引用本文: 連子碩, 張曉莉, 李肖, 郭芪良, 賈天明. PPM1D基因突變致Jansen-de Vries綜合征一例并文獻復習. 癲癇雜志, 2024, 10(4): 353-359. doi: 10.7507/2096-0247.202405008 復制
版權信息: ?四川大學華西醫院華西期刊社《癲癇雜志》版權所有,未經授權不得轉載、改編
Jansen-de Vries綜合征又稱為“具有胃腸功能紊亂和高疼痛閾值的智力發育障礙”(Intellectual developmental disorder with gastrointestinal difficulties and high pain threshold,IDDGIP)[(OMIM):617450],是一種由PPM1D基因突變導致多系統受累的常染色體顯性遺傳(Autosomal dominant,AD)或常染色體隱性遺傳(Autosomal recessive,AR)的罕見疾病,由Jansen等[1]于2017年首先報道,主要臨床特征:嘴寬、薄上唇紅、寬前額、后旋耳、低位耳、鼻孔前傾、斜視、遠視、強迫行為、自閉癥行為/孤獨癥樣行為、焦慮、說話和語言發育延遲、短指畸形綜合征、智力障礙、全面性發育遲緩、全身性肌張力減退、短足、小甲、嘔吐、便秘、胃食管反流、寬基步態、脊柱前凸過度、身材矮小、注意力缺陷多動癥、喂養困難、小手、先天性心臟發育畸形、肝腫大、左腹股溝疝、肌張力減退、高疼痛閾值、對聲音的超敏反應、隱睪、小生殖器、喉軟骨發育不良、少見的指甲發育不良和反復感染等相關表型[1,2]。
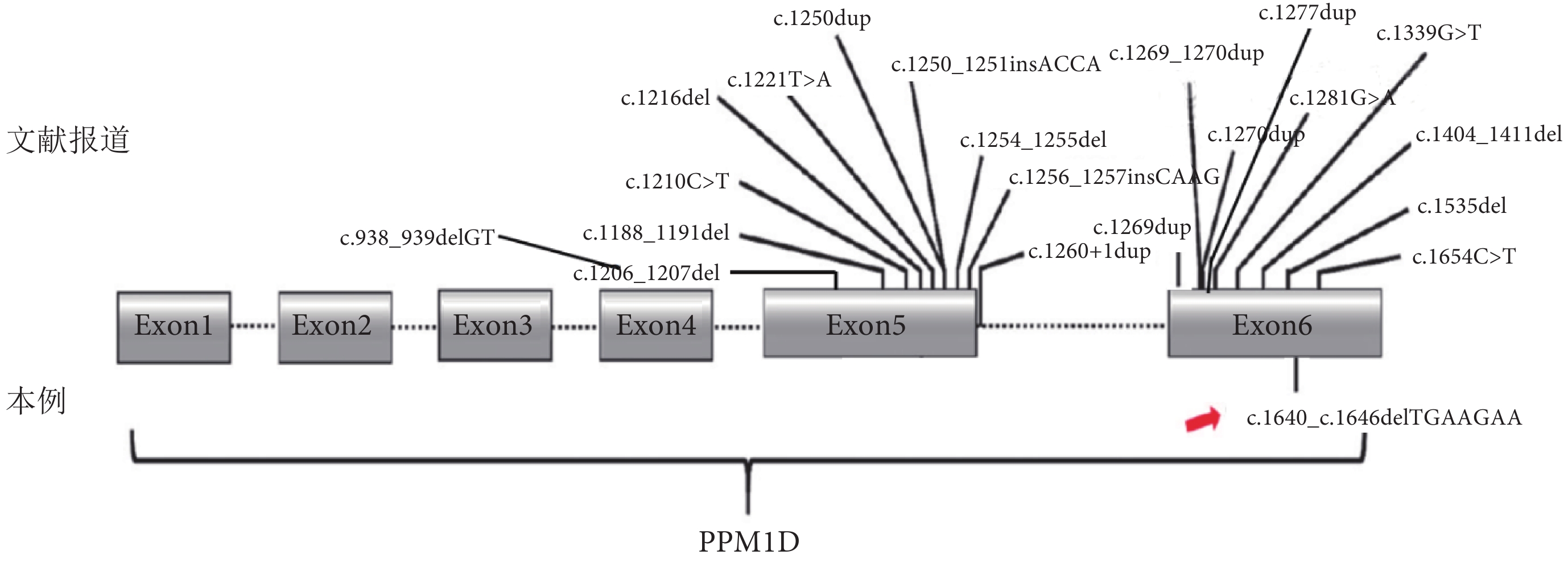
PPM1D基因定位于人染色體17q23上,編碼p53誘導的蛋白磷酸酶1(Wild-type p53-indduced phosphatase 1,Wip 1)[OMIM:605100],該蛋白磷酸酶屬于蛋白磷酸酶2C家族(Type 2C protein phosphatase,PP2C),包含605個氨基酸,分子量大約為66 000。PPM1D編碼的mRNA在很多器官及組織上表達,發揮著調節細胞周期、抑制細胞凋亡、抑制DNA修復和抑制炎癥反應等功能[1-5]。鄭州大學第三附屬醫院于2022年10月收治1例4歲10月齡女性Jansen-de Vries綜合征患兒,基因檢測結果提示患兒為PPM1D基因c.1640(exon6)_c.1646(exon6)delTGAAGAA,p.M547fs*7雜合移碼突變,根據美國醫學遺傳學與基因組學學會(American College of Medical Genetics and Genomics,ACMG)指南(2019年)[6],生物學致病評級為致病(Pathogenic):PVS1_Strong+PS2+PM2_Supporting。檢索文獻發現,國內外共報道23例PPM1D基因導致Jansen-de Vries綜合征中外文文獻病例[1,2,7-12],其中僅1例合并癲癇發作,其變異位點為[c.938(exon4)_939(exon4)delGT,p.S313Rfs*28][7];國內僅報道4例[2,10,12],致病性位點分別是[c.1216(exon5)del,p. Thr406Profs*3][2],[c.1254(exon5)_1255(exon5)del,p. Val419Glnfs*14][10],[c.1277(exon6)dupC,p.Trp427MetfsTer7][12],[c.1269(exon6)dupG,p.Glu424GlyfsTer10][12]。本例的突變位點[c.1640(exon6)_c.1646(exon6)delTGAAGAA(NM_003620),p.M547fs*7]為新突變(novel mutation),是首次報道,其臨床表現并未或尚未出現胃腸功能紊亂和高疼痛閾值等表型且合并有癲癇發作,現對其臨床資料進行總結分析。
病例資料 患兒 女,4歲10月齡。因“間斷抽搐4年1個月余并全面性發育遲緩” 于2022年10月12日就診于我院。患兒為G3P2,足月順產,出生體重3 050 g,出生后無缺血缺氧、病理性黃疸等,母孕期體健。患兒發育持續落后,3月齡抬頭不穩,6月齡不能獨坐,7月齡不能翻身,9月齡不會爬行,2歲會走、會叫“爸爸媽媽”,說話和語言發育較同齡人落后,未做過量表評價。4年1個月余(9月齡)前無明顯誘因出現抽搐發作,表現為:意識喪失,雙眼上翻,雙手握拳,四肢強直抖動,無口唇發紺、大小便失禁等,持續約1 min緩解,緩解后入睡,遂至當地醫院就診,查腦電圖、頭顱核磁共振均未見明顯異常(未見報告單),給予口服苯巴比妥片(具體劑量不詳)。后每于發熱時出現抽搐發作(體溫波動于37.5~39°C),表現同前,共抽搐8次。3年10個月余前至某三甲醫院醫院就診,腦電圖示:正常兒童腦電圖,查全外顯子家系測序(trio-Whole exome sequencing,trio-WES):無確定致病基因,囑繼續口服苯巴比妥片,患兒未再出現抽搐。1年7個月余前來我院就診,停用苯巴比妥片,給予丙戊酸鈉口服液5 mL/次 ,2次/d,定期于我院門診復診,未再出現抽搐發作,目前仍在服用丙戊酸鈉口服液。本研究為回顧性病例研究,已征得患兒家長知情同意并簽署知情同意書。
家族史:父母體健,否認近親結婚,患兒姑奶(父親的姑姑)幼時有1次抽搐病史,未處理,后未在發作,智力正常。余可追溯的家族中無認知障礙、行為缺陷、不明原因夭亡史。
體格檢查:身高115 cm(90~97th),體重21 kg(75~90th),體格檢查配合,回避眼神交流,能聽從簡單指令,可以語言表達基本需求,語流慢且邏輯性、連貫性差,能正確使用你我人稱代詞,無無意識多動、刻板性行為等。前額寬闊、鼻梁低平(圖1),無耳廓外旋,低位耳;雙手指及足趾長短均正常,無小手、短腳等,心、肺聽診無異常,腹部平坦,無臍疝,腸鳴音正常。
 圖1
患兒發育情況
圖1
患兒發育情況
a. 前額寬闊、鼻梁低平;b. 無耳廓外旋,低位耳;c,d. 雙手指及足趾長短均正常,無小手、短腳等
專科檢查:神志清,精神可,走路不穩,不會單腳跳,運動能力差,自我描述欠佳,玩耍時不合群,但孤獨癥表現不明顯,不能復述10字節句子,理解及表達能力落后,語言能力差,手部精細動作及雙手協調配合能力差,觀察能力及空間視覺感知力欠佳,推理能力差,疼痛閾值及強刺激后痛覺潛伏期均為正常水平,雙側瞳孔等大等圓、對光反射靈敏,雙側眼球活動正常,四肢肌力5級,肌張力正常,雙上肢腱反射可引出,雙側膝腱反射正常,雙側跟腱反射正常,腹壁反射可引出,雙側巴氏征陰性,腦膜刺激征陰性。
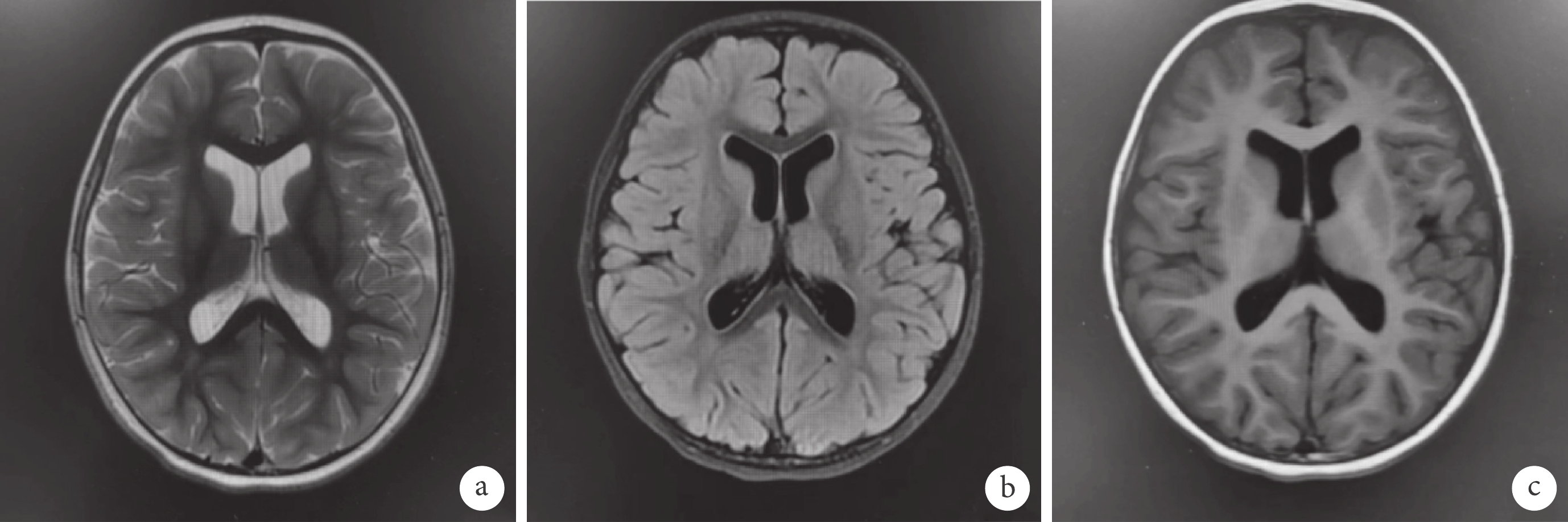
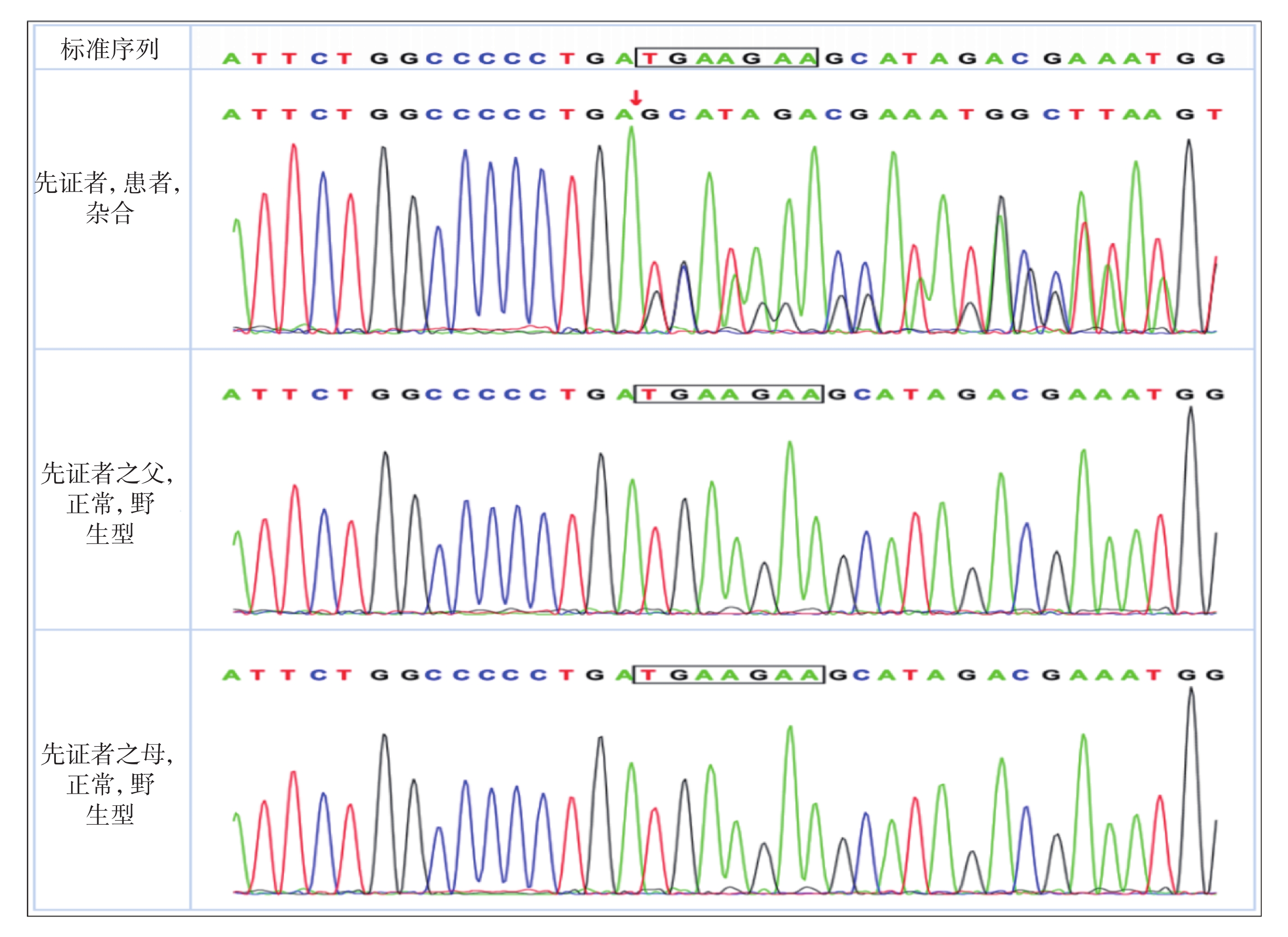
輔助檢查:血生化、血氨、血乳酸、同型半胱氨酸、銅藍蛋白、甲狀腺功能、血、尿遺傳代謝篩查均無異常。2歲7月齡頭顱磁共振成像(Magnetic resonance imaging,MRI)示:① 雙側顳前極蛛網膜下腔增寬;② 雙側側腦室擴張;③ 雙側額葉內點狀脫髓鞘首先考慮;④ 提示腺樣體肥大(圖2)。4歲10月齡Griffiths神經發育評分示:運動發育相當于3歲0.5月齡,百分位為<1%;個人與社會發育相當于3歲9.5月齡,百分位為7.5%;語言發育相當于3歲11.5月齡,百分位為10%;手眼協調發育相當于3歲9.5月齡,百分位為2.5%;表現發育相當于4歲0.5月齡,百分位為10%;推理發育相當于3歲9月齡,百分位為5%。4歲10月齡長程視頻腦電圖(Video-electroencephalography,VEEG):界線兒童腦電圖:睡眠期雙側中央及中線區少量不典型棘波散發。復測基因檢測:經患兒父母知情同意后,對先證者及其父母進行trio-WES,并采用一代Sanger測序對可疑變異進行家系驗證。基因檢測結果顯示患者在PPM1D基因的6號外顯子上[c.1640_c.1646delTGAAGAA(NM_003620),p.M547fs*7]發生移碼雜合變異,患兒父親、母親經一代Sanger測序檢測,該位點表現為野生型(圖3),根據2019年美國醫學遺傳學與基因組學學會(American College of Medical Genetics and Genomics,ACMG)《ACMG遺傳變異分類標準與指南》[6],生物學致病評級為致病(Pathogenic):PVS1_Strong+PS2+PM2_Supporting。強致病證據PVS1_Strong:已確定LOF是疾病致病機制的基因上的Null變異移碼導致蛋白功能缺失,受累區域為蛋白功能關鍵區域或超過蛋白總長的10%,逃脫NMD;強致病證據PS2:已在1個或多個表型相關且經親緣關系確定的家系中檢測到該變異為新發突變(de novo mutation),達到2≤PS2-Case_Score<4;支持致病證據PM2_Supporting:正常人群數據庫頻率<0.0005(AD/XL)或<0.001(AR)。先證者為雜合denovo突變,符合AD疾病發病機制,先證者符合與其家系成員表型及基因型的共分離。經過WES數據分析表明,尚未發現其他與患者臨床表型相關的變異基因。
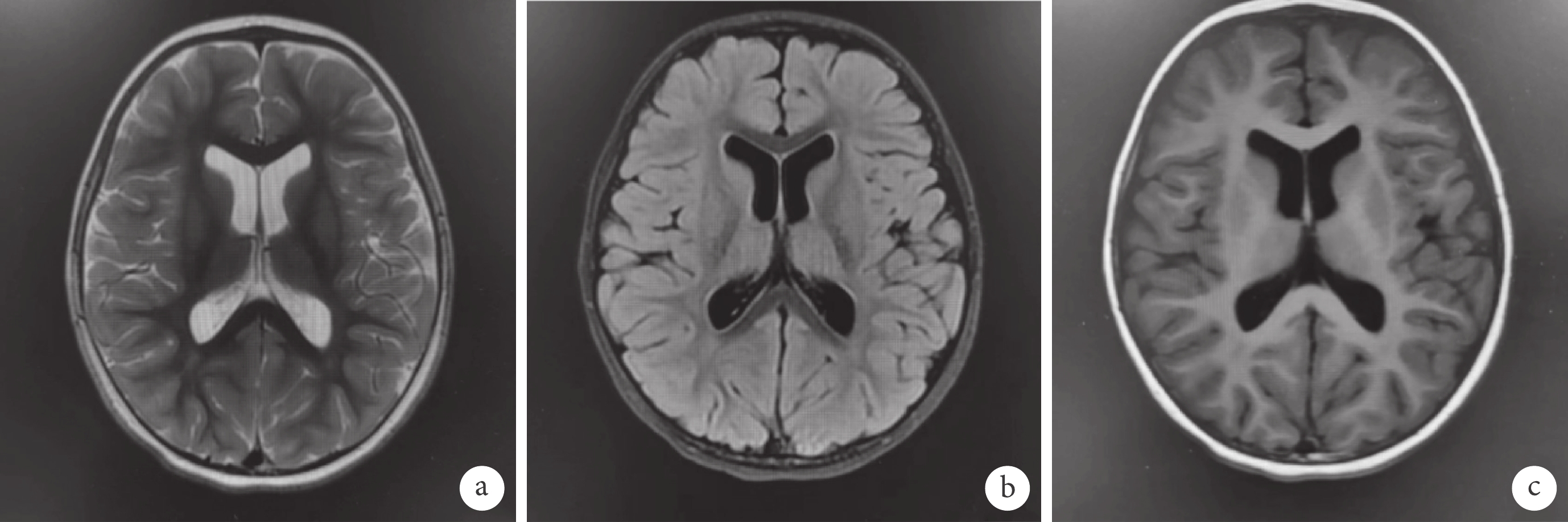 圖2
患兒的頭顱MRI影像學表現
圖2
患兒的頭顱MRI影像學表現
雙側顳前極蛛網膜下腔增寬,左側顳前極較寬處約12.3 mm,雙側側腦室擴張,三、四腦室不大,雙側額葉內可見少許稍長T2信號、在壓水T2像(b)呈稍高信號,腦中線結構居中,幕下小腦及腦干機構可;垂體、視交叉及雙側聽神經干未見明顯異常。鼻咽頂后壁軟組織厚約13.8 mm(a. T2WI; b. T2flair;c. T1WI)
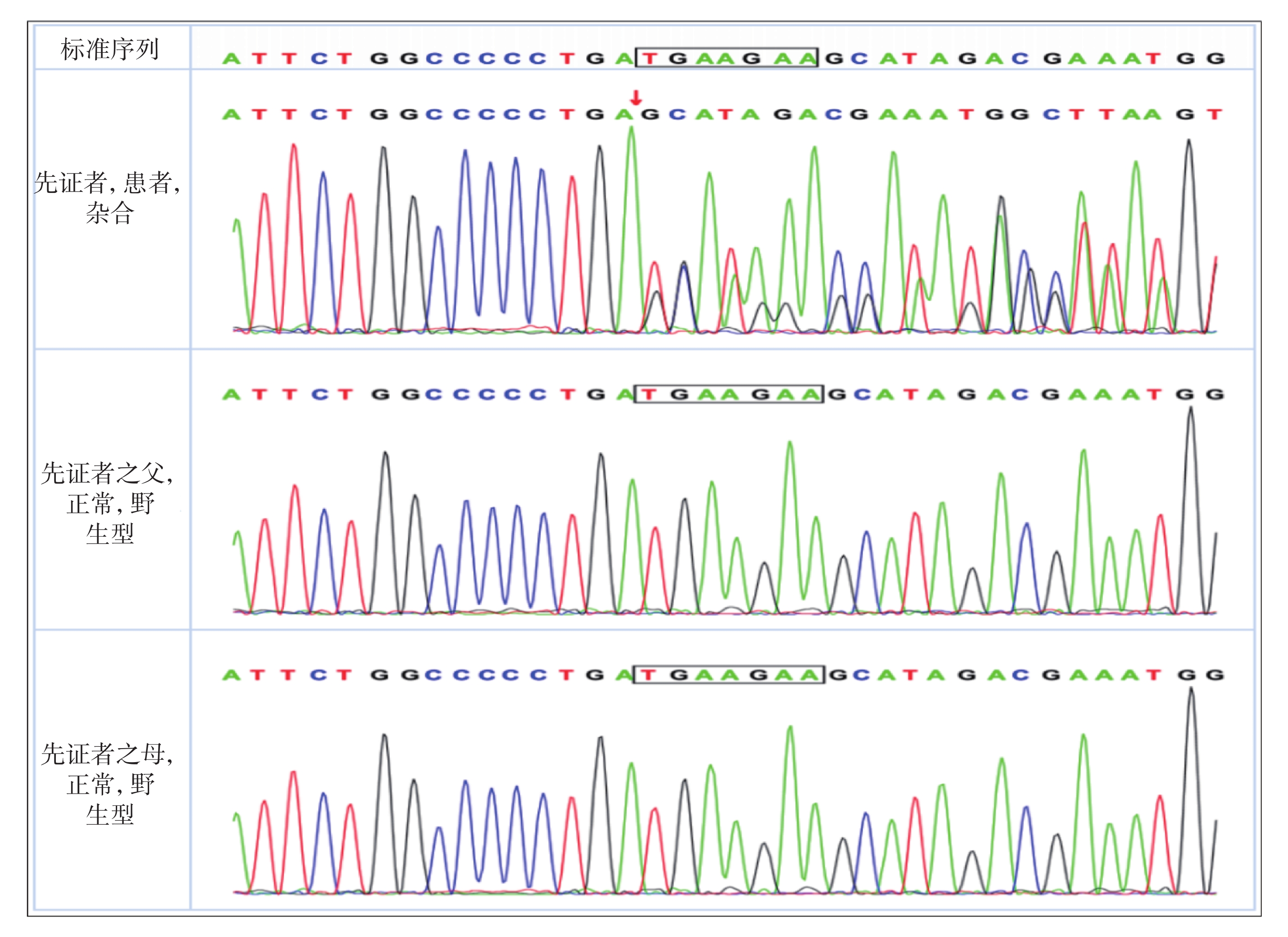 圖3
基因測序結果
圖3
基因測序結果
先證者
討論 Jansen-de Vries綜合征又稱為“具有胃腸功能紊亂和高疼痛閾值的智力發育障礙”,是一種異常罕見的由PPM1D基因突變導致多系統受累的AD或AR神經發育障礙性疾病[1,2,7,14-16],已確診患者的PPM1D基因突變全都位于4號、5號或6號外顯子之間,其臨床特征如前文所述,在所有表型中最核心的癥狀是智力低下。至今為止,Jansen-de Vries綜合征準確的發病率尚不清楚。
PPM1D基因定位于人染色體17q23上,有6個外顯子,基因突變全都位于4號、5號或6號外顯子之間,該基因編碼p53誘導的蛋白磷酸酶1(Wip 1,也稱為PPM1D)[OMIM:605100]。該蛋白磷酸酶屬于絲/蘇氨酸蛋白磷酸酶中的PPM家族蛋白磷酸酶(金屬離子依賴性蛋白磷酸酶,metal-dependent protein phosphatases,也稱為 PP2C家族蛋白磷酸酶),是一種Mg2+/Mn2+依賴性1D的絲氨酸/蘇氨酸磷酸酶,具有致癌活性,包含605個氨基酸,分子量大約為66 000[1,2,4]。PPM1D在腫瘤發生和DNA損傷反應中起到重要作用,也是全部異染色質基因沉默的重要調節劑,對維持基因組完整性至關重要。獲得性PPM1D基因突變已在卵巢透明細胞癌、神經母細胞瘤、髓母細胞瘤、乳腺癌、肝細胞癌、前列腺癌、膀胱尿路上皮癌、胰腺癌、甲狀腺癌、胃癌、結直腸癌和肺癌患者中被發現,這些研究表明PPM1D與腫瘤的發生發展密切相關,在正常情況下,暴露于電離輻射(Ionising radiation,IR)會導致p53水平的上調和組蛋白H2A的變體(H2A histone family member X,H2AX)的磷酸化,而PPM1D突變時此分子反應則會被抑制[1,17]。PPM1D磷酸蛋白酶則通過去磷酸化這些蛋白抑制p53和其他腫瘤抑制因子(p38、ATM、Chk1和Chk2),從而調節DNA損傷反應(DNA-damage response,DDR)途徑,有助于抑制應激和細胞生長,并誘導細胞凋亡。PPM1D磷酸蛋白酶參與細胞周期過程,協助信號轉導、基因轉錄、蛋白翻譯以及翻譯后修飾[1]。相關文獻認為,PPM1D基因變異會減少相關磷酸酶的合成,影響細胞增殖活性或mRNA降解,從而通過減少細胞增殖而導致Jansen-de Vries綜合征[1,2,18]。本例患兒為PPM1D新發移碼突變,基因突變位置在6號外顯子上,與相關文獻報道相吻合[1,8,12]。
分別以“PPM1D基因”、“Jansen-de Vries綜合征”、“具有胃腸功能紊亂和高疼痛閾值的智力發育障礙”為關鍵詞查閱萬方數據知識服務平臺(萬方數據庫)、中國知網(CNKI數據庫)建庫至2022年11月相關文獻,共檢索到1篇1例關于Jansen-de Vries綜合征患者的中文文獻報道(2021年梅道啟等[2]);分別以“PPM1D”、“Jansen-de Vries syndrome”為關鍵詞查閱OMIM和Pubmed數據庫、Web of Science、人類基因突變數據庫(HGMD)、在線人類孟德爾遺傳數據庫(OMIM)建庫至2022年11月相關文獻,共檢索到7篇22例報道關于Jansen-de Vries綜合征患者的外文文獻(2017年Jansen等[1],2018年Almaghthawi等[7],2018年Porrmann等[8],2019年Kuroda等[9],2020年Li等[10],2021年Martin等[11],2022年Tsai等[12]),全部23例病例報道中21例為新發突變,1例在文獻中未聲明其基因突變類型[1],1例為遺傳性突變(父母雙方均為無癥狀雜合子攜帶者)[7]。迄今為止,國內有4例報道,突變位點分別是[c.1216(exon5)delA,p. Thr406Pro fs*3][2]、[c.1254(exon5)_1255(exon5)del,p.V419Qfs*14][10]、[c.1277(exon6)dupC,p.Trp427MetfsTer7][12]、[c.1269(exon6)dupG,p.Glu424GlyfsTer10][12]。2018年國外報道1例3歲4月齡的女孩PPM1D基因自發截短突變:c.938(exon 4)_939(exon 4)delGT,p.S313Rfs *28導致合并癲癇的Jansen-de Vries綜合征的外文文獻[7]。本例先證者的PPM1D基因雜合變體[c.1640(exon6)_c.1646(exon6)delTGAAGAA(NM_003620),p.M547fs*7]在國內外均無本位點的相關文獻報道,這是在中國人群中不同位點第5次報道的Jansen-de Vries綜合征。
PPM1D基因變異致Jansen-de Vries綜合征相關文獻報道的基因突變位點和臨床特征總結見圖4和表1。
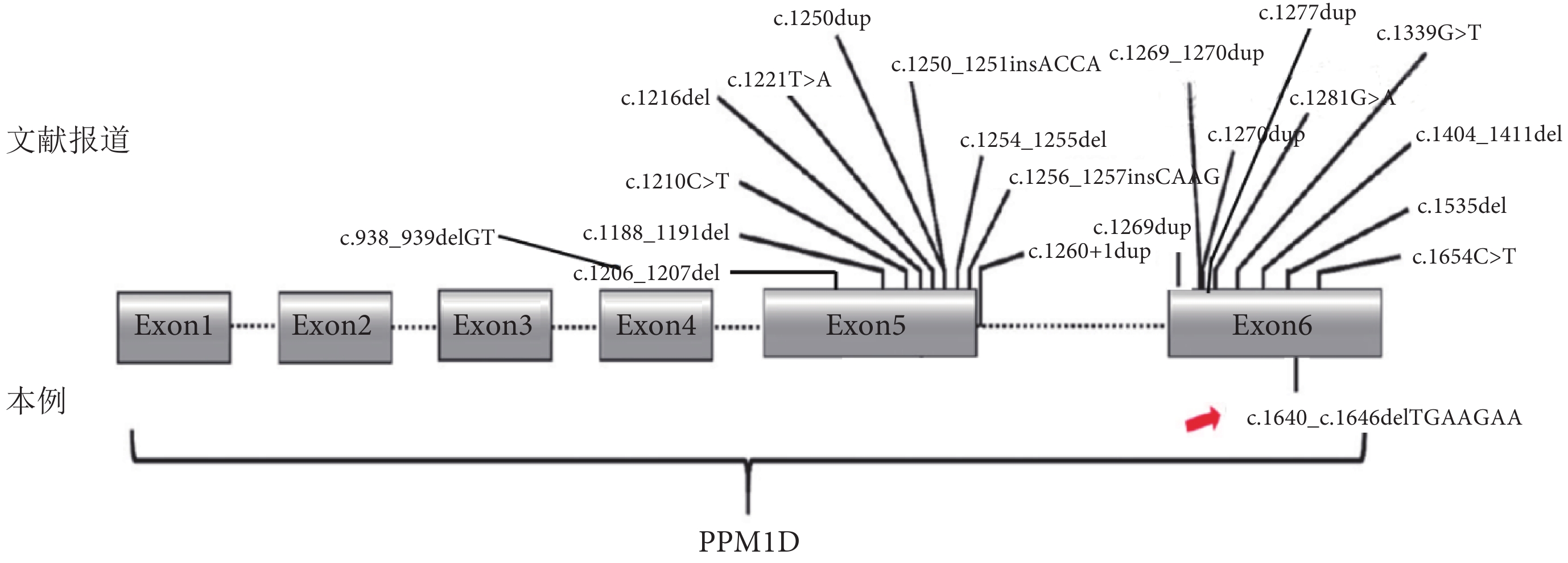 圖4
Jansen-de Vries綜合征相關文獻報道基因突變位點及本例患兒基因突變位點示意圖
圖4
Jansen-de Vries綜合征相關文獻報道基因突變位點及本例患兒基因突變位點示意圖
24例Jansen-de Vries綜合征患者中男10例、女14例,除1例外,所有患者均有輕度到重度智力障礙(95.8%),大部分患者都存在肌張力障礙[21/23(91.3%)]、小手[18/20(90.0%)]、小腳[15/17(88.2%)]、高疼痛閾值[15/17(88.2%)]、上唇薄[17/21(81.0%)]、低位后旋耳[14/18(77.8%)]、寬前額[17/22(77.3%)]、身高[17/22(77.3%)]或體重[15/23(65.2%)]低于同齡人水平、喂養困難[14/22(63.6%)]或便秘[11/18(61.1%)]等特征。值得注意的是,11例患有先天畸形[11/22(50.0%)],11例[11/24(45.8%)]對聲音極度敏感,9例[9/19(47.4%)]面部觀察到嘴寬,9例[9/23(39.1%)]外生殖器發育畸形,7例頭圍小于同齡例[7/22(31.8%)]、6例患者[6/16(37.5%)]出現反復感染的臨床表現,有6例[6/19(31.6%)]出現了如近視、遠視和斜視等視力障礙。此外,僅有1例[7]與本例患兒[2/24(8.4%)]在臨床上出現癲癇發作。本例患兒僅存在智力障礙、寬前額、癲癇發作等表型,并沒有肌張力障礙、小手、小腳、高疼痛閾值、上唇薄、低位后旋耳、身高或體重低于同齡人水平、喂養困難、便秘等特征,其對聲音敏感、視力障礙等癥狀暫不明顯,不排除年齡較小臨床表型輕微,需進一步長期隨訪。而另一例存在癲癇發作的患兒也為女性,其存在智力障礙,肌張力障礙,高疼痛閾值,喂養困難,先天畸形(小頭畸形)等表型,其所患癲癇為難治性癲癇,基因型為[c.938(exon 4)_939(exon 4)delGT,p.S313Rfs * 28][7]。本例患兒應用抗癲癇發作藥物效果則良好,基因型為[c.1640(exon6)_c.1646(exon6)delTGAAGAA(NM_003620),p.M547fs*7],因此PPM1D基因突變是否與癲癇發作相關仍需更多病例和更完備的實驗來驗證。總之,這些表明PPM1D基因突變致Jansen-de Vries綜合征患者的表型存在一定差異,需要更多的病例來補充并完善其臨床表型。
智力障礙(Intellectual disability,ID)是一種高度異質性的神經發育障礙性疾病,其遺傳學病因有染色體異常、基因突變、先天性代謝異常等,而PPM1D基因突變致Jansen-de Vries綜合征則是智力障礙的可能原因之一[14-16]。
在臨床工作中,如果觀察到智力障礙的患者(尤其是9月齡~21歲的患者)有肌張力障礙、小手、小腳、高疼痛閾值、上唇薄、低位后旋耳、寬前額、身高或體重低于同齡人水平、喂養困難或便秘等表型時,則需要高度懷疑為Jansen-de Vries綜合征,若是有先天畸形、對聲音敏感、嘴寬、外生殖器發育畸形、頭圍小于同齡人、反復感染、視力障礙、癲癇發作等特征,也需要進行Jansen-de Vries綜合征的鑒別診斷。而在診斷方面需要注意的是,盡管患者存在相同的面部特征,包括上唇薄、低位后旋耳、寬前額和寬嘴,但并未觀察到明顯一致的面容[1],所以并不能用特殊面容來診斷。且神經影像學及腦電圖多無異常發現,Jansen等[1]進行多中心回顧性研究,發現患者頭顱MRI正常率達64%,而國內外文獻均無統計腦電圖正常率的相關回顧性研究。查本例患兒頭顱MRI示:雙側顳前極蛛網膜下腔增寬;雙側側腦室擴張;提示雙側額葉內點狀脫髓鞘,為非特異性結果;長程VEEG示:界線兒童腦電圖:睡眠期雙側中央及中線區少量不典型棘波散發,行基因檢測后方確診Jansen-de Vries綜合征。因此,神經影像學及腦電圖檢查對診斷本病的作用也較小,而基因檢測是最重要的診斷方法。
Jansen-de Vries綜合征與黏多糖貯積癥臨床特點具有相似性:都具有額頭寬大、鼻梁低平和發育不良等表型[19],但患兒腦部MRI檢查及相關溶酶體酶值均正常,且父母體健,無近親關系,說明這些是Jansen-de Vries綜合征的臨床表型。患兒也符合部分Dravet綜合征[20](Dravet syndrome,DS,原稱嬰兒嚴重肌陣攣性癲癇)的特征:有熱性驚厥和癲癇家族史,1歲以內起病,病初腦電圖正常,存在精神運動發育遲緩,特別是語言發育遲緩。但患兒發病前智力運動發育便落后,首次發作非發熱所誘發,起病后發作形式固定,行為障礙和認知能力下降并非是漸進性的,近期腦電圖表現為界限腦電圖,抗癲癇發作藥物較理想,患兒基因檢測到PPM1D基因突變,而80%的DS患兒為SCN1A基因的突變,少數患者為PCDH19、GABRG2、GABRA1以及SCN2A等基因的突變,說明患兒所患疾病為Jansen-de Vries綜合征。
值得注意的是,該患兒有向熱性感染相關癲癇綜合征(Febrile infection related epilepsy syndrome,FIRES)表型轉變的風險[21],目前患兒存在部分相似表型:癲癇發作前出現非特異性發熱,癲癇發作形式為全身性癲癇發作,癲癇發作時伴有意識障礙,智力障礙。FIRES是發作于3~15歲的嚴重性癲癇綜合征,患者會出現非特異性發熱性疾病,通常在癲癇發作前1~14 d消失,癲癇發作后會迅速發展為癲癇持續狀態,或變得非常頻繁,每天癲癇發作數十次至數百次,并伴有意識喪失。經研究發現,65%的患者存在全身性癲癇發作,觀察到所有存活患者(n = 6)在初次發病5~17年后出現慢性難治性癲癇[21]。經統計,18%的患者認知水平正常,16%處于邊緣狀態,14%患有輕度ID,24%患有中度ID,12%患有重度ID,16%處于植物人狀態。FIRES的病因尚不清楚,有理論認為,免疫來源、遺傳傾向和炎癥介導過程可能是引起FIRES的原因。據報道,只有少數病例沒有嚴重的后遺癥,66%~100%的患者出現難治性癲癇和彌漫性腦病伴ID,高達30%的患者死亡[21]。所以臨床醫生應注意到患兒向FIRES表型轉變的風險,長期隨訪,避免嚴重后果的發生。
目前,癌癥患者的PPM1D基因突變均為獲得性突變,癌癥個體的PPM1D遺傳性突變尚未被報道[1]。現又已知一些基因的遺傳性突變會導致智力障礙,而獲得性突變會導致癌癥,例如編碼RAS-MAPK通路組成部分的基因、SETBP1[SET結合蛋白1(MIM:611060)]和CTNNB1[連環蛋白β1(MIM:116806)],且這些基因的遺傳性突變有些會增加患癌癥的風險,有些則不會。一些基因的遺傳性突變則會導致智力障礙及增高患良性腫瘤的風險,如NF1[神經纖維蛋白1(MIM:613113)][1]。且本研究中24例PPM1D突變的患者[1,2,7-12]均沒有發展為良性腫瘤或癌癥。因此,不能排除或證實智力障礙患者的PPM1D突變易患良性腫瘤或癌癥的可能性,即Jansen-de Vries綜合征患者患良性腫瘤或癌癥的風險不能確定。
目前患兒診斷Jansen-de Vries綜合征明確,但目前尚無有效的方法,現主要以加強康復與功能訓練為主,盡量改善智力、語言及運動發育情況,及時隨訪。回顧該患兒治療管理過程,隨年齡增長,運動和認知緩慢進步,癲癇發作控制較為順利。
利益沖突聲明 所有作者無利益沖突。
Jansen-de Vries綜合征又稱為“具有胃腸功能紊亂和高疼痛閾值的智力發育障礙”(Intellectual developmental disorder with gastrointestinal difficulties and high pain threshold,IDDGIP)[(OMIM):617450],是一種由PPM1D基因突變導致多系統受累的常染色體顯性遺傳(Autosomal dominant,AD)或常染色體隱性遺傳(Autosomal recessive,AR)的罕見疾病,由Jansen等[1]于2017年首先報道,主要臨床特征:嘴寬、薄上唇紅、寬前額、后旋耳、低位耳、鼻孔前傾、斜視、遠視、強迫行為、自閉癥行為/孤獨癥樣行為、焦慮、說話和語言發育延遲、短指畸形綜合征、智力障礙、全面性發育遲緩、全身性肌張力減退、短足、小甲、嘔吐、便秘、胃食管反流、寬基步態、脊柱前凸過度、身材矮小、注意力缺陷多動癥、喂養困難、小手、先天性心臟發育畸形、肝腫大、左腹股溝疝、肌張力減退、高疼痛閾值、對聲音的超敏反應、隱睪、小生殖器、喉軟骨發育不良、少見的指甲發育不良和反復感染等相關表型[1,2]。
PPM1D基因定位于人染色體17q23上,編碼p53誘導的蛋白磷酸酶1(Wild-type p53-indduced phosphatase 1,Wip 1)[OMIM:605100],該蛋白磷酸酶屬于蛋白磷酸酶2C家族(Type 2C protein phosphatase,PP2C),包含605個氨基酸,分子量大約為66 000。PPM1D編碼的mRNA在很多器官及組織上表達,發揮著調節細胞周期、抑制細胞凋亡、抑制DNA修復和抑制炎癥反應等功能[1-5]。鄭州大學第三附屬醫院于2022年10月收治1例4歲10月齡女性Jansen-de Vries綜合征患兒,基因檢測結果提示患兒為PPM1D基因c.1640(exon6)_c.1646(exon6)delTGAAGAA,p.M547fs*7雜合移碼突變,根據美國醫學遺傳學與基因組學學會(American College of Medical Genetics and Genomics,ACMG)指南(2019年)[6],生物學致病評級為致病(Pathogenic):PVS1_Strong+PS2+PM2_Supporting。檢索文獻發現,國內外共報道23例PPM1D基因導致Jansen-de Vries綜合征中外文文獻病例[1,2,7-12],其中僅1例合并癲癇發作,其變異位點為[c.938(exon4)_939(exon4)delGT,p.S313Rfs*28][7];國內僅報道4例[2,10,12],致病性位點分別是[c.1216(exon5)del,p. Thr406Profs*3][2],[c.1254(exon5)_1255(exon5)del,p. Val419Glnfs*14][10],[c.1277(exon6)dupC,p.Trp427MetfsTer7][12],[c.1269(exon6)dupG,p.Glu424GlyfsTer10][12]。本例的突變位點[c.1640(exon6)_c.1646(exon6)delTGAAGAA(NM_003620),p.M547fs*7]為新突變(novel mutation),是首次報道,其臨床表現并未或尚未出現胃腸功能紊亂和高疼痛閾值等表型且合并有癲癇發作,現對其臨床資料進行總結分析。
病例資料 患兒 女,4歲10月齡。因“間斷抽搐4年1個月余并全面性發育遲緩” 于2022年10月12日就診于我院。患兒為G3P2,足月順產,出生體重3 050 g,出生后無缺血缺氧、病理性黃疸等,母孕期體健。患兒發育持續落后,3月齡抬頭不穩,6月齡不能獨坐,7月齡不能翻身,9月齡不會爬行,2歲會走、會叫“爸爸媽媽”,說話和語言發育較同齡人落后,未做過量表評價。4年1個月余(9月齡)前無明顯誘因出現抽搐發作,表現為:意識喪失,雙眼上翻,雙手握拳,四肢強直抖動,無口唇發紺、大小便失禁等,持續約1 min緩解,緩解后入睡,遂至當地醫院就診,查腦電圖、頭顱核磁共振均未見明顯異常(未見報告單),給予口服苯巴比妥片(具體劑量不詳)。后每于發熱時出現抽搐發作(體溫波動于37.5~39°C),表現同前,共抽搐8次。3年10個月余前至某三甲醫院醫院就診,腦電圖示:正常兒童腦電圖,查全外顯子家系測序(trio-Whole exome sequencing,trio-WES):無確定致病基因,囑繼續口服苯巴比妥片,患兒未再出現抽搐。1年7個月余前來我院就診,停用苯巴比妥片,給予丙戊酸鈉口服液5 mL/次 ,2次/d,定期于我院門診復診,未再出現抽搐發作,目前仍在服用丙戊酸鈉口服液。本研究為回顧性病例研究,已征得患兒家長知情同意并簽署知情同意書。
家族史:父母體健,否認近親結婚,患兒姑奶(父親的姑姑)幼時有1次抽搐病史,未處理,后未在發作,智力正常。余可追溯的家族中無認知障礙、行為缺陷、不明原因夭亡史。
體格檢查:身高115 cm(90~97th),體重21 kg(75~90th),體格檢查配合,回避眼神交流,能聽從簡單指令,可以語言表達基本需求,語流慢且邏輯性、連貫性差,能正確使用你我人稱代詞,無無意識多動、刻板性行為等。前額寬闊、鼻梁低平(圖1),無耳廓外旋,低位耳;雙手指及足趾長短均正常,無小手、短腳等,心、肺聽診無異常,腹部平坦,無臍疝,腸鳴音正常。
圖1
患兒發育情況
a. 前額寬闊、鼻梁低平;b. 無耳廓外旋,低位耳;c,d. 雙手指及足趾長短均正常,無小手、短腳等
專科檢查:神志清,精神可,走路不穩,不會單腳跳,運動能力差,自我描述欠佳,玩耍時不合群,但孤獨癥表現不明顯,不能復述10字節句子,理解及表達能力落后,語言能力差,手部精細動作及雙手協調配合能力差,觀察能力及空間視覺感知力欠佳,推理能力差,疼痛閾值及強刺激后痛覺潛伏期均為正常水平,雙側瞳孔等大等圓、對光反射靈敏,雙側眼球活動正常,四肢肌力5級,肌張力正常,雙上肢腱反射可引出,雙側膝腱反射正常,雙側跟腱反射正常,腹壁反射可引出,雙側巴氏征陰性,腦膜刺激征陰性。
輔助檢查:血生化、血氨、血乳酸、同型半胱氨酸、銅藍蛋白、甲狀腺功能、血、尿遺傳代謝篩查均無異常。2歲7月齡頭顱磁共振成像(Magnetic resonance imaging,MRI)示:① 雙側顳前極蛛網膜下腔增寬;② 雙側側腦室擴張;③ 雙側額葉內點狀脫髓鞘首先考慮;④ 提示腺樣體肥大(圖2)。4歲10月齡Griffiths神經發育評分示:運動發育相當于3歲0.5月齡,百分位為<1%;個人與社會發育相當于3歲9.5月齡,百分位為7.5%;語言發育相當于3歲11.5月齡,百分位為10%;手眼協調發育相當于3歲9.5月齡,百分位為2.5%;表現發育相當于4歲0.5月齡,百分位為10%;推理發育相當于3歲9月齡,百分位為5%。4歲10月齡長程視頻腦電圖(Video-electroencephalography,VEEG):界線兒童腦電圖:睡眠期雙側中央及中線區少量不典型棘波散發。復測基因檢測:經患兒父母知情同意后,對先證者及其父母進行trio-WES,并采用一代Sanger測序對可疑變異進行家系驗證。基因檢測結果顯示患者在PPM1D基因的6號外顯子上[c.1640_c.1646delTGAAGAA(NM_003620),p.M547fs*7]發生移碼雜合變異,患兒父親、母親經一代Sanger測序檢測,該位點表現為野生型(圖3),根據2019年美國醫學遺傳學與基因組學學會(American College of Medical Genetics and Genomics,ACMG)《ACMG遺傳變異分類標準與指南》[6],生物學致病評級為致病(Pathogenic):PVS1_Strong+PS2+PM2_Supporting。強致病證據PVS1_Strong:已確定LOF是疾病致病機制的基因上的Null變異移碼導致蛋白功能缺失,受累區域為蛋白功能關鍵區域或超過蛋白總長的10%,逃脫NMD;強致病證據PS2:已在1個或多個表型相關且經親緣關系確定的家系中檢測到該變異為新發突變(de novo mutation),達到2≤PS2-Case_Score<4;支持致病證據PM2_Supporting:正常人群數據庫頻率<0.0005(AD/XL)或<0.001(AR)。先證者為雜合denovo突變,符合AD疾病發病機制,先證者符合與其家系成員表型及基因型的共分離。經過WES數據分析表明,尚未發現其他與患者臨床表型相關的變異基因。
圖2
患兒的頭顱MRI影像學表現
雙側顳前極蛛網膜下腔增寬,左側顳前極較寬處約12.3 mm,雙側側腦室擴張,三、四腦室不大,雙側額葉內可見少許稍長T2信號、在壓水T2像(b)呈稍高信號,腦中線結構居中,幕下小腦及腦干機構可;垂體、視交叉及雙側聽神經干未見明顯異常。鼻咽頂后壁軟組織厚約13.8 mm(a. T2WI; b. T2flair;c. T1WI)
圖3
基因測序結果
先證者
討論 Jansen-de Vries綜合征又稱為“具有胃腸功能紊亂和高疼痛閾值的智力發育障礙”,是一種異常罕見的由PPM1D基因突變導致多系統受累的AD或AR神經發育障礙性疾病[1,2,7,14-16],已確診患者的PPM1D基因突變全都位于4號、5號或6號外顯子之間,其臨床特征如前文所述,在所有表型中最核心的癥狀是智力低下。至今為止,Jansen-de Vries綜合征準確的發病率尚不清楚。
PPM1D基因定位于人染色體17q23上,有6個外顯子,基因突變全都位于4號、5號或6號外顯子之間,該基因編碼p53誘導的蛋白磷酸酶1(Wip 1,也稱為PPM1D)[OMIM:605100]。該蛋白磷酸酶屬于絲/蘇氨酸蛋白磷酸酶中的PPM家族蛋白磷酸酶(金屬離子依賴性蛋白磷酸酶,metal-dependent protein phosphatases,也稱為 PP2C家族蛋白磷酸酶),是一種Mg2+/Mn2+依賴性1D的絲氨酸/蘇氨酸磷酸酶,具有致癌活性,包含605個氨基酸,分子量大約為66 000[1,2,4]。PPM1D在腫瘤發生和DNA損傷反應中起到重要作用,也是全部異染色質基因沉默的重要調節劑,對維持基因組完整性至關重要。獲得性PPM1D基因突變已在卵巢透明細胞癌、神經母細胞瘤、髓母細胞瘤、乳腺癌、肝細胞癌、前列腺癌、膀胱尿路上皮癌、胰腺癌、甲狀腺癌、胃癌、結直腸癌和肺癌患者中被發現,這些研究表明PPM1D與腫瘤的發生發展密切相關,在正常情況下,暴露于電離輻射(Ionising radiation,IR)會導致p53水平的上調和組蛋白H2A的變體(H2A histone family member X,H2AX)的磷酸化,而PPM1D突變時此分子反應則會被抑制[1,17]。PPM1D磷酸蛋白酶則通過去磷酸化這些蛋白抑制p53和其他腫瘤抑制因子(p38、ATM、Chk1和Chk2),從而調節DNA損傷反應(DNA-damage response,DDR)途徑,有助于抑制應激和細胞生長,并誘導細胞凋亡。PPM1D磷酸蛋白酶參與細胞周期過程,協助信號轉導、基因轉錄、蛋白翻譯以及翻譯后修飾[1]。相關文獻認為,PPM1D基因變異會減少相關磷酸酶的合成,影響細胞增殖活性或mRNA降解,從而通過減少細胞增殖而導致Jansen-de Vries綜合征[1,2,18]。本例患兒為PPM1D新發移碼突變,基因突變位置在6號外顯子上,與相關文獻報道相吻合[1,8,12]。
分別以“PPM1D基因”、“Jansen-de Vries綜合征”、“具有胃腸功能紊亂和高疼痛閾值的智力發育障礙”為關鍵詞查閱萬方數據知識服務平臺(萬方數據庫)、中國知網(CNKI數據庫)建庫至2022年11月相關文獻,共檢索到1篇1例關于Jansen-de Vries綜合征患者的中文文獻報道(2021年梅道啟等[2]);分別以“PPM1D”、“Jansen-de Vries syndrome”為關鍵詞查閱OMIM和Pubmed數據庫、Web of Science、人類基因突變數據庫(HGMD)、在線人類孟德爾遺傳數據庫(OMIM)建庫至2022年11月相關文獻,共檢索到7篇22例報道關于Jansen-de Vries綜合征患者的外文文獻(2017年Jansen等[1],2018年Almaghthawi等[7],2018年Porrmann等[8],2019年Kuroda等[9],2020年Li等[10],2021年Martin等[11],2022年Tsai等[12]),全部23例病例報道中21例為新發突變,1例在文獻中未聲明其基因突變類型[1],1例為遺傳性突變(父母雙方均為無癥狀雜合子攜帶者)[7]。迄今為止,國內有4例報道,突變位點分別是[c.1216(exon5)delA,p. Thr406Pro fs*3][2]、[c.1254(exon5)_1255(exon5)del,p.V419Qfs*14][10]、[c.1277(exon6)dupC,p.Trp427MetfsTer7][12]、[c.1269(exon6)dupG,p.Glu424GlyfsTer10][12]。2018年國外報道1例3歲4月齡的女孩PPM1D基因自發截短突變:c.938(exon 4)_939(exon 4)delGT,p.S313Rfs *28導致合并癲癇的Jansen-de Vries綜合征的外文文獻[7]。本例先證者的PPM1D基因雜合變體[c.1640(exon6)_c.1646(exon6)delTGAAGAA(NM_003620),p.M547fs*7]在國內外均無本位點的相關文獻報道,這是在中國人群中不同位點第5次報道的Jansen-de Vries綜合征。
PPM1D基因變異致Jansen-de Vries綜合征相關文獻報道的基因突變位點和臨床特征總結見圖4和表1。
圖4
Jansen-de Vries綜合征相關文獻報道基因突變位點及本例患兒基因突變位點示意圖
24例Jansen-de Vries綜合征患者中男10例、女14例,除1例外,所有患者均有輕度到重度智力障礙(95.8%),大部分患者都存在肌張力障礙[21/23(91.3%)]、小手[18/20(90.0%)]、小腳[15/17(88.2%)]、高疼痛閾值[15/17(88.2%)]、上唇薄[17/21(81.0%)]、低位后旋耳[14/18(77.8%)]、寬前額[17/22(77.3%)]、身高[17/22(77.3%)]或體重[15/23(65.2%)]低于同齡人水平、喂養困難[14/22(63.6%)]或便秘[11/18(61.1%)]等特征。值得注意的是,11例患有先天畸形[11/22(50.0%)],11例[11/24(45.8%)]對聲音極度敏感,9例[9/19(47.4%)]面部觀察到嘴寬,9例[9/23(39.1%)]外生殖器發育畸形,7例頭圍小于同齡例[7/22(31.8%)]、6例患者[6/16(37.5%)]出現反復感染的臨床表現,有6例[6/19(31.6%)]出現了如近視、遠視和斜視等視力障礙。此外,僅有1例[7]與本例患兒[2/24(8.4%)]在臨床上出現癲癇發作。本例患兒僅存在智力障礙、寬前額、癲癇發作等表型,并沒有肌張力障礙、小手、小腳、高疼痛閾值、上唇薄、低位后旋耳、身高或體重低于同齡人水平、喂養困難、便秘等特征,其對聲音敏感、視力障礙等癥狀暫不明顯,不排除年齡較小臨床表型輕微,需進一步長期隨訪。而另一例存在癲癇發作的患兒也為女性,其存在智力障礙,肌張力障礙,高疼痛閾值,喂養困難,先天畸形(小頭畸形)等表型,其所患癲癇為難治性癲癇,基因型為[c.938(exon 4)_939(exon 4)delGT,p.S313Rfs * 28][7]。本例患兒應用抗癲癇發作藥物效果則良好,基因型為[c.1640(exon6)_c.1646(exon6)delTGAAGAA(NM_003620),p.M547fs*7],因此PPM1D基因突變是否與癲癇發作相關仍需更多病例和更完備的實驗來驗證。總之,這些表明PPM1D基因突變致Jansen-de Vries綜合征患者的表型存在一定差異,需要更多的病例來補充并完善其臨床表型。
智力障礙(Intellectual disability,ID)是一種高度異質性的神經發育障礙性疾病,其遺傳學病因有染色體異常、基因突變、先天性代謝異常等,而PPM1D基因突變致Jansen-de Vries綜合征則是智力障礙的可能原因之一[14-16]。
在臨床工作中,如果觀察到智力障礙的患者(尤其是9月齡~21歲的患者)有肌張力障礙、小手、小腳、高疼痛閾值、上唇薄、低位后旋耳、寬前額、身高或體重低于同齡人水平、喂養困難或便秘等表型時,則需要高度懷疑為Jansen-de Vries綜合征,若是有先天畸形、對聲音敏感、嘴寬、外生殖器發育畸形、頭圍小于同齡人、反復感染、視力障礙、癲癇發作等特征,也需要進行Jansen-de Vries綜合征的鑒別診斷。而在診斷方面需要注意的是,盡管患者存在相同的面部特征,包括上唇薄、低位后旋耳、寬前額和寬嘴,但并未觀察到明顯一致的面容[1],所以并不能用特殊面容來診斷。且神經影像學及腦電圖多無異常發現,Jansen等[1]進行多中心回顧性研究,發現患者頭顱MRI正常率達64%,而國內外文獻均無統計腦電圖正常率的相關回顧性研究。查本例患兒頭顱MRI示:雙側顳前極蛛網膜下腔增寬;雙側側腦室擴張;提示雙側額葉內點狀脫髓鞘,為非特異性結果;長程VEEG示:界線兒童腦電圖:睡眠期雙側中央及中線區少量不典型棘波散發,行基因檢測后方確診Jansen-de Vries綜合征。因此,神經影像學及腦電圖檢查對診斷本病的作用也較小,而基因檢測是最重要的診斷方法。
Jansen-de Vries綜合征與黏多糖貯積癥臨床特點具有相似性:都具有額頭寬大、鼻梁低平和發育不良等表型[19],但患兒腦部MRI檢查及相關溶酶體酶值均正常,且父母體健,無近親關系,說明這些是Jansen-de Vries綜合征的臨床表型。患兒也符合部分Dravet綜合征[20](Dravet syndrome,DS,原稱嬰兒嚴重肌陣攣性癲癇)的特征:有熱性驚厥和癲癇家族史,1歲以內起病,病初腦電圖正常,存在精神運動發育遲緩,特別是語言發育遲緩。但患兒發病前智力運動發育便落后,首次發作非發熱所誘發,起病后發作形式固定,行為障礙和認知能力下降并非是漸進性的,近期腦電圖表現為界限腦電圖,抗癲癇發作藥物較理想,患兒基因檢測到PPM1D基因突變,而80%的DS患兒為SCN1A基因的突變,少數患者為PCDH19、GABRG2、GABRA1以及SCN2A等基因的突變,說明患兒所患疾病為Jansen-de Vries綜合征。
值得注意的是,該患兒有向熱性感染相關癲癇綜合征(Febrile infection related epilepsy syndrome,FIRES)表型轉變的風險[21],目前患兒存在部分相似表型:癲癇發作前出現非特異性發熱,癲癇發作形式為全身性癲癇發作,癲癇發作時伴有意識障礙,智力障礙。FIRES是發作于3~15歲的嚴重性癲癇綜合征,患者會出現非特異性發熱性疾病,通常在癲癇發作前1~14 d消失,癲癇發作后會迅速發展為癲癇持續狀態,或變得非常頻繁,每天癲癇發作數十次至數百次,并伴有意識喪失。經研究發現,65%的患者存在全身性癲癇發作,觀察到所有存活患者(n = 6)在初次發病5~17年后出現慢性難治性癲癇[21]。經統計,18%的患者認知水平正常,16%處于邊緣狀態,14%患有輕度ID,24%患有中度ID,12%患有重度ID,16%處于植物人狀態。FIRES的病因尚不清楚,有理論認為,免疫來源、遺傳傾向和炎癥介導過程可能是引起FIRES的原因。據報道,只有少數病例沒有嚴重的后遺癥,66%~100%的患者出現難治性癲癇和彌漫性腦病伴ID,高達30%的患者死亡[21]。所以臨床醫生應注意到患兒向FIRES表型轉變的風險,長期隨訪,避免嚴重后果的發生。
目前,癌癥患者的PPM1D基因突變均為獲得性突變,癌癥個體的PPM1D遺傳性突變尚未被報道[1]。現又已知一些基因的遺傳性突變會導致智力障礙,而獲得性突變會導致癌癥,例如編碼RAS-MAPK通路組成部分的基因、SETBP1[SET結合蛋白1(MIM:611060)]和CTNNB1[連環蛋白β1(MIM:116806)],且這些基因的遺傳性突變有些會增加患癌癥的風險,有些則不會。一些基因的遺傳性突變則會導致智力障礙及增高患良性腫瘤的風險,如NF1[神經纖維蛋白1(MIM:613113)][1]。且本研究中24例PPM1D突變的患者[1,2,7-12]均沒有發展為良性腫瘤或癌癥。因此,不能排除或證實智力障礙患者的PPM1D突變易患良性腫瘤或癌癥的可能性,即Jansen-de Vries綜合征患者患良性腫瘤或癌癥的風險不能確定。
目前患兒診斷Jansen-de Vries綜合征明確,但目前尚無有效的方法,現主要以加強康復與功能訓練為主,盡量改善智力、語言及運動發育情況,及時隨訪。回顧該患兒治療管理過程,隨年齡增長,運動和認知緩慢進步,癲癇發作控制較為順利。
利益沖突聲明 所有作者無利益沖突。