Krüppel樣因子4(Krüppel-like factor 4,KLF4)是Kruppel樣轉錄因子蛋白家族的一員,是一種進化保守的含鋅指轉錄因子,參與調節多種細胞過程,如細胞生長、增殖、分化和侵襲等,KLF4在體內多種組織細胞中均有表達,在許多生理及病理情況下都有廣泛的作用。諸多研究表明KLF4參與神經炎性反應、氧化應激、細胞凋亡和軸突再生等多種神經生物過程,與癲癇、腦卒中、阿爾茲海默病等多種神經系統疾病有著密切聯系。現就KLF4在神經系統疾病發生發展中的作用進行綜述,有助于深入了解疾病的發病機制,并為神經系統疾病提供潛在的臨床治療靶點。
引用本文: 楊陽, 孫洪英. Krüppel樣因子4在神經系統疾病中的作用研究進展. 癲癇雜志, 2024, 10(2): 141-146. doi: 10.7507/2096-0247.202312003 復制
版權信息: ?四川大學華西醫院華西期刊社《癲癇雜志》版權所有,未經授權不得轉載、改編
Krüppel樣因子4(Krüppel-like factor 4,KLF4)最初發現是在胃腸上皮細胞中表達,是含有鋅指結構的轉錄因子,曾被稱為胃腸富集KLF(Gut-enriched KLF)或表皮鋅指因子(Epi-thelial zinc finger,EZF) [1]。人類KLF4由513個氨基酸組成,基因定位于染色體9q31,長度為5.6 kb,含有轉錄激活調節結構域、轉錄抑制調節結構域、DNA結合結構域、核定位序列四個主要功能結構域,這種結構決定了KLF4可通過與其他因子相互作用和調節DNA結合效率從而影響轉錄調節活性的特異性。KLF4是一種多功能轉錄因子,參與調節細胞的生長、增殖、分化和侵襲等,通過磷酸化、乙酰化、甲基化和泛素化介導基因激活在多個水平上受到調控[2]。KLF4的表達在轉錄和轉錄后水平均受到調控,其具有雙重調節作用,通過激活或抑制多個基因的轉錄活性參與多種細胞功能[3],因此在腫瘤研究中成為熱門話題。大量的研究表明KLF4是一種抑癌基因,然而在乳腺癌和膠質母細胞瘤起到促癌作用。在病理生理學中,KLF4也是環境依賴性的抗炎和促炎因子,這使得KLF4在動脈粥樣硬化中的促進或是抑制作用依賴于靶基因和靶細胞決定[4],因此,KLF4在疾病中的具體作用依賴于具體的信號通路及細胞環境等多種復雜因素,目前的研究表明KLF4下游重要的分子靶點包括P21、P53、Cyclin-D等。在神經系統中,KLF4不僅調控神經元的增殖和分化,也對軸突生長的調控起關鍵作用,提示其可能介導了多種神經系統疾病的發生,近年來逐漸成為神經學領域的研究熱點。
1 KLF4在神經系統疾病中的獨特作用
KLF4在各種神經系統疾病的發生和發展中起著重要作用,包括癲癇、阿爾茲海默病、帕金森病、腦卒中、精神分裂癥和腦積水等多種疾病。然而KLF4在疾病中起到保護作用還是促進作用尚未可知。KLF4自身就具有雙重調節作用,其在疾病中的調控作用很難明確。腦發育過程中KLF4過表達可誘導腦積水,帕金森病中過表達KLF4可促進1-甲基-4-苯基吡啶離子(1-methyl-4-phenylpyridinium,MPP+)的神經毒性,增加細胞易感性和氧化應激;而在癲癇模型中過表達KLF4對癲癇有抑制作用。KLF4的同種作用也可以體現在不同的疾病狀態下,例如:KLF4參與的炎癥反應涉及多種疾病,其可以產生不同的疾病狀態離不開炎癥與疾病本身病理基礎的關系。癲癇的發生通常伴隨著膠質細胞的增生,KLF4介導膠質細胞產生炎癥介質導致神經組織微環境中持續的炎癥狀態加重了癲癇后腦損害;阿爾茲海默病的病理基礎與淀粉樣蛋白-β (Amyloid β,Aβ)沉積密切相關,而KLF4參與Aβ沉積可導致小膠質細胞和星形膠質細胞產生炎癥介質;缺血性腦卒中是由于腦血管病變所致的腦組織缺血、缺氧,血管內皮細胞富集的KLF4介導的血管炎癥與腦血管病變有著深入的聯系。KLF4參與調節氧化應激、神經炎癥、神經元凋亡和突觸再生等多種細胞過程,這表明KLF4作為一種潛在的靶點對神經系統疾病的發生發展有很大影響。以下重點討論KLF4參與癲癇、阿爾茲海默病、缺血性腦卒中可能的機制和相關通路。
1.1 KLF4與癲癇
癲癇是一種常見的神經系統疾病,表現為腦內神經元同步異常放電所引起的突發性、反復性、短暫性的中樞神經系統功能紊亂,以反復發作和進行性加重為主要特點,通常伴有不可預知的癇性發作,給患者的正常生活和工作造成了極大的影響[5]。因此,尋找潛在的原因和更有效的治療方法以提高患者的生活質量就變得越來越緊迫。然而,癲癇的病因十分復雜,關于其確切的發病機制的研究仍在進行中。據報道癲癇的發生是由氧化應激、細胞凋亡、突觸傳遞和突觸可塑性、離子轉運、通道和受體功能、神經遞質代謝和轉錄因子等多種因素引起的[6]。研究發現,癲癇小鼠海馬中KLF4表達下調,而上調KLF4對癲癇的發作有抑制作用[7]。KLF4參與癲癇發生的具體機制可能是開發新的癲癇治療策略的重要靶點。
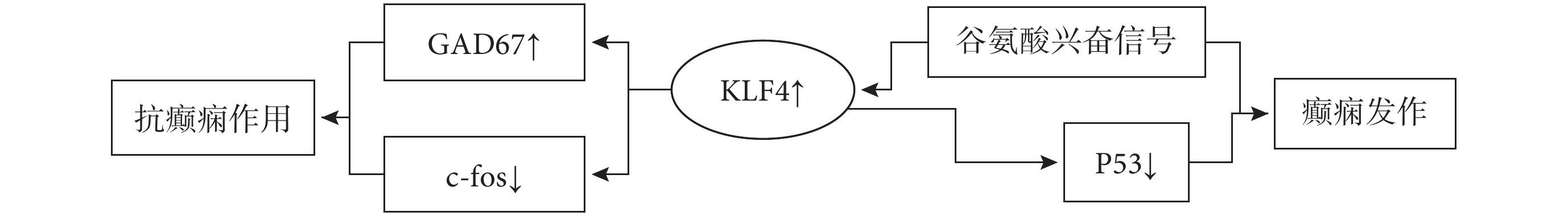
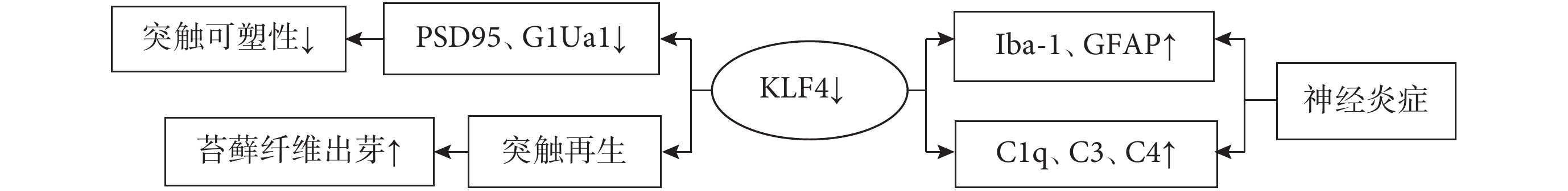
結合國內外研究發現,KLF4參與癲癇的機制可能與神經炎癥、軸突再生、神經保護作用和神經遞質及其受體功能異常等相關。炎癥在癲癇的發生和發展中起重要作用,神經炎癥可能會對神經興奮性和癲癇易感性產生影響。小膠質細胞免疫炎癥功能失調是誘發或促成癲癇發作的常見因素[8]。許文博等人研究發現,KLF4敲除小鼠海馬中,小膠質細胞標志物(Iba-1)、星形膠質細胞標志物(GFAP)顯著升高并且補體信號通路(C1q、C3、C4)亢進,這表明KLF4敲除可能會參與誘導炎癥反應發作,正是這種腦部炎癥促進了神經元的高興奮性和癲癇發作。癲癇較常見的一種病理變化是突觸重塑。突觸相關蛋白PSD95是評價突觸可塑性的關鍵指標。KLF4敲除小鼠的海馬中檢測到PSD95和G1Ua1蛋白也呈現顯著性降低,KLF4敲除會降低突觸可塑性[9]。KLF4上調后導致海馬神經元軸突再生能力下降,而下調KLF4可顯著增加海馬神經元軸突再生,苔蘚纖維出芽顯著增加[10]。KLF4是P53的上游蛋白,P53對軸突生長有重要作用,但其蛋白表達與苔蘚纖維出芽無關[11]。研究表明KLF4表達增加可直接引起P53表達下調,因此KLF4/P53信號通路參與癲癇誘發和抗癲癇作用[12]。P53可能通過升高神經生長因子活性起到神經保護作用。目前研究認為谷氨酸作為興奮性神經遞質可導致癲癇發作,其毒性也與繼發性腦損害密切相關。體外研究發現,谷氨酸能刺激觸發神經元中KLF4 mRNA和蛋白水平的快速升高[13]。KLF4可能通過調控谷氨酸興奮性信號傳遞等影響癲癇發作。 c-fos是參與細胞活動的重要基因,c-fos被過度激活后即可病理性調控多個下游基因表達,通過級聯放大效應將致癇因素的刺激轉換為中樞神經系統內細胞的病理增殖、分化或凋亡,最終形成癲癇[14]。海馬和梨狀皮質是癲癇相關的重要腦區。KLF4過表達對戊四唑(PTZ)誘導的癲癇小鼠產生抗癲癇作用,顯著減少海馬和梨狀皮層c-fos表達[15]。PTZ主要作用于GABA-A受體,所以過表達KLF4可能通過活化海馬的GABA系統而起到抗癲癇作用。GABA是最重要的抑制性神經遞質之一,而GAD67參與GABA的形成。KLF4過表達后GAD67的表達增加,上調的 GAD67導致癲癇小鼠發作次數明顯減少,海馬神經元c-fos表達也顯著下降。神經系統外源的GAD67也可以產生抗癲癇作用[16],由此猜測KLF4的抗癲癇作用可能是通過激活GAD67介導的。KLF4也是神經系統發育時期的重要調控因子,因此,KLF4在胚胎時期的調控是否是原發性癲癇的發病機制之一,這可能對癲癇的預防起到重要作用。KLF4參與癲癇的調控是通過何種通路和分子機制尚未明確,關于其下游作用靶點及具體作用通路值得進一步深入研究(圖1、2)。
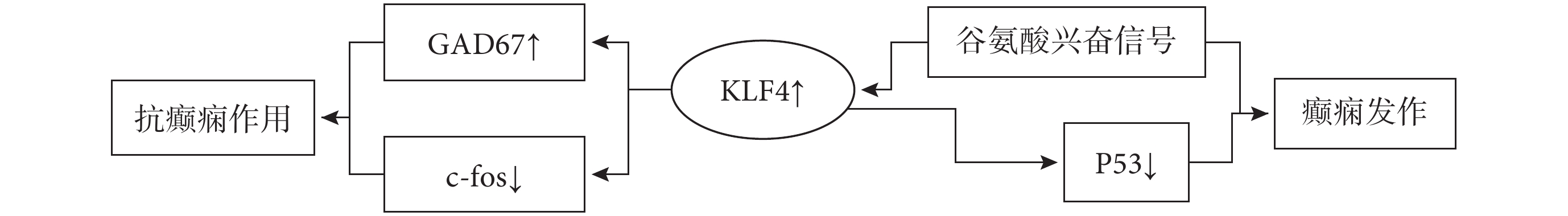 圖1
KLF4↑參與癲癇的作用通路
圖1
KLF4↑參與癲癇的作用通路
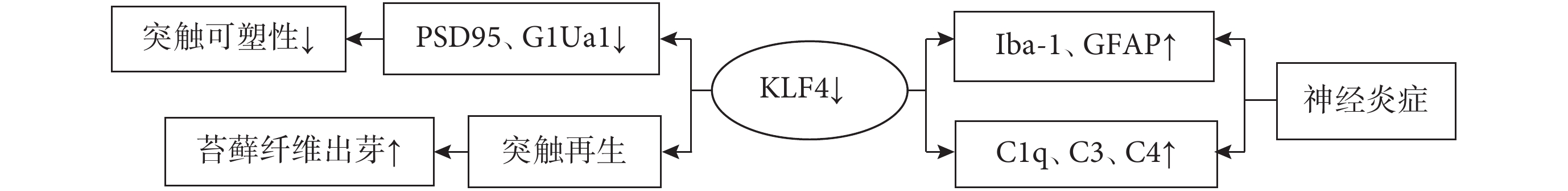 圖2
KLF4↓參與癲癇的作用通路及病理基礎
圖2
KLF4↓參與癲癇的作用通路及病理基礎
1.2 KLF4與阿爾茲海默病
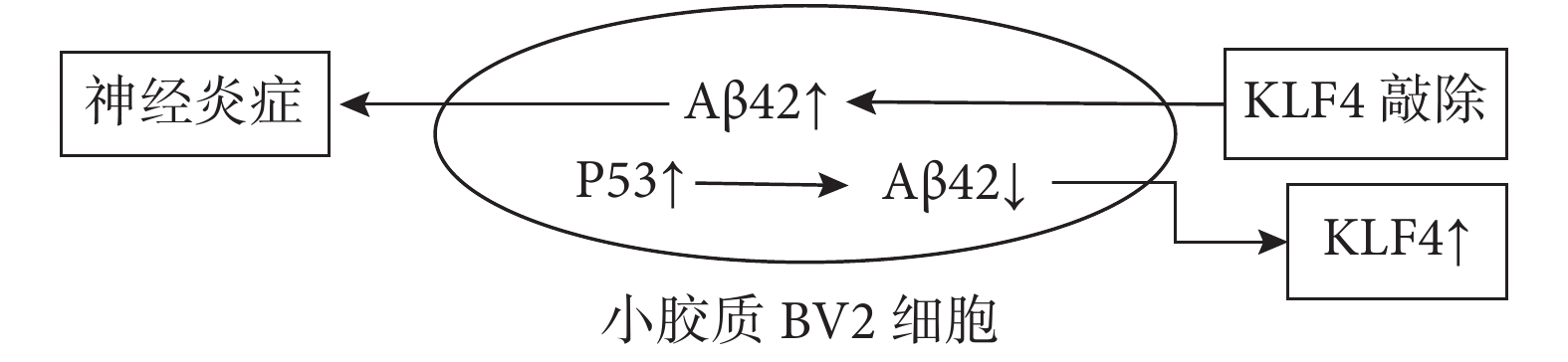
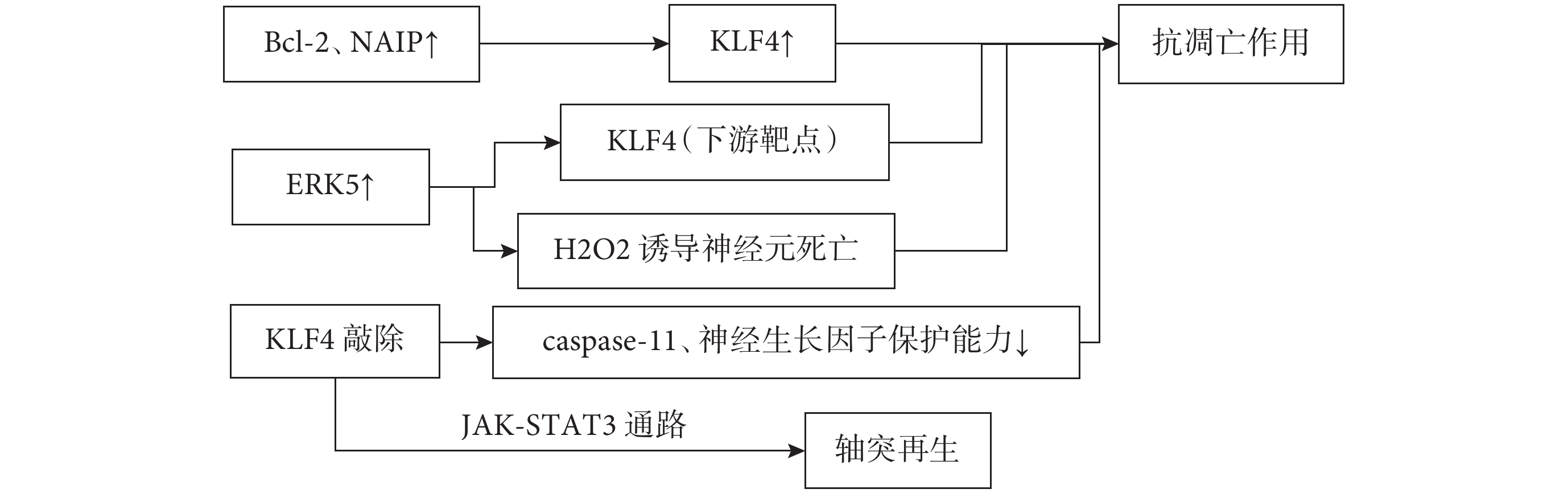
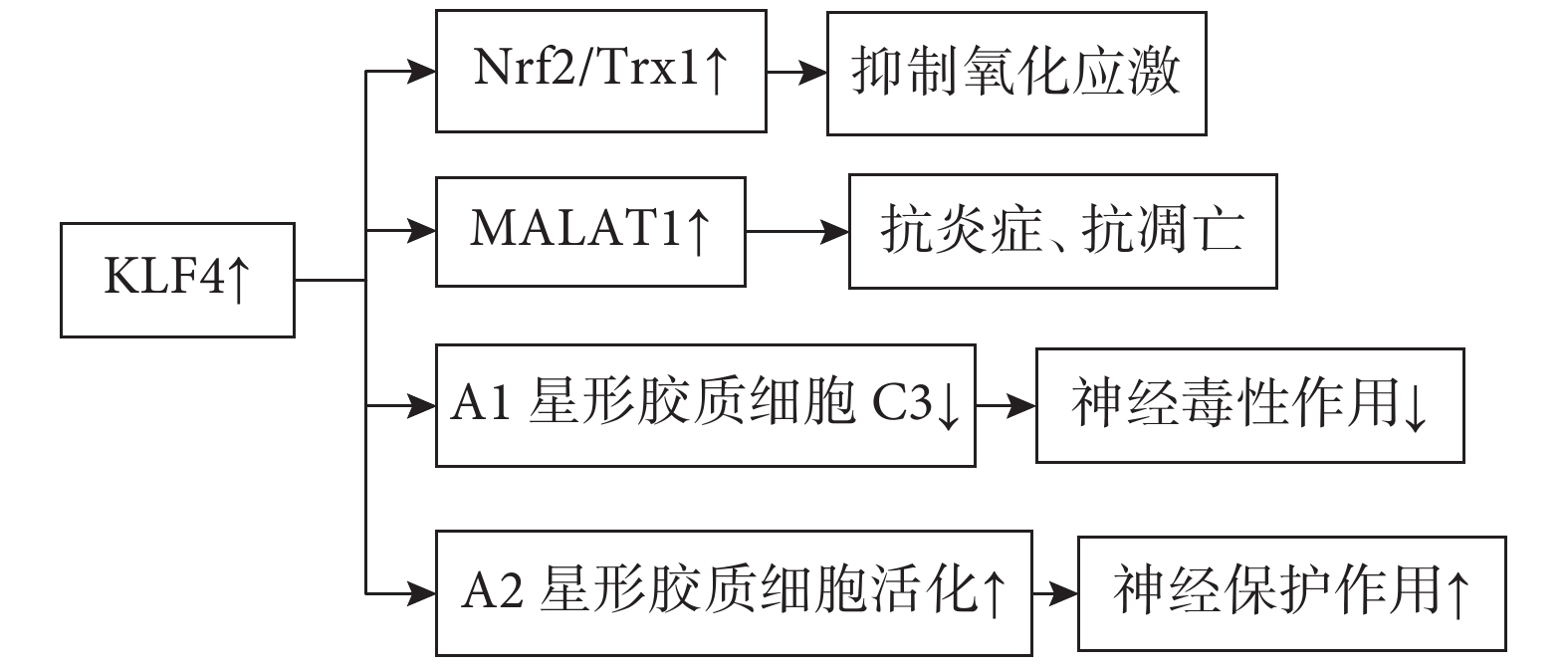
阿爾茲海默病 (Alzheimer’s disease,AD)是一種進行性神經退行性疾病,包括認知和記憶功能障礙,各種精神癥狀和行為異常,進行性癡呆是最常見的臨床特征[17]。其最主要病理特征包括Aβ異常沉積引起的老年斑形成和tau積聚引起的神經原纖維纏結[18]。近年來陸續提出氧化應激、炎癥反應、胰島素信號傳導通路障礙、線粒體功能障礙等多種假說,但其具體發病機制目前并不明確。Aβ沉積、氧化應激和鐵積累的累積損傷可導致阿爾茲海默病患者神經元功能障礙和細胞凋亡。阿爾茲海默病患者腦中的氧化應激現象非常明顯,氧化應激是Aβ積累和tau過度磷酸化的重要因素。過量的Aβ沉積會刺激小膠質細胞和星形膠質細胞釋放促炎介質,如細胞因子、趨化因子、活性氧(Reactive oxygen species,ROS)和補體蛋白等影響因素導致氧化應激,而氧化應激可以刺激Aβ的產生[19]。因此,Aβ和氧化應激可以相互作用并影響阿爾茲海默病的進展。此外,炎癥的惡性循環進一步加重了神經細胞的功能障礙和細胞凋亡,導致了阿爾茲海默病的加重。抗氧化應激和抗炎作用在阿爾茲海默病的治療中就顯得尤為重要。近年來,有研究表明激活的P53在小膠質BV2細胞中介導寡聚物Aβ42可以增加KLF4的表達,KLF4的敲除可恢復Aβ42介導的神經炎癥[20],這表明KLF4表達可能抑制Aβ42誘導的神經炎癥(圖3)。KLF4可能通過抑制炎癥信號參與了阿爾茲海默病的氧化應激。持續的氧化應激也可導致神經元的凋亡,越來越多證據表明阿爾茲海默病患者腦中神經元丟失的機制是由神經元凋亡引起,而神經元凋亡反過來又可加速阿爾茲海默病進展。KLF4表達增加可能通過增加B細胞淋巴瘤-2(B cell lymphoma-2,Bcl-2)及神經元凋亡抑制蛋白(NAIP)的表達介導神經元的抗凋亡作用[21]。而在白血病中,KLF4通過下調Bcl-2和上調Bax促進過氧化氫誘導的細胞凋亡[22]。相同的結論是Zhu等[13]在體外實驗中發現KLF4的過表達使神經元對胱天蛋白酶-3(cysteine aspartic acid specific protease-3,Caspase-3)活性敏感。因此,KLF4被證明既可抑制細胞凋亡,也可促進細胞凋亡。目前已證實關于KLF4在細胞中的作用是雙向的。在阿爾茲海默病中,更傾向于它的抗凋亡作用,Su等[23]研究發現,敲除KLF4后caspase-1增加、神經生長因子保護神經元的能力下降,KLF4是絲裂原活化蛋白激酶5 (ERK5)新的下游靶點,ERK5的激活可以部分減少過氧化氫誘導的海馬神經元死亡,這些結果證實了KLF4具有抗凋亡的特性。此外,AD也是一種突觸功能障礙。KLF4在抑制軸突生長中起重要作用,敲除KLF4可增強軸突再生,加快軸突生長速度,KLF4表達降低通過酪氨酸激酶/信號轉導和轉錄活化因子3(Janus kinase/signal transducers and activators of transcription 3,JAK/STAT3)通路促進軸突再生,這表明KLF4可能通過JAK-STAT3通路促進軸突再生來影響阿爾茲海默病的發生發展[24](圖4)。另一方面,大腦中鐵負荷的增加加速了Aβ和過度磷酸化的tau纏結的形成,同時也增強了它們的毒性,鐵平衡失調可能導致毒性病理特征[25]。研究發現, KLF4介導了糖皮質激素對血紅素載體蛋白1(HCP1)的上調,上調的HCP1能夠促進血紅素的吸收[26],血紅素是人體功能鐵的主要組成成分,因此,可能加劇了腦內鐵的積累,增加細胞凋亡或功能障礙,加重腦損傷(圖5)。綜上所述,KLF4可能參與了AD抗炎、抗凋亡、軸突再生和鐵積累等過程,這些發現表明KLF4是AD的潛在治療靶點。然而,KLF4對AD影響的深層細胞和分子機制尚不清楚,需要進一步研究。
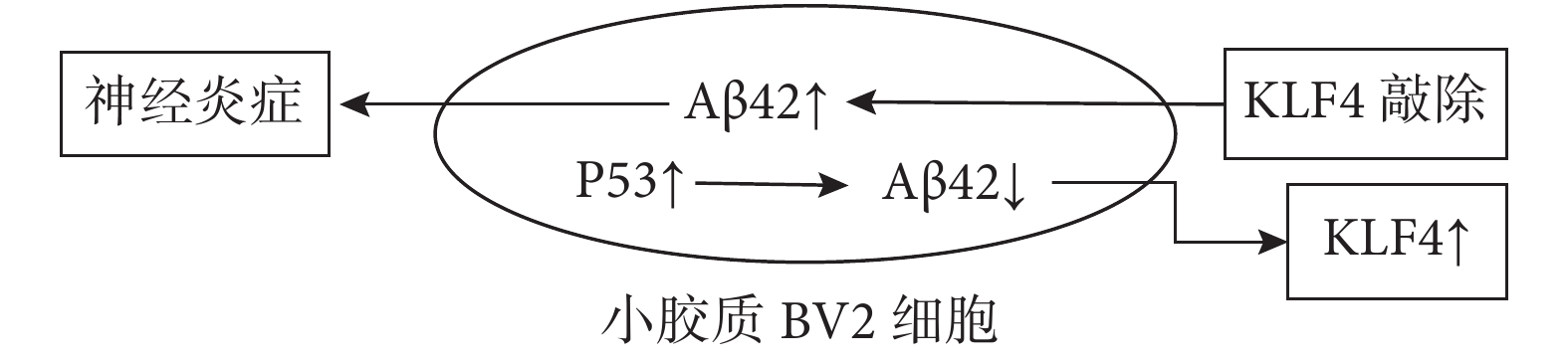 圖3
KLF4與Aβ42介導的神經炎癥
圖3
KLF4與Aβ42介導的神經炎癥
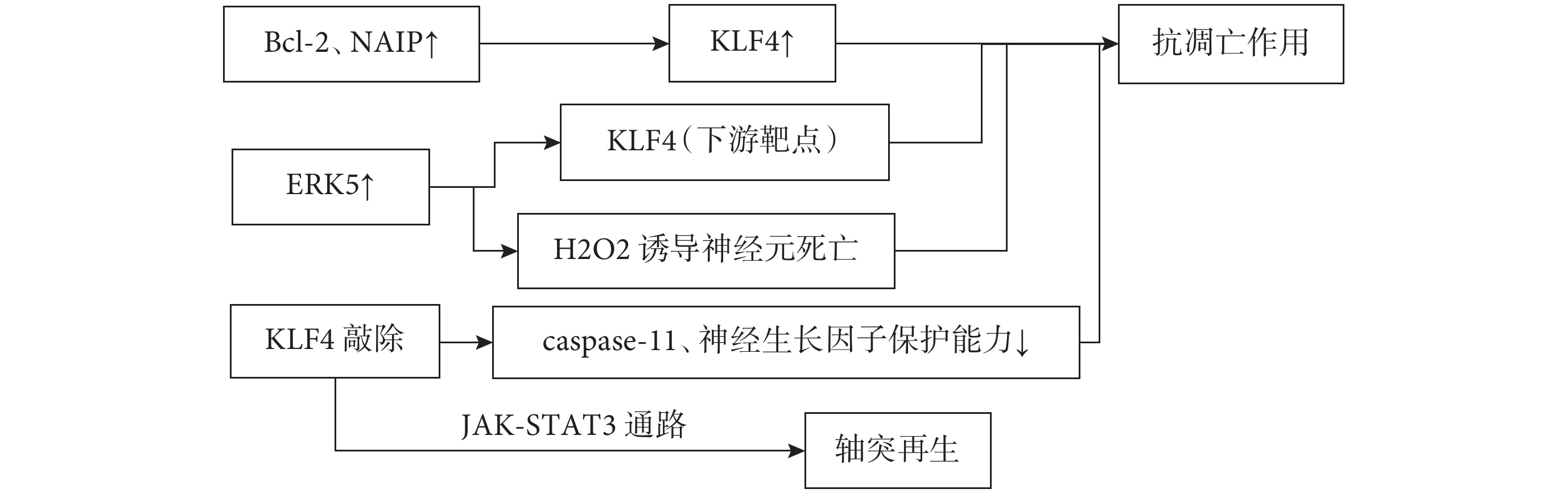 圖4
KLF4在AD中的抗凋亡作用和軸突生長
圖4
KLF4在AD中的抗凋亡作用和軸突生長
 圖5
KLF4與鐵代謝的關系
圖5
KLF4與鐵代謝的關系
1.3 KLF4與缺血性腦卒中
缺血性腦卒中是指因腦部血液循環障礙,缺血、缺氧所致的局限性腦組織的缺血性壞死或軟化,進而導致腦進行性梗死和神經功能受損,是全世界致殘、致死的主要原因之一[27]。腦梗死后遺留的偏癱、失語、認知障礙等癥狀給家庭和社會帶來沉重的負擔,雖然目前大多數腦卒中患者可獲得溶栓和血管內血栓切除術等有效治療方法,但效果仍然欠佳[28],因此對腦梗死發病機制的深入研究至關重要。缺血性腦卒中病理過程在腦動脈閉合后數秒鐘至數分鐘觸發,腦細胞缺血、缺氧導致細胞內乳酸增加,毒性興奮性神經遞質增加,自由基產生,細胞發生凋亡、水腫。
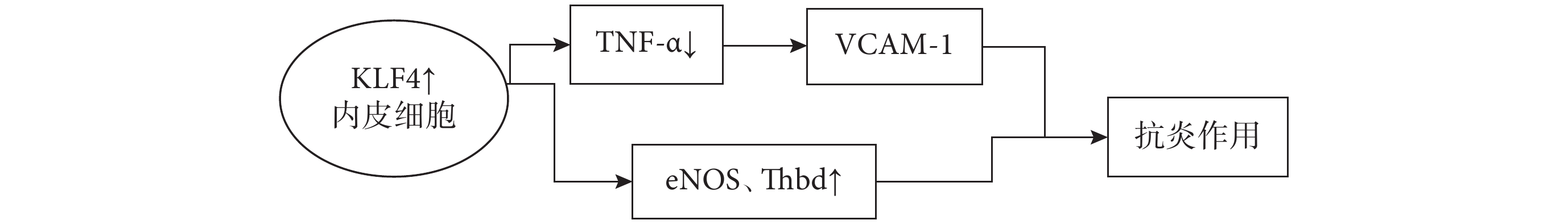
炎癥過程已被證明參與動脈粥樣硬化的過程以及腦血管疾病的發病機制,腦缺血損傷后產生的炎癥介質觸發的復雜信號級聯反應也決定著腦卒中結局[29]。KLF4富集于內皮細胞中且抑制炎癥信號的激活,在內皮細胞中過表達可誘導多種內皮因子,包括內皮型一氧化氮合酶(eNOS)和血栓調節蛋白(Thbd),起到抗炎和抗血栓的作用;KLF4也可抑制腫瘤壞死因子-α(TNF-α)誘導的血管細胞粘附分子1 (VCAM-1)的表達,起到抑制動脈炎癥,調節血管損傷后新內膜形成的作用[30](圖6)。重要的是,KLF4可以保護腦微血管內皮細胞免受缺血性卒中誘導的凋亡。KLF4對血腦屏障的完整性具有保護作用可顯著減輕血腦屏障滲漏,其是通過上調核因子紅細胞系2相關因子2(Nuclear Factor erythroid2-Related Factor 2,Nrf2)/硫氧還蛋白1(Thioredoxin1,Trx1)通路,提供血腦屏障完整性的神經保護作用。Nrf2是抗氧化系統的關鍵分子,可以啟動抗氧化酶的表達,抑制氧化應激[31]。KLF4的神經保護作用也可能與缺血性腦卒中急性期氧化應激抑制有關。在缺血性卒中急性期,線粒體功能障礙、炎癥級聯和能量代謝紊亂會導致腦梗死、神經功能損傷和血腦屏障損傷等,這使得多種自發保護機制立即被激活以維持細胞內穩態。KLF4在腦缺血損傷后被發現上調,KLF4通過改善缺血性卒中后血管內皮炎癥和調節緊密連接蛋白表達來減輕腦血管損傷,表明KLF4對缺血性腦損傷具有血管保護作用[30]。此外,KLF4可能是MALA T1啟動子的一個結合位點,其上調直接增加MALA T1的表達,通過減少炎癥反應和細胞凋亡來激活自發保護機制,MALA T1是KLF4在腦缺血損傷后發揮保護作用的轉錄靶點[32]。除了腦血管內皮細胞外,還有大量反應性星形膠質細胞表達KLF4, 腦卒中發生后,星形膠質細胞立即被激活。經典激活的星形膠質細胞(A1亞型)通過釋放促炎介質發揮神經毒性作用,而選擇性激活的星形膠質細胞(A2亞型)通過分泌抗炎介質發揮神經保護作用。C3和S100A10被認為是A1和A2星形膠質細胞的標記物,增強的KLF4可以抑制腦缺血后A1星形膠質細胞C3的表達,促進A2型星形膠質細胞的活化[33](圖7)。KLF4可抑制腦缺血后星形膠質細胞的神經毒性極化,促進其神經保護性極化。急性缺血性腦卒中患者血清中KLF4水平與梗死面積呈負相關,這表明KLF4水平可以反映缺血性腦卒中的嚴重程度且在發病機制中發揮著保護作用,循環KLF4可能作為預測急性缺血性腦卒中預后的潛在生物標志物。綜上所述,在缺血性腦卒中中,KLF4通過減少梗死面積,抑制氧化應激,恢復血腦屏障功能和促進神經長期恢復等功能來起到神經保護作用。然而,還需要進一步的研究來闡明KLF4在缺血性腦卒中的具體分子機制,更好地了解KLF4在這一過程中所起的作用可以為未來缺血性腦卒中的預防和治療提供了新的概念證明和潛在靶點。
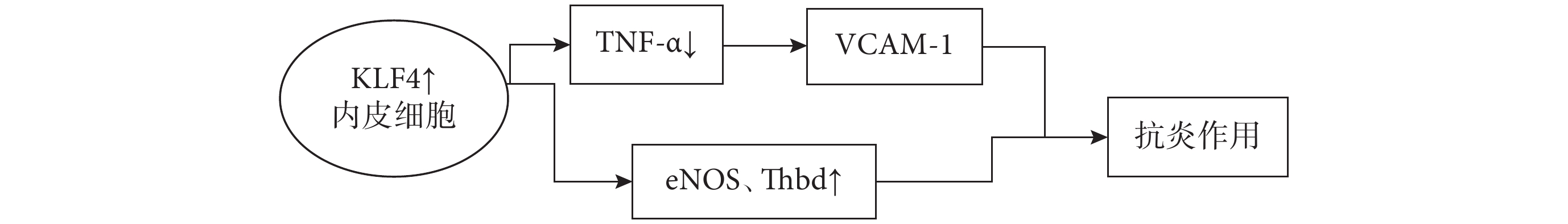 圖6
KLF4↑在內皮細胞中的抗炎作用通路
圖6
KLF4↑在內皮細胞中的抗炎作用通路
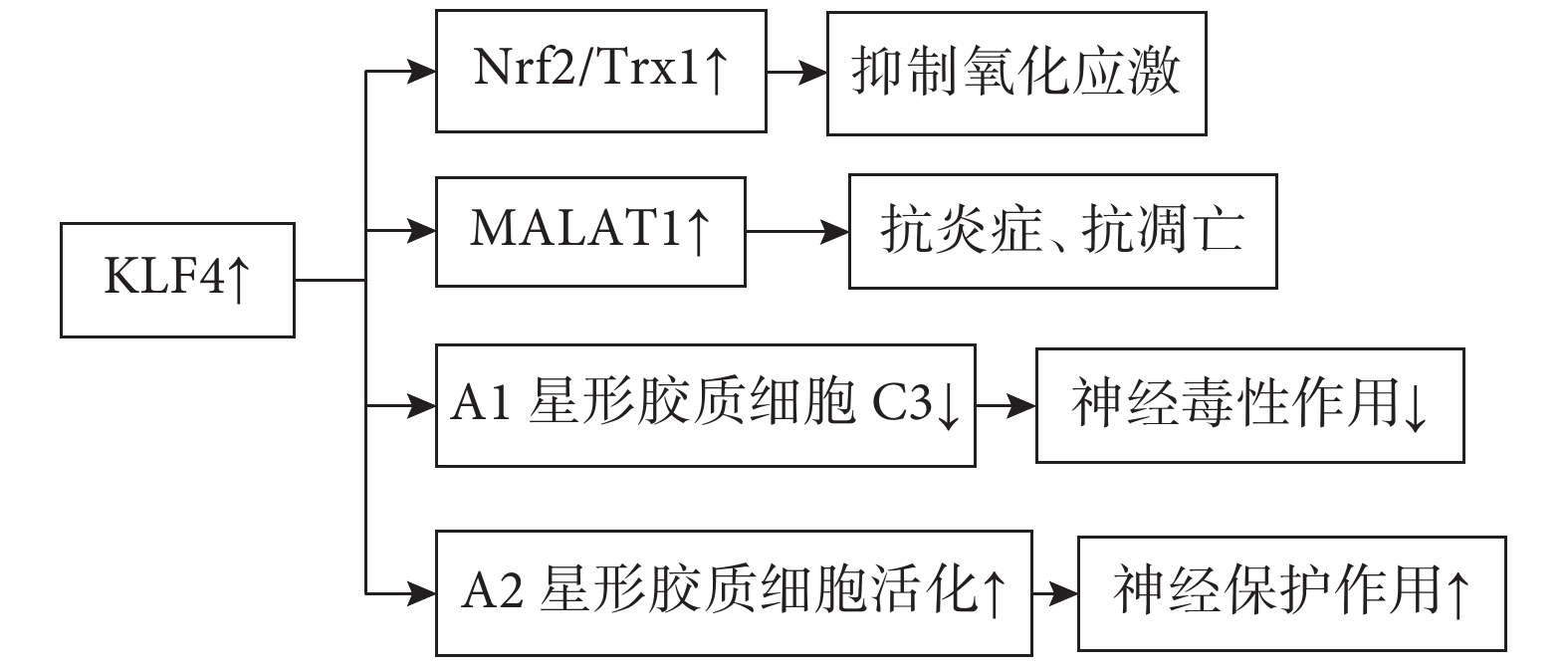 圖7
KLF4↑參與缺血性腦卒中的作用
圖7
KLF4↑參與缺血性腦卒中的作用
2 小結與展望
文章對KLF4在神經系統疾病中可能的作用機制和途徑進行了綜述,涉及到多種分子及信號通路共同調控,但其具體調控機制尚不能完全闡明。KLF4在不同的細胞環境及細胞類型中又通過不同的通路和分子機制發揮著同種或者相反的作用,因而是否可以通過KLF4而闡明相關疾病的內部關聯,這些問題都將有待進一步研究。隨著對KLF4越來越多的研究,證實了其在神經系統的發育階段和相關疾病的調節中具有重要意義,KLF4有望成為特定的生物標志物或治療的潛在干預靶點,更具體的機制及靶點將進一步明確,未來通過KLF4對于神經系統疾病的預防及治療或將成為一種可行的方案。
利益沖突聲明 所有作者無利益沖突。
Krüppel樣因子4(Krüppel-like factor 4,KLF4)最初發現是在胃腸上皮細胞中表達,是含有鋅指結構的轉錄因子,曾被稱為胃腸富集KLF(Gut-enriched KLF)或表皮鋅指因子(Epi-thelial zinc finger,EZF) [1]。人類KLF4由513個氨基酸組成,基因定位于染色體9q31,長度為5.6 kb,含有轉錄激活調節結構域、轉錄抑制調節結構域、DNA結合結構域、核定位序列四個主要功能結構域,這種結構決定了KLF4可通過與其他因子相互作用和調節DNA結合效率從而影響轉錄調節活性的特異性。KLF4是一種多功能轉錄因子,參與調節細胞的生長、增殖、分化和侵襲等,通過磷酸化、乙酰化、甲基化和泛素化介導基因激活在多個水平上受到調控[2]。KLF4的表達在轉錄和轉錄后水平均受到調控,其具有雙重調節作用,通過激活或抑制多個基因的轉錄活性參與多種細胞功能[3],因此在腫瘤研究中成為熱門話題。大量的研究表明KLF4是一種抑癌基因,然而在乳腺癌和膠質母細胞瘤起到促癌作用。在病理生理學中,KLF4也是環境依賴性的抗炎和促炎因子,這使得KLF4在動脈粥樣硬化中的促進或是抑制作用依賴于靶基因和靶細胞決定[4],因此,KLF4在疾病中的具體作用依賴于具體的信號通路及細胞環境等多種復雜因素,目前的研究表明KLF4下游重要的分子靶點包括P21、P53、Cyclin-D等。在神經系統中,KLF4不僅調控神經元的增殖和分化,也對軸突生長的調控起關鍵作用,提示其可能介導了多種神經系統疾病的發生,近年來逐漸成為神經學領域的研究熱點。
1 KLF4在神經系統疾病中的獨特作用
KLF4在各種神經系統疾病的發生和發展中起著重要作用,包括癲癇、阿爾茲海默病、帕金森病、腦卒中、精神分裂癥和腦積水等多種疾病。然而KLF4在疾病中起到保護作用還是促進作用尚未可知。KLF4自身就具有雙重調節作用,其在疾病中的調控作用很難明確。腦發育過程中KLF4過表達可誘導腦積水,帕金森病中過表達KLF4可促進1-甲基-4-苯基吡啶離子(1-methyl-4-phenylpyridinium,MPP+)的神經毒性,增加細胞易感性和氧化應激;而在癲癇模型中過表達KLF4對癲癇有抑制作用。KLF4的同種作用也可以體現在不同的疾病狀態下,例如:KLF4參與的炎癥反應涉及多種疾病,其可以產生不同的疾病狀態離不開炎癥與疾病本身病理基礎的關系。癲癇的發生通常伴隨著膠質細胞的增生,KLF4介導膠質細胞產生炎癥介質導致神經組織微環境中持續的炎癥狀態加重了癲癇后腦損害;阿爾茲海默病的病理基礎與淀粉樣蛋白-β (Amyloid β,Aβ)沉積密切相關,而KLF4參與Aβ沉積可導致小膠質細胞和星形膠質細胞產生炎癥介質;缺血性腦卒中是由于腦血管病變所致的腦組織缺血、缺氧,血管內皮細胞富集的KLF4介導的血管炎癥與腦血管病變有著深入的聯系。KLF4參與調節氧化應激、神經炎癥、神經元凋亡和突觸再生等多種細胞過程,這表明KLF4作為一種潛在的靶點對神經系統疾病的發生發展有很大影響。以下重點討論KLF4參與癲癇、阿爾茲海默病、缺血性腦卒中可能的機制和相關通路。
1.1 KLF4與癲癇
癲癇是一種常見的神經系統疾病,表現為腦內神經元同步異常放電所引起的突發性、反復性、短暫性的中樞神經系統功能紊亂,以反復發作和進行性加重為主要特點,通常伴有不可預知的癇性發作,給患者的正常生活和工作造成了極大的影響[5]。因此,尋找潛在的原因和更有效的治療方法以提高患者的生活質量就變得越來越緊迫。然而,癲癇的病因十分復雜,關于其確切的發病機制的研究仍在進行中。據報道癲癇的發生是由氧化應激、細胞凋亡、突觸傳遞和突觸可塑性、離子轉運、通道和受體功能、神經遞質代謝和轉錄因子等多種因素引起的[6]。研究發現,癲癇小鼠海馬中KLF4表達下調,而上調KLF4對癲癇的發作有抑制作用[7]。KLF4參與癲癇發生的具體機制可能是開發新的癲癇治療策略的重要靶點。
結合國內外研究發現,KLF4參與癲癇的機制可能與神經炎癥、軸突再生、神經保護作用和神經遞質及其受體功能異常等相關。炎癥在癲癇的發生和發展中起重要作用,神經炎癥可能會對神經興奮性和癲癇易感性產生影響。小膠質細胞免疫炎癥功能失調是誘發或促成癲癇發作的常見因素[8]。許文博等人研究發現,KLF4敲除小鼠海馬中,小膠質細胞標志物(Iba-1)、星形膠質細胞標志物(GFAP)顯著升高并且補體信號通路(C1q、C3、C4)亢進,這表明KLF4敲除可能會參與誘導炎癥反應發作,正是這種腦部炎癥促進了神經元的高興奮性和癲癇發作。癲癇較常見的一種病理變化是突觸重塑。突觸相關蛋白PSD95是評價突觸可塑性的關鍵指標。KLF4敲除小鼠的海馬中檢測到PSD95和G1Ua1蛋白也呈現顯著性降低,KLF4敲除會降低突觸可塑性[9]。KLF4上調后導致海馬神經元軸突再生能力下降,而下調KLF4可顯著增加海馬神經元軸突再生,苔蘚纖維出芽顯著增加[10]。KLF4是P53的上游蛋白,P53對軸突生長有重要作用,但其蛋白表達與苔蘚纖維出芽無關[11]。研究表明KLF4表達增加可直接引起P53表達下調,因此KLF4/P53信號通路參與癲癇誘發和抗癲癇作用[12]。P53可能通過升高神經生長因子活性起到神經保護作用。目前研究認為谷氨酸作為興奮性神經遞質可導致癲癇發作,其毒性也與繼發性腦損害密切相關。體外研究發現,谷氨酸能刺激觸發神經元中KLF4 mRNA和蛋白水平的快速升高[13]。KLF4可能通過調控谷氨酸興奮性信號傳遞等影響癲癇發作。 c-fos是參與細胞活動的重要基因,c-fos被過度激活后即可病理性調控多個下游基因表達,通過級聯放大效應將致癇因素的刺激轉換為中樞神經系統內細胞的病理增殖、分化或凋亡,最終形成癲癇[14]。海馬和梨狀皮質是癲癇相關的重要腦區。KLF4過表達對戊四唑(PTZ)誘導的癲癇小鼠產生抗癲癇作用,顯著減少海馬和梨狀皮層c-fos表達[15]。PTZ主要作用于GABA-A受體,所以過表達KLF4可能通過活化海馬的GABA系統而起到抗癲癇作用。GABA是最重要的抑制性神經遞質之一,而GAD67參與GABA的形成。KLF4過表達后GAD67的表達增加,上調的 GAD67導致癲癇小鼠發作次數明顯減少,海馬神經元c-fos表達也顯著下降。神經系統外源的GAD67也可以產生抗癲癇作用[16],由此猜測KLF4的抗癲癇作用可能是通過激活GAD67介導的。KLF4也是神經系統發育時期的重要調控因子,因此,KLF4在胚胎時期的調控是否是原發性癲癇的發病機制之一,這可能對癲癇的預防起到重要作用。KLF4參與癲癇的調控是通過何種通路和分子機制尚未明確,關于其下游作用靶點及具體作用通路值得進一步深入研究(圖1、2)。
圖1
KLF4↑參與癲癇的作用通路
圖2
KLF4↓參與癲癇的作用通路及病理基礎
1.2 KLF4與阿爾茲海默病
阿爾茲海默病 (Alzheimer’s disease,AD)是一種進行性神經退行性疾病,包括認知和記憶功能障礙,各種精神癥狀和行為異常,進行性癡呆是最常見的臨床特征[17]。其最主要病理特征包括Aβ異常沉積引起的老年斑形成和tau積聚引起的神經原纖維纏結[18]。近年來陸續提出氧化應激、炎癥反應、胰島素信號傳導通路障礙、線粒體功能障礙等多種假說,但其具體發病機制目前并不明確。Aβ沉積、氧化應激和鐵積累的累積損傷可導致阿爾茲海默病患者神經元功能障礙和細胞凋亡。阿爾茲海默病患者腦中的氧化應激現象非常明顯,氧化應激是Aβ積累和tau過度磷酸化的重要因素。過量的Aβ沉積會刺激小膠質細胞和星形膠質細胞釋放促炎介質,如細胞因子、趨化因子、活性氧(Reactive oxygen species,ROS)和補體蛋白等影響因素導致氧化應激,而氧化應激可以刺激Aβ的產生[19]。因此,Aβ和氧化應激可以相互作用并影響阿爾茲海默病的進展。此外,炎癥的惡性循環進一步加重了神經細胞的功能障礙和細胞凋亡,導致了阿爾茲海默病的加重。抗氧化應激和抗炎作用在阿爾茲海默病的治療中就顯得尤為重要。近年來,有研究表明激活的P53在小膠質BV2細胞中介導寡聚物Aβ42可以增加KLF4的表達,KLF4的敲除可恢復Aβ42介導的神經炎癥[20],這表明KLF4表達可能抑制Aβ42誘導的神經炎癥(圖3)。KLF4可能通過抑制炎癥信號參與了阿爾茲海默病的氧化應激。持續的氧化應激也可導致神經元的凋亡,越來越多證據表明阿爾茲海默病患者腦中神經元丟失的機制是由神經元凋亡引起,而神經元凋亡反過來又可加速阿爾茲海默病進展。KLF4表達增加可能通過增加B細胞淋巴瘤-2(B cell lymphoma-2,Bcl-2)及神經元凋亡抑制蛋白(NAIP)的表達介導神經元的抗凋亡作用[21]。而在白血病中,KLF4通過下調Bcl-2和上調Bax促進過氧化氫誘導的細胞凋亡[22]。相同的結論是Zhu等[13]在體外實驗中發現KLF4的過表達使神經元對胱天蛋白酶-3(cysteine aspartic acid specific protease-3,Caspase-3)活性敏感。因此,KLF4被證明既可抑制細胞凋亡,也可促進細胞凋亡。目前已證實關于KLF4在細胞中的作用是雙向的。在阿爾茲海默病中,更傾向于它的抗凋亡作用,Su等[23]研究發現,敲除KLF4后caspase-1增加、神經生長因子保護神經元的能力下降,KLF4是絲裂原活化蛋白激酶5 (ERK5)新的下游靶點,ERK5的激活可以部分減少過氧化氫誘導的海馬神經元死亡,這些結果證實了KLF4具有抗凋亡的特性。此外,AD也是一種突觸功能障礙。KLF4在抑制軸突生長中起重要作用,敲除KLF4可增強軸突再生,加快軸突生長速度,KLF4表達降低通過酪氨酸激酶/信號轉導和轉錄活化因子3(Janus kinase/signal transducers and activators of transcription 3,JAK/STAT3)通路促進軸突再生,這表明KLF4可能通過JAK-STAT3通路促進軸突再生來影響阿爾茲海默病的發生發展[24](圖4)。另一方面,大腦中鐵負荷的增加加速了Aβ和過度磷酸化的tau纏結的形成,同時也增強了它們的毒性,鐵平衡失調可能導致毒性病理特征[25]。研究發現, KLF4介導了糖皮質激素對血紅素載體蛋白1(HCP1)的上調,上調的HCP1能夠促進血紅素的吸收[26],血紅素是人體功能鐵的主要組成成分,因此,可能加劇了腦內鐵的積累,增加細胞凋亡或功能障礙,加重腦損傷(圖5)。綜上所述,KLF4可能參與了AD抗炎、抗凋亡、軸突再生和鐵積累等過程,這些發現表明KLF4是AD的潛在治療靶點。然而,KLF4對AD影響的深層細胞和分子機制尚不清楚,需要進一步研究。
圖3
KLF4與Aβ42介導的神經炎癥
圖4
KLF4在AD中的抗凋亡作用和軸突生長
圖5
KLF4與鐵代謝的關系
1.3 KLF4與缺血性腦卒中
缺血性腦卒中是指因腦部血液循環障礙,缺血、缺氧所致的局限性腦組織的缺血性壞死或軟化,進而導致腦進行性梗死和神經功能受損,是全世界致殘、致死的主要原因之一[27]。腦梗死后遺留的偏癱、失語、認知障礙等癥狀給家庭和社會帶來沉重的負擔,雖然目前大多數腦卒中患者可獲得溶栓和血管內血栓切除術等有效治療方法,但效果仍然欠佳[28],因此對腦梗死發病機制的深入研究至關重要。缺血性腦卒中病理過程在腦動脈閉合后數秒鐘至數分鐘觸發,腦細胞缺血、缺氧導致細胞內乳酸增加,毒性興奮性神經遞質增加,自由基產生,細胞發生凋亡、水腫。
炎癥過程已被證明參與動脈粥樣硬化的過程以及腦血管疾病的發病機制,腦缺血損傷后產生的炎癥介質觸發的復雜信號級聯反應也決定著腦卒中結局[29]。KLF4富集于內皮細胞中且抑制炎癥信號的激活,在內皮細胞中過表達可誘導多種內皮因子,包括內皮型一氧化氮合酶(eNOS)和血栓調節蛋白(Thbd),起到抗炎和抗血栓的作用;KLF4也可抑制腫瘤壞死因子-α(TNF-α)誘導的血管細胞粘附分子1 (VCAM-1)的表達,起到抑制動脈炎癥,調節血管損傷后新內膜形成的作用[30](圖6)。重要的是,KLF4可以保護腦微血管內皮細胞免受缺血性卒中誘導的凋亡。KLF4對血腦屏障的完整性具有保護作用可顯著減輕血腦屏障滲漏,其是通過上調核因子紅細胞系2相關因子2(Nuclear Factor erythroid2-Related Factor 2,Nrf2)/硫氧還蛋白1(Thioredoxin1,Trx1)通路,提供血腦屏障完整性的神經保護作用。Nrf2是抗氧化系統的關鍵分子,可以啟動抗氧化酶的表達,抑制氧化應激[31]。KLF4的神經保護作用也可能與缺血性腦卒中急性期氧化應激抑制有關。在缺血性卒中急性期,線粒體功能障礙、炎癥級聯和能量代謝紊亂會導致腦梗死、神經功能損傷和血腦屏障損傷等,這使得多種自發保護機制立即被激活以維持細胞內穩態。KLF4在腦缺血損傷后被發現上調,KLF4通過改善缺血性卒中后血管內皮炎癥和調節緊密連接蛋白表達來減輕腦血管損傷,表明KLF4對缺血性腦損傷具有血管保護作用[30]。此外,KLF4可能是MALA T1啟動子的一個結合位點,其上調直接增加MALA T1的表達,通過減少炎癥反應和細胞凋亡來激活自發保護機制,MALA T1是KLF4在腦缺血損傷后發揮保護作用的轉錄靶點[32]。除了腦血管內皮細胞外,還有大量反應性星形膠質細胞表達KLF4, 腦卒中發生后,星形膠質細胞立即被激活。經典激活的星形膠質細胞(A1亞型)通過釋放促炎介質發揮神經毒性作用,而選擇性激活的星形膠質細胞(A2亞型)通過分泌抗炎介質發揮神經保護作用。C3和S100A10被認為是A1和A2星形膠質細胞的標記物,增強的KLF4可以抑制腦缺血后A1星形膠質細胞C3的表達,促進A2型星形膠質細胞的活化[33](圖7)。KLF4可抑制腦缺血后星形膠質細胞的神經毒性極化,促進其神經保護性極化。急性缺血性腦卒中患者血清中KLF4水平與梗死面積呈負相關,這表明KLF4水平可以反映缺血性腦卒中的嚴重程度且在發病機制中發揮著保護作用,循環KLF4可能作為預測急性缺血性腦卒中預后的潛在生物標志物。綜上所述,在缺血性腦卒中中,KLF4通過減少梗死面積,抑制氧化應激,恢復血腦屏障功能和促進神經長期恢復等功能來起到神經保護作用。然而,還需要進一步的研究來闡明KLF4在缺血性腦卒中的具體分子機制,更好地了解KLF4在這一過程中所起的作用可以為未來缺血性腦卒中的預防和治療提供了新的概念證明和潛在靶點。
圖6
KLF4↑在內皮細胞中的抗炎作用通路
圖7
KLF4↑參與缺血性腦卒中的作用
2 小結與展望
文章對KLF4在神經系統疾病中可能的作用機制和途徑進行了綜述,涉及到多種分子及信號通路共同調控,但其具體調控機制尚不能完全闡明。KLF4在不同的細胞環境及細胞類型中又通過不同的通路和分子機制發揮著同種或者相反的作用,因而是否可以通過KLF4而闡明相關疾病的內部關聯,這些問題都將有待進一步研究。隨著對KLF4越來越多的研究,證實了其在神經系統的發育階段和相關疾病的調節中具有重要意義,KLF4有望成為特定的生物標志物或治療的潛在干預靶點,更具體的機制及靶點將進一步明確,未來通過KLF4對于神經系統疾病的預防及治療或將成為一種可行的方案。
利益沖突聲明 所有作者無利益沖突。