯示無腸桿菌科菌尿組糞便標本中糞桿菌屬和羅氏菌屬的相對豐度高于腸桿菌科菌尿組(P<0.01)。糞桿菌和羅氏菌屬的組合相對豐度與腸桿菌科的相對豐度呈負相關(r=?0.13,P=0.003)。表明糞桿菌和羅氏菌屬的相對豐度高與腎移植受者腸桿菌科菌尿和UTI發生的風險降低相關。遺憾的是,本研究沒有發現上述研究中發現的UTI患者與正常患者腸道中豐度存在統計學差異的菌種在遺傳學上與UTI存在明確的因果關系,這有可能是既往研究中個體差異所導致,也可能是本研究所使用的GWAS數據樣本量不足。
Eggerthella菌屬為伊格爾茲氏菌科中的一個屬,既往的一些研究發現放線菌屬的Eggerthellalenta在某些疾病或暴露個體的腸道菌群中顯著富集[26-28],同時是導致多種感染的致病因素[29,30]。有研究發現人類腸道放線菌屬的Eggerthella lenta可以通過解除對Th17轉錄因子Ror γ t的抑制作用,進而誘導了腸道Th17的激活,從而促進腸道炎癥[31]。腸道細菌導致泌尿系統炎癥的過程中是否存在同樣的機制目前不得而知,需要進一步的探索。Ruminococcaceae(UCG005)是瘤胃球菌科中的一個細菌屬,瘤胃球菌是最早發現的胃部細菌之一,在新陳代謝中起著至關重要的作用。瘤胃球菌通過分解宿主消化系統的纖維素來獲取營養。也能夠發酵葡萄糖和木糖。但許多研究也揭示了其在炎癥發生發展過程中的作用,例如一項關于瘤胃球菌炎癥特性的研究發現,它以葡甘露聚糖多糖的形式產生代謝物。瘤胃球菌多糖可以激發免疫系統細胞,如腫瘤壞死因子TNFα[32]。而TNFα是克羅恩病的炎癥生物標志物之一。然而,關于瘤胃球菌對UTI影響的研究有限。Defluviitaleaceae(UCG011)是本研究中發現的唯一對UTI易感性具有保護作用的一個細菌屬,Tong等[33]發現與健康者相比,類風濕關節炎高風險個體表現出口腔微生物群中Defluviitaleaceae(UCG011)屬豐度的降低。顯示出了Defluviitaleaceae(UCG011)在炎癥發生過程中的某種保護作用。然而,目前少有研究探討Defluviitaleaceae(UCG011)和UTI之間的潛在關系。
腸道微生物群通過影響多種生理過程影響宿主健康,包括器官發育、免疫力和疾病易感性[4]。腸道生態失調可以改變腸道遠端器官的信號傳導,包括大腦(定義為腸-腦軸)[34]和肺(定義為腸-肺軸)[35]。而Worby等[36]研究證明,腸道微生物群的變化可以增強對復發性UTI的易感性,從而定義了腸道-膀胱軸。Magruder等[37]的研究發現發生UTI的腎移植患者糞便中的大腸埃希氏菌和腸球菌的腸道豐度分別與大腸埃希氏菌和腸球菌菌尿癥的未來發展相關,以及腸道大腸埃希氏菌的豐度與癥狀性大腸埃希氏菌UTI的發展有關。該研究結果支持了腸道微生物群-泌尿道感染軸,表明調節腸道微生物群可能是預防UTI的潛在新策略。
使用廣譜抗生素是治療UTI最主要的方法,但不合理的臨床抗生素使用正在加劇細菌耐藥性問題。同時,腸-膀胱軸突出了現有抗生素治療的局限性。抗生素治療不僅是難治性復發UTI的已知風險因素[38,39],還可能導致顯著和持久地擾動腸道微生物組[36]。如果UTI的抗生素治療確實產生或維持腸道生態失調的狀態,那么這可能導致治療的惡性循環,并增加了再次感染的風險及對未來感染的易感性[40]。因此,為降低尿路感染復發的風險,并盡量減少抗微生物藥物耐藥性,開發新的預防及治療UTI的方法顯得尤為重要。
既往研究已證實糞便微生物群移植在治療某些病原體導致的難治性復發UTI中的可行性[41,42],Tariq等[43]的糞菌微生物移植實驗表明腸道共生細菌可以通過競爭營養物質、改變腸道pH值、形成抗菌代謝物和阻止多重耐藥菌(MDROs)進入黏膜壁上的結合位點來減少MDROs的泌尿道定植,從而提高UTI患者的抗生素敏感性。顯示出了糞菌移植的極大潛力,但仍需要進一步探索更佳的實驗方案及潛在的病理機制。
在觀察性研究中已經報道了腸道菌群和UTI之間的關聯。然而,由于方法上的限制,確定因果關系可能具有挑戰性。本MR研究的一個關鍵優勢是我們的研究結果避免了反向因果關系,并最大限度地減少了殘余混雜因素,同時避免了既往觀察性研究中的各種偏倚。本MR分析結果可以為選擇個體腸道細菌以研究腸道微生物群在UTI發病機制中的作用提供指導。然而,本研究也存在局限性:① 盡管我們采用了迄今為止最大的腸道微生物組數據集,但樣本量仍需要擴大,這可能會進一步增強統計能力。另外數據只測量到屬水平,限制了在種水平上進一步探討腸道菌群與UTI之間的聯系。② 由于使用了摘要級數據,腸道細菌豐度和UTI風險之間的非線性關聯無法探索。③ 研究數據均采集于歐洲個體,由于種族群體之間固有的遺傳差異,本結果外推到其他種族時應當謹慎。
綜上所述,本項雙樣本MR研究發現遺傳水平上腸道細菌屬Eggerthella、Ruminococcaceae(UCG005)和Defluviitaleaceae(UCG011)對UTI風險具有潛在的因果影響。本研究可為腸道微生物群介導的UTI發生發展機制提供新的見解,但仍需更多的研究來探究腸道細菌群與UTI之間的生物學聯系。
泌尿道感染(UTI)是腎臟、輸尿管、膀胱、尿道等泌尿系統各個部位感染的總稱,是尿路上皮對細菌等病原體侵入的炎癥反應,UTI的癥狀可以從輕微的尿頻和尿急到嚴重的腹痛、發熱和尿液混濁,根據感染的部位和嚴重程度而異[1]。全球范圍內,UTI廣泛分布,每年有約1.3億至1.75億人患病,位居第二大感染性疾病。UTI的復發率極高,大約27%的患者在6個月內會再次發生尿路感染,而3%的患者在同一期間內可能會經歷超過3次感染[2]。這些情況導致了大量抗菌藥物的使用和醫療開支增加,因此需要更多的研究來深入了解UTI的發生和發展,以減輕相關疾病負擔。尿道感染可由來源于陰道菌群和會陰部皮膚的表皮葡萄球菌和白念珠菌等引起,但大多數尿路感染是由來源于腸道菌群的兼性厭氧菌菌群引起的,所以本質上是一種內源性感染[3]。腸道微生物組,是人體腸道內的微生物生態系統,是人體內最多樣化的微生物群落之一,這些微生物與宿主的互動對于多個方面的生理和病理過程至關重要[4]。近年來,許多研究表明腸道微生物組生態失調與尿路感染之間存在著各種各樣的聯系[5,6]。但腸道微生物群與UTI之間的關聯是否為因果關系以及因果關系的方向仍然未知。
本研究基于全基因組關聯研究(GWAS)的數據,采用雙樣本孟德爾隨機化(MR)方法[7],探討腸道菌群與UTI之間的潛在因果關系,并確定特定的致病菌分類群。同時進行反向MR分析檢查UTI風險的遺傳易感性是否會影響腸道微生物群。試圖闡明腸道微生物群在UTI發生發展中的作用,以最終幫助開發新的UTI治療策略。
1 資料與方法
1.1 暴露數據
使用來自國際聯盟MiBioGen的GWAS數據集中與人腸道微生物組組成相關的單核苷酸多態性(SNP)作為工具變量(IV)[8]。MiBioGen的GWAS數據集來自一項多種族大規模GWAS,協調了來自美國、加拿大、以色列、韓國、德國、丹麥、荷蘭、比利時、瑞典、芬蘭和英國的24個隊列的18 340名參與者的16S核糖體RNA基因測序譜和基因分型數據,以探索常染色體人類遺傳變異與腸道微生物組之間的關聯。共包括9門16綱20目35科131屬211個分類群。在本研究中,最低的分類學分類是在屬的水平[9]。研究結果表明,131個屬被鑒定為平均豐度超過1%,其中12個屬無法鑒定[10]。因此,本次分析限于可鑒定的119個細菌屬。
1.2 結局數據
UTI的遺傳匯總統計量來自UK Biobank(數據集:ieu-b-
1.3 IV的選擇
為了滿足MR的3個關鍵假設[11]:① 相關性假設:基因工具與感興趣的暴露顯著相關;② 獨立性假設:遺傳工具與暴露-結果關聯的任何混雜因素均不相關;③ 排除假設:遺傳工具僅通過暴露影響結果[12],確保關于腸道菌群對UTI的因果影響結果的準確性,實施了一系列質量控制程序以選擇與腸道微生物組特征相關的遺傳預測因子。首先,選擇用于分析的IV必須證明與暴露因子顯著相關。在本研究中,為獲取一定數量的SNP以供分析,將基因座范圍顯著性閾值定為P值低于1×10?5[13,14],并以0.01的閾值排除次要等位基因頻率(MAF)。第二,由于強連鎖不平衡可能導致結果偏倚,為確保所有IV的獨立性,我們以r2<0.000 1為閾值,以10 000 kb為聚集窗口進行連鎖不平衡分析,這種方法最小化了連鎖不平衡的影響,避免了違反隨機化等位基因分配。第三,MR風險分析中的重要步驟是確保SNP對暴露的影響對應于與對結果的影響相同的等位基因。為了避免鏈取向或等位基因編碼的失真,在協調暴露和結果的影響后,我們去除了模糊和重復的SNP。并采用SNP的F統計量來確定IV和暴露因素之間聯系的強度和穩定性,只有F>10的SNP才會被用于分析[15]。F=R2/(1?R2)×(N?K?1)/K。其中N是樣本量,K是有效SNP的數量,R2=2×MAF×(1?MAF)×β2/[2×MAF×(1?MAF)×β2+2×MAF×(1?MAF)×N×SE2][16]。MAF通常為0至0.5之間的值。β代表回歸系數,表示遺傳變異和表型特征之間的關聯強度。SE代表標準誤差。它們通常用于衡量回歸系數估計的準確性。
1.4 MR分析
使用6種方法來研究腸道微生物群對UTI的因果關系,包括逆方差加權法(IVW)[17]、MR-Egger[18]、加權中位數法[19]、加權模型法[20]、簡單模型法[21]和最大似然法(ML)[22]。在IVW模型中,所有IV都被假定為有效IV,然后使用元分析技術進行組合,該方法被用作主要分析[19]。當存在異質性時,采用隨機效應的IVW模型;否則,將固定效應的IVW估計作為主要分析。MR-Egger和加權中位數方法在即使存在多效性的情況下也能產生較為穩健的結果。此外,為了減少因有限樣本而引起的偏倚,提高結果的準確性和可信度,還使用了ML方法,ML方法通過最大化給定數據條件下參數的似然函數來估計因果效應。
敏感性分析方面,采用Cochran’s Q檢驗評價IV的異質性,P<0.05的Q統計表明存在異質性[17]。使用MR-Egger回歸,我們可以通過判斷截距是否顯著偏離零點來評估水平多效性的存在。同時采用留一法,系統地每次消除一個納入的SNP,并重新計算其余IV所產生的結果,以檢驗是否有單個離群SNP導致結果產生偏倚[23]。而MR-PRESSO方法可以通過比較IVW模型和MR-Egger模型的殘差來檢測是否存在多效性的同時可以識別并排除可能由于單個SNP導致的異常值,這些異常值可能會扭曲因果估計[24]。
此外,為了驗證不存在多效性和反向因果關系,使用UTI作為暴露,腸道微生物群作為結果進行反向MR分析,并使用MR Steiger方向性檢驗[25]來檢查暴露是否與結局有方向性因果關系。
所有統計分析均使用R 4.3.1版本進行。使用“Two Sample MR”“MR-PRESSO”和“Mendelian Randomization”函數包進行數據分析。使用“ggplot 2”函數包繪制圖。
2 結果
經過IVW方法分析后,三種腸道微生物細菌屬:Eggerthella、Ruminococcaceae(UCG005)和Defluviitaleaceae(UCG011)與UTI之間存在關聯。IVW分析確定了細菌屬Eggerthella和Ruminococcaceae(UCG005)豐度增加與UTI風險升高有關。相反,細菌屬Defluviitaleaceae(UCG011)對UTI有保護作用。其中在對Eggerthella菌屬的所有MR分析中觀察到了一致的方向:IVW[OR=1.08,95%CI(1.01,1.16),P=0.03]、ML[OR=1.08,95%CI(1.00,1.16),P=0.04]、MR-Egger[OR=1.22,95%CI(0.89,1.67),P=0.26]、簡單模型法[OR=1.07,95%CI(0.93,1.23),P=0.39]、加權模型法[OR=1.06,95%CI(0.93,1.23),P=0.42]、加權中位數法[OR=1.07,95%CI(0.98,1.17),P=0.15]。對Defluviitaleaceae(UCG011)菌屬的所有MR分析中同樣觀察到了一致的方向:IVW[OR=0.90,95%CI(0.82,0.99),P=0.02]、ML[OR=0.90,95%CI(0.82,0.99),P=0.03]、MR-Egger[OR=0.79,95%CI(0.59,1.07),P=0.19]、簡單模型法[OR=0.89,95%CI(0.74,1.06),P=0.24]、加權模型法[OR=0.89,95%CI(0.75,1.06),P=0.22]、加權中位數法[OR=0.90,95%CI(0.80,1.01),P=0.08]。對Ruminococcaceae(UCG005)菌屬的MR分析中:IVW[OR=1.10,95%CI(1.01,1.20),P=0.02]、ML[OR=1.11,95%CI(1.02,1.21),P=0.02]、MR-Egger[OR=0.98,95%CI(0.77,1.24),P=0.87]、簡單模型法[OR=1.03,95%CI(0.86,1.22),P=0.64]、加權模型法[OR=1.02,95%CI(0.88,1.19),P=0.71]、加權中位數法[OR=1.04,95%CI(0.93,1.17),P=0.41]。
敏感性分析中,Cochran’s Q檢驗證明了IV間的均勻性,未檢測到明顯異質性(P>0.05)。MR-Egger(P-intecept>0.05)和MR-PRESSO(P-global test>0.05)分析進一步驗證了這一點,這在很大程度上排除了這些IV中的水平多效性效應。MR-PRESSO分析沒有將任何SNP鑒定為離群值,并且在仔細檢查數據圖(圖1和圖2)時,沒有發現明顯的偏差或異常。MR Steiger方向性測試進一步加強了所有評價結果之間的穩健連接。詳細結果見表1。
圖1
腸道菌群和泌尿道感染之間因果關系的散點圖
該圖每個SNP對腸道菌群和UTI風險的效應大小和95%可信區間的散點圖。橫軸反映每個SNP對腸道菌群的遺傳效應。縱軸代表每個SNP對UTI風險的遺傳效應。MR:孟德爾隨機化;UTI:泌尿道感染;SNP:單核苷酸多態性。
圖2
單個SNP對腸道菌群和泌尿道感染風險之間關聯的影響的留一法分析
MR:孟德爾隨機化;SNP:單核苷酸多態性;UTI:泌尿道感染。
反向MR分析評估了三種細菌性狀與UTI的潛在反向關聯。篩選SNP時,我們嘗試使用傳統的P<5×10?8作為篩選門檻,但未篩選到任何SNP,選用P<5×10?7作為篩選門檻時,經和諧后獲取到2個SNP以進行MR分析,SNP數量過少可能導致IV偏倚,同時導致部分敏感性分析無法進行,故我們最終選擇P<5×10?6作為篩選門檻,經和諧匹配后獲取到了8個SNP,使用IVW方法進行分析后,我們沒有發現UTI與這三種腸道細菌屬中的任何一種之間存在統計學關聯。敏感性分析結果均顯示不存在顯著異質性或水平多效性。
3 討論
本研究使用了基于超過50萬個歐洲個體的綜合遺傳數據,并從中鑒定出了強相關的基因變異,在基因預測水平上對腸道菌群和UTI之間的因果關系進行了初步的廣泛的MR研究,我們發現遺傳預測的腸道細菌屬Eggerthella和Ruminococcaceae(UCG005)與患UTI的風險正相關,而腸道細菌屬Defluviitaleaceae(UCG011)與患UTI的風險呈負相關。反向MR分析沒有觀察到任何證據表明UTI對腸道各菌群豐度有因果作用。
腸道微生物群被認為是尿路致病性腸道菌的來源,腸道菌群可定植于尿道周圍空間并上升至膀胱,導致UTI。越來越多的研究發現,本研究中選擇的腸道菌群與UTI之間可能存在其他聯系。如Paalanne等[5]進行的一項比較37名急性UTI住院兒童與69名健康對照兒童的腸道微生物組的病例-對照研究中,發現腸桿菌屬在UTI兒童腸道微生物組中的富集,以及消化鏈球菌科大芬戈爾德菌和梭菌屬在健康受試者中的富集,后二者均為厭氧菌,揭示了較高的厭氧菌相對豐度可能是UTI的保護因素。Magruder等[6]使用V4~V5高變區的16S rRNA基因測序對168名腎移植受者的510份糞便標本進行了腸道微生物組分析,結果顯示無腸桿菌科菌尿組糞便標本中糞桿菌屬和羅氏菌屬的相對豐度高于腸桿菌科菌尿組(P<0.01)。糞桿菌和羅氏菌屬的組合相對豐度與腸桿菌科的相對豐度呈負相關(r=?0.13,P=0.003)。表明糞桿菌和羅氏菌屬的相對豐度高與腎移植受者腸桿菌科菌尿和UTI發生的風險降低相關。遺憾的是,本研究沒有發現上述研究中發現的UTI患者與正常患者腸道中豐度存在統計學差異的菌種在遺傳學上與UTI存在明確的因果關系,這有可能是既往研究中個體差異所導致,也可能是本研究所使用的GWAS數據樣本量不足。
Eggerthella菌屬為伊格爾茲氏菌科中的一個屬,既往的一些研究發現放線菌屬的Eggerthellalenta在某些疾病或暴露個體的腸道菌群中顯著富集[26-28],同時是導致多種感染的致病因素[29,30]。有研究發現人類腸道放線菌屬的Eggerthella lenta可以通過解除對Th17轉錄因子Ror γ t的抑制作用,進而誘導了腸道Th17的激活,從而促進腸道炎癥[31]。腸道細菌導致泌尿系統炎癥的過程中是否存在同樣的機制目前不得而知,需要進一步的探索。Ruminococcaceae(UCG005)是瘤胃球菌科中的一個細菌屬,瘤胃球菌是最早發現的胃部細菌之一,在新陳代謝中起著至關重要的作用。瘤胃球菌通過分解宿主消化系統的纖維素來獲取營養。也能夠發酵葡萄糖和木糖。但許多研究也揭示了其在炎癥發生發展過程中的作用,例如一項關于瘤胃球菌炎癥特性的研究發現,它以葡甘露聚糖多糖的形式產生代謝物。瘤胃球菌多糖可以激發免疫系統細胞,如腫瘤壞死因子TNFα[32]。而TNFα是克羅恩病的炎癥生物標志物之一。然而,關于瘤胃球菌對UTI影響的研究有限。Defluviitaleaceae(UCG011)是本研究中發現的唯一對UTI易感性具有保護作用的一個細菌屬,Tong等[33]發現與健康者相比,類風濕關節炎高風險個體表現出口腔微生物群中Defluviitaleaceae(UCG011)屬豐度的降低。顯示出了Defluviitaleaceae(UCG011)在炎癥發生過程中的某種保護作用。然而,目前少有研究探討Defluviitaleaceae(UCG011)和UTI之間的潛在關系。
腸道微生物群通過影響多種生理過程影響宿主健康,包括器官發育、免疫力和疾病易感性[4]。腸道生態失調可以改變腸道遠端器官的信號傳導,包括大腦(定義為腸-腦軸)[34]和肺(定義為腸-肺軸)[35]。而Worby等[36]研究證明,腸道微生物群的變化可以增強對復發性UTI的易感性,從而定義了腸道-膀胱軸。Magruder等[37]的研究發現發生UTI的腎移植患者糞便中的大腸埃希氏菌和腸球菌的腸道豐度分別與大腸埃希氏菌和腸球菌菌尿癥的未來發展相關,以及腸道大腸埃希氏菌的豐度與癥狀性大腸埃希氏菌UTI的發展有關。該研究結果支持了腸道微生物群-泌尿道感染軸,表明調節腸道微生物群可能是預防UTI的潛在新策略。
使用廣譜抗生素是治療UTI最主要的方法,但不合理的臨床抗生素使用正在加劇細菌耐藥性問題。同時,腸-膀胱軸突出了現有抗生素治療的局限性。抗生素治療不僅是難治性復發UTI的已知風險因素[38,39],還可能導致顯著和持久地擾動腸道微生物組[36]。如果UTI的抗生素治療確實產生或維持腸道生態失調的狀態,那么這可能導致治療的惡性循環,并增加了再次感染的風險及對未來感染的易感性[40]。因此,為降低尿路感染復發的風險,并盡量減少抗微生物藥物耐藥性,開發新的預防及治療UTI的方法顯得尤為重要。
既往研究已證實糞便微生物群移植在治療某些病原體導致的難治性復發UTI中的可行性[41,42],Tariq等[43]的糞菌微生物移植實驗表明腸道共生細菌可以通過競爭營養物質、改變腸道pH值、形成抗菌代謝物和阻止多重耐藥菌(MDROs)進入黏膜壁上的結合位點來減少MDROs的泌尿道定植,從而提高UTI患者的抗生素敏感性。顯示出了糞菌移植的極大潛力,但仍需要進一步探索更佳的實驗方案及潛在的病理機制。
在觀察性研究中已經報道了腸道菌群和UTI之間的關聯。然而,由于方法上的限制,確定因果關系可能具有挑戰性。本MR研究的一個關鍵優勢是我們的研究結果避免了反向因果關系,并最大限度地減少了殘余混雜因素,同時避免了既往觀察性研究中的各種偏倚。本MR分析結果可以為選擇個體腸道細菌以研究腸道微生物群在UTI發病機制中的作用提供指導。然而,本研究也存在局限性:① 盡管我們采用了迄今為止最大的腸道微生物組數據集,但樣本量仍需要擴大,這可能會進一步增強統計能力。另外數據只測量到屬水平,限制了在種水平上進一步探討腸道菌群與UTI之間的聯系。② 由于使用了摘要級數據,腸道細菌豐度和UTI風險之間的非線性關聯無法探索。③ 研究數據均采集于歐洲個體,由于種族群體之間固有的遺傳差異,本結果外推到其他種族時應當謹慎。
綜上所述,本項雙樣本MR研究發現遺傳水平上腸道細菌屬Eggerthella、Ruminococcaceae(UCG005)和Defluviitaleaceae(UCG011)對UTI風險具有潛在的因果影響。本研究可為腸道微生物群介導的UTI發生發展機制提供新的見解,但仍需更多的研究來探究腸道細菌群與UTI之間的生物學聯系。