引用本文: 鄭小紅, 范佛養, 周有峰, 胡春輝. DPAGT1基因變異相關先天性糖基化障礙一例并文獻復習. 癲癇雜志, 2024, 10(5): 454-458. doi: 10.7507/2096-0247.202406002 復制
版權信息: ?四川大學華西醫院華西期刊社《癲癇雜志》版權所有,未經授權不得轉載、改編
先天性糖基化紊亂(Congenital disorders of glycosylation ,CDG)是一組由蛋白質和脂質糖基化缺陷引起的罕見遺傳病,涉及160多種亞型,與103余種不同的基因相關[1,2]。在這些病癥中,多乙酰磷酸N-乙酰葡萄糖胺-1-磷酸轉移酶(Dolichylphosphate N-acetylglucosamine phosphotransferase 1,DPAGT1)基因的變異會引起相關的先天性糖基化障礙(DPAGT1-CDG)。該病以多系統異常為特征,包括精神運動遲滯、癲癇發作、共濟失調和視網膜病變,其中部分出現肝功能異常和凝血功能障礙。發育畸形特征包括斜視、面部異常、乳頭內陷和皮下脂肪墊[3-5]。文章報道一例福建省兒童醫院2023年11月6日入院治療的DPAGT1-CDG患兒的臨床特點,并文獻復習,以提高對該病的臨床認識。本研究經我院倫理委員會批準(2023ETKLR11006)及患兒監護人知情同意。
病例資料 患者 女,5月齡23天齡。因“反復抽搐12天”于2023年11月至福建省兒童醫院神經內科就診。患兒入院前12天無明顯誘因出現反復抽搐,具體表現為點頭擁抱樣動作,成串發作,每串發作8~9下,持續1~2 min自行緩解,緩解后精神倦怠,每天發作8~9串,偶伴吐奶,無發熱、腹瀉,無大小便失禁,為進一步診治來我院。自發病以來,精神欠佳,食納、睡眠一般。患兒系其母第 2胎第 2 產,足月剖宮產娩出,出生體重2 800 g,否認出生時缺氧窒息史。目前尚不會抬頭。父母健康,非近親婚配,姐姐18歲,體健。既往患兒生后發育遲緩,營養不良,肌無力,目前不能抬頭,平素吃奶差,容易呼吸道感染,既往4月齡3天齡時,因“嗆奶”后導致“重癥肺炎、呼吸衰竭”于我院兒科重癥監護病房(Pediatric intensive care unit,PICU)治療,治療上予氣管插管、呼吸機輔助通氣、抗感染等處理,病情好轉后出院,平素予雞尾酒療法口服改善肌無力癥狀。此次入院體格檢查:神志清楚,精神差,表情淡漠,哭聲低弱,營養不良,自主體位,消瘦外觀,皮下脂肪消失,皮膚彈性稍差。追視不佳,眼球水平震顫,直接對光反射遲鈍,間接對光反射尚可。口唇紅,咽充血,頸軟,呼吸節律不規則,吸凹征陰性,雙肺呼吸音粗,聞及少許痰鳴。心音有力律齊,腹平軟。四肢自主活動少,頸軟無力,四肢肌力Ⅲ級,肌張力低下。跟腱、膝腱反射可引出。腦膜刺激征陰性,病理征陰性。入院輔檢:血常規、腦脊液常規、生化、培養、甲功、凝血等均大致正常。血生化提示:丙氨酸氨基轉移酶149 U/L、天門冬氨酸氨基轉移酶81 U/L,均偏高。長程腦電圖提示異常嬰兒腦電圖,出現高度失律(圖1a),后頭部顯著,監測到清醒期3次成串痙攣發作(圖1b)。頭顱核磁共振成像3.0T薄層平掃提示雙側額部腦外間隙略增寬。診斷嬰兒癲癇性痙攣綜合征,入院后予托吡酯、左乙拉西坦口服抗癲癇治療,患兒抽搐好轉后出院。同時行全外顯子基因測序,出院后結果回報DPAGT1復合雜合變異:DPAGT1 c.764G>T:p.Cys255Phe雜合變異,變異來自母親,父親為野生型;DPAGT1c.315C>G:p.lle105Met雜合變異,為新發變異,父母均為野生型(圖2);根據ACMG評級、腦表達、基因多態性、基因功能、基因保守性、等位基因頻率等評估致病性,考慮為可能致病性變異,結合患兒有肌無力、癲癇發作、發育遲緩、呼吸功能不全、肝功能異常等表現,最終診斷為DPAGT1基因變異相關先天性糖基化障礙。出院后1個月至我院神經內科門診隨診,未再抽搐。但患兒肌無力癥狀未見明顯改善,考慮DPAGT1基因變異相關先天性糖基化障礙部分患兒口服溴比斯的明有效,遂加用溴比斯的明口服,逐漸加量至5 mg/(kg·d),4個月后隨診患兒肌無力癥狀較前改善,未見抽搐。
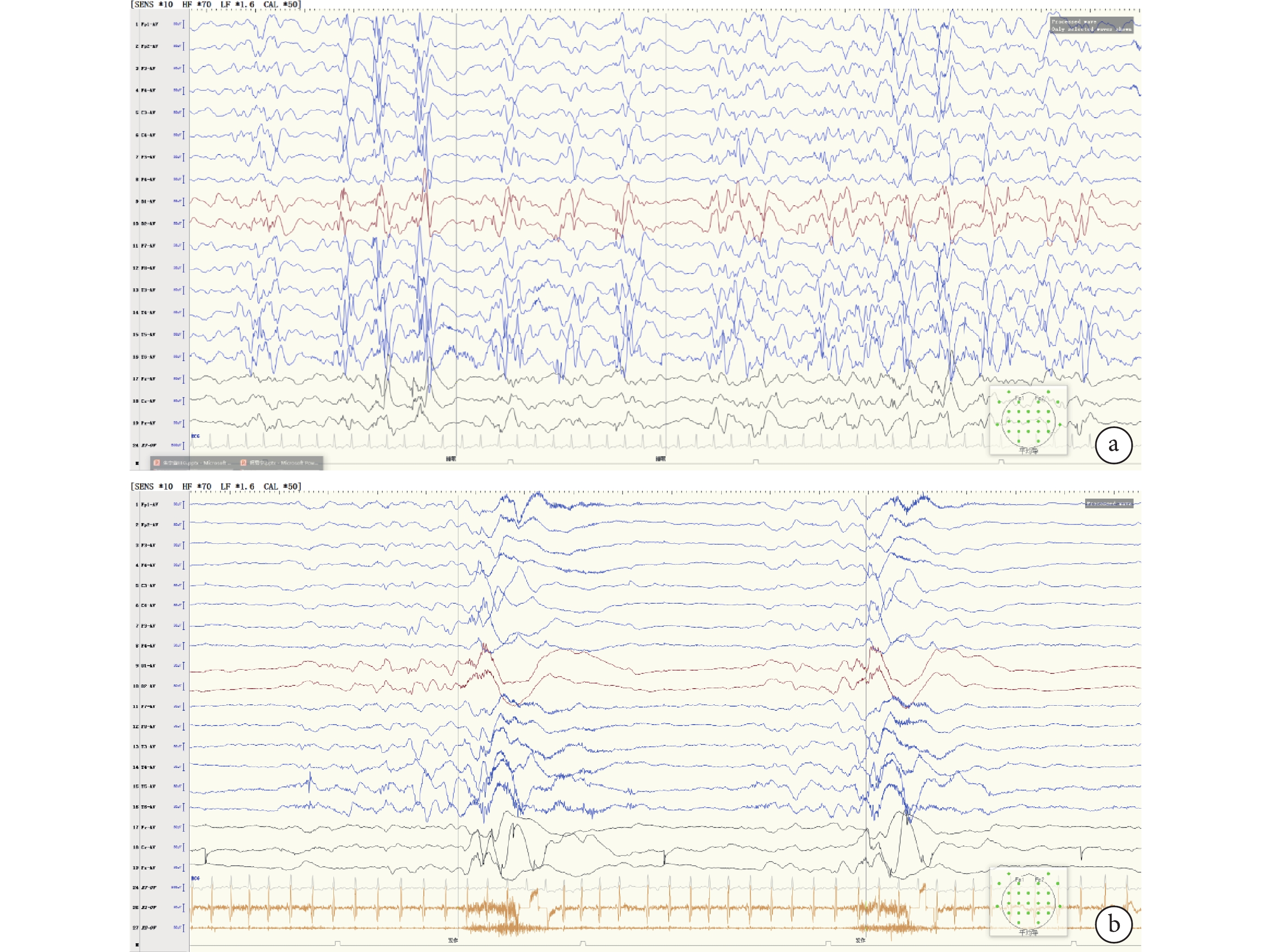
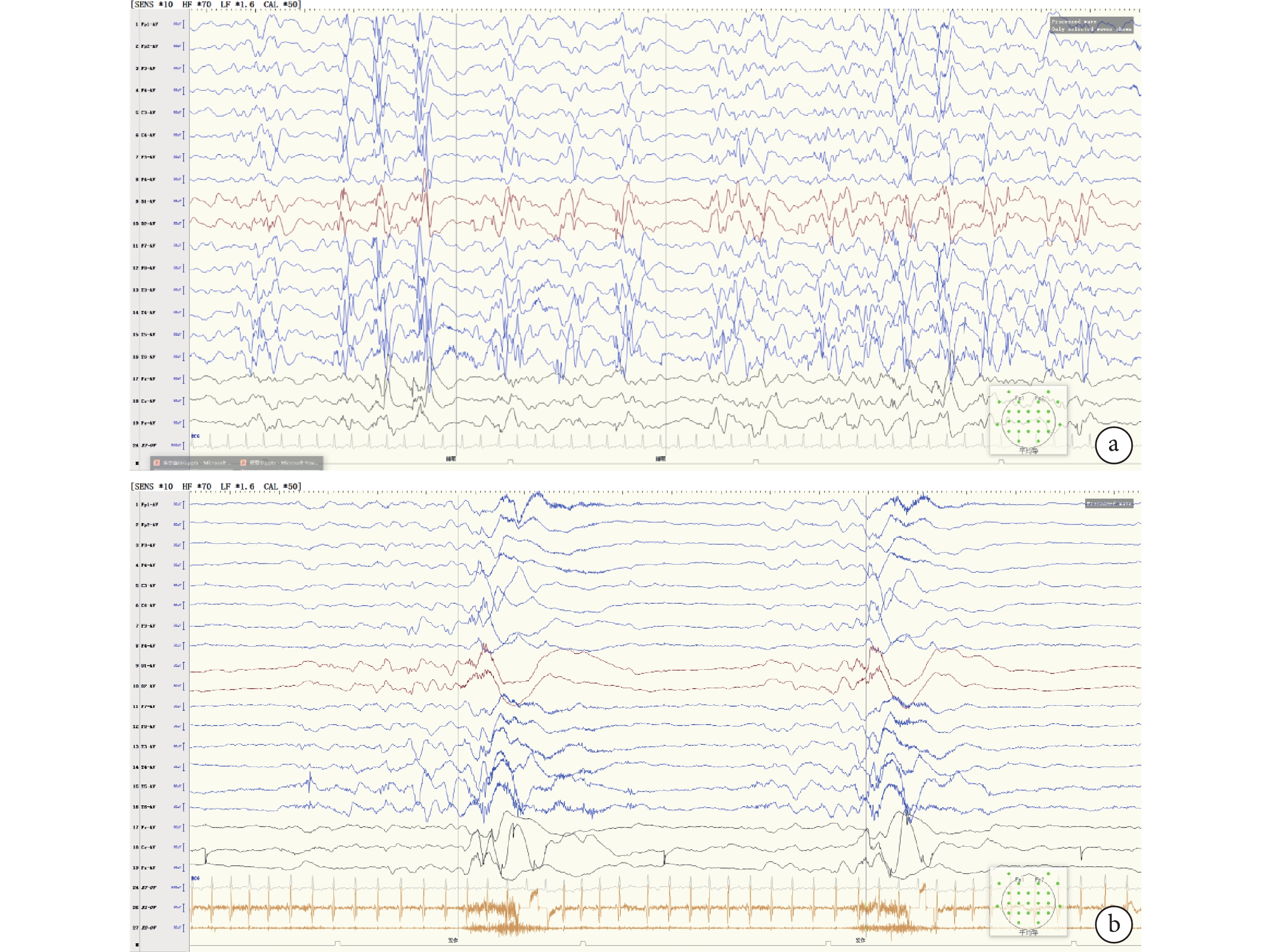 圖1
患兒腦電圖
圖1
患兒腦電圖
a. 睡眠期高度失律,后頭部顯著;b. 腦電監測到成串痙攣發作
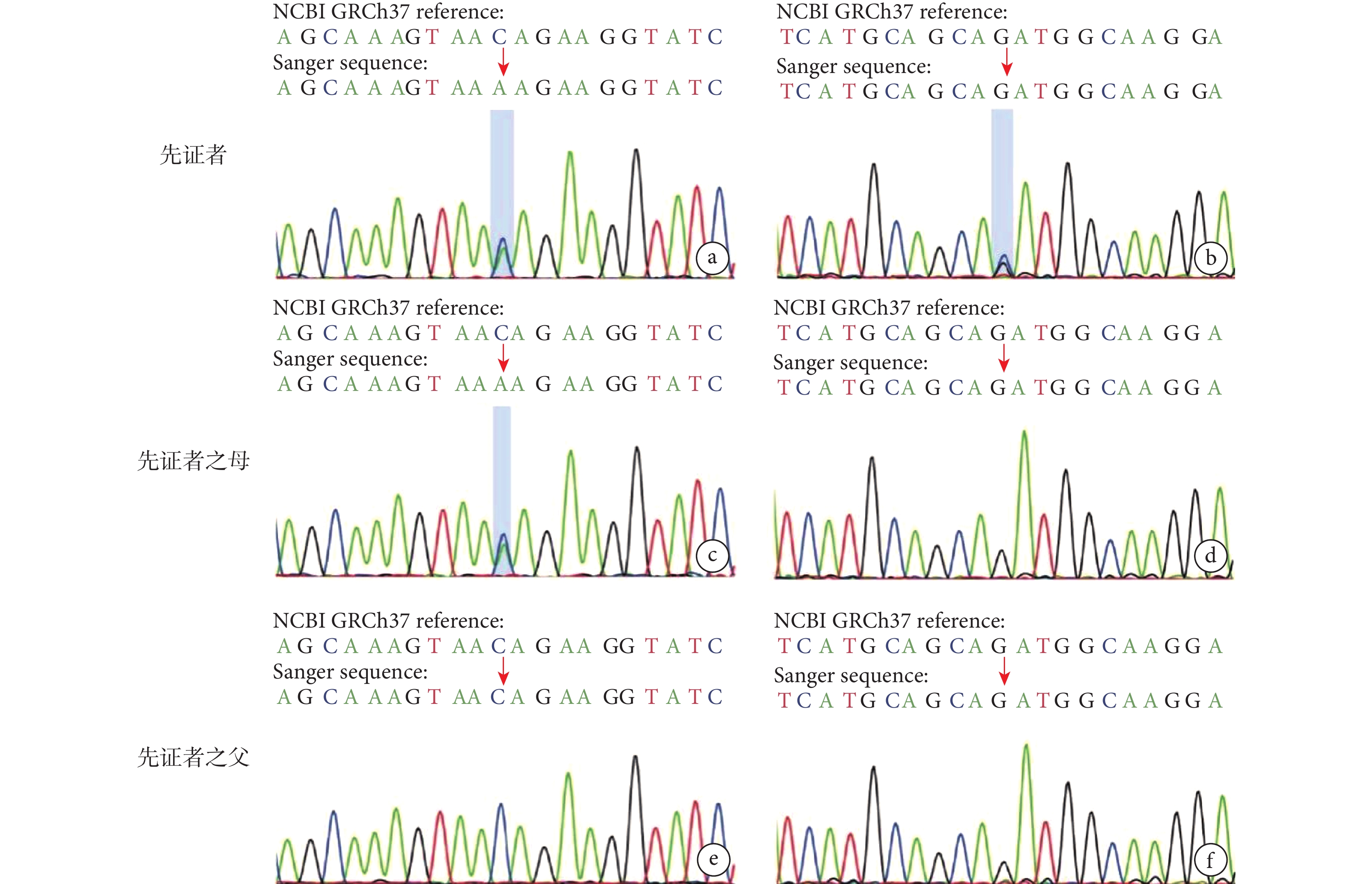
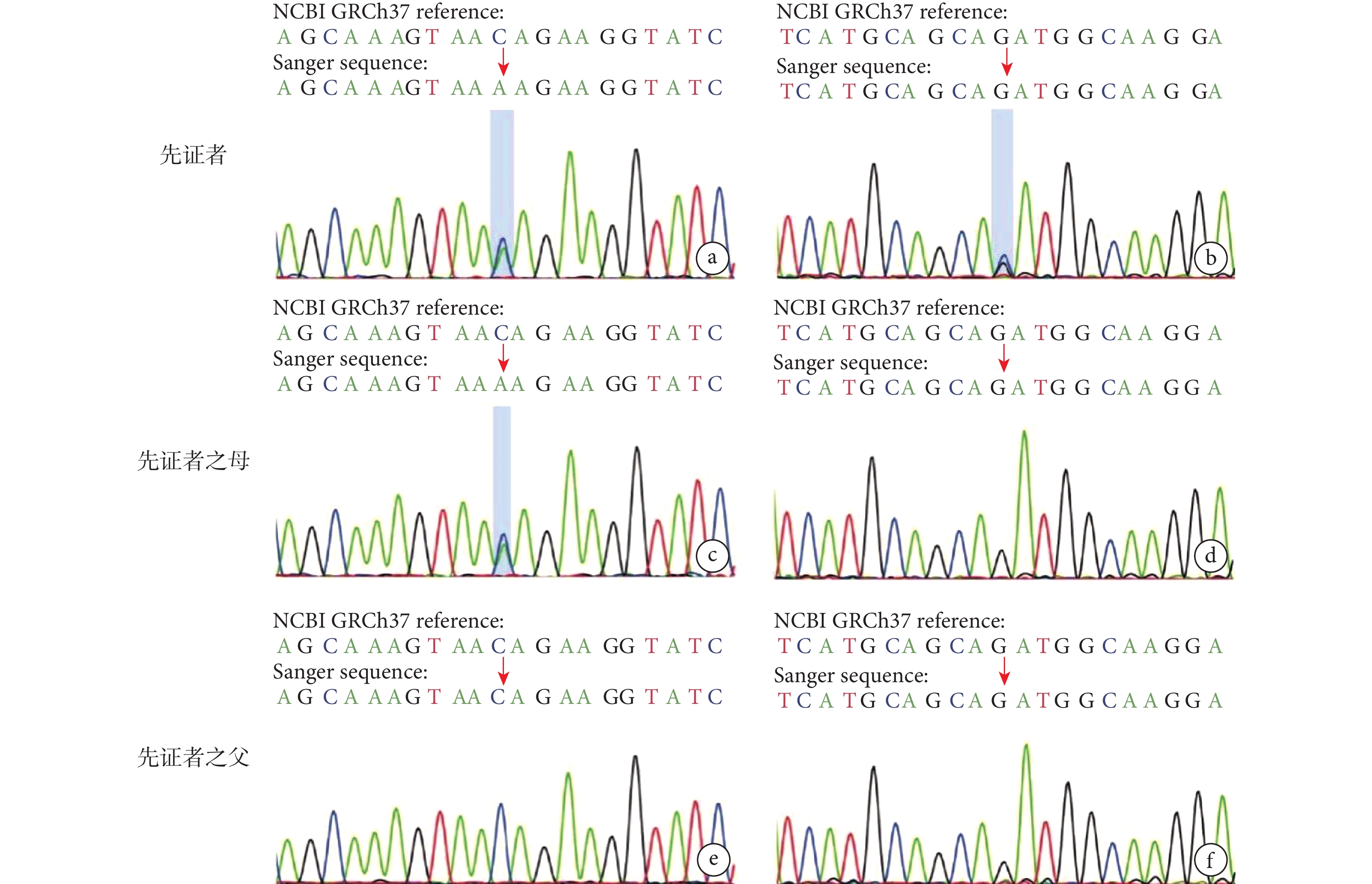 圖2
DPAGT1復合雜合變異
圖2
DPAGT1復合雜合變異
文獻復習 分別以“先天性糖基化、DPAGT1、DPAGT1-CDG、Congenital disorders of glycosylation” 為檢索詞檢索 PubMed、中國知網、萬方醫學網等數據庫(建庫至2024年5月),發現檢索符合條件的中文文獻 0 篇、英文文獻 13篇共32例DPAGT1-CDG患兒[4-16]。結合本組 1 例,共 33例DPAGT1-CDG患兒的臨床資料進行總結。
33例患兒中6例患兒父母親近結婚,14例在5歲前死亡,包括2例宮內死亡和1例流產胎兒。患兒從出生即可出現異常表現,如呼吸衰竭、肌張力下降、白內障、多關節攣縮等。除宮內死亡胎兒,均出現不同程度的運動發育遲緩或語言發育遲緩、智力低下。33例患兒中有24例出現肌張力低下,1例出現肌張力增高;20例出現發育遲緩;17例出現眼部異常(包括8例白內障以及眼震顫、斜視、夜盲癥、視神經萎縮、散光、視網膜色素變性、乳突萎縮等);8例出現癲癇發作,其中3例為藥物難治性癲癇,大部分未明確發作類型;8例出現肝臟腫大或者轉氨酶升高;6例出現關節攣縮,3例出現關節畸形(第2和第3趾聯合畸形等)。與文獻類似,本例患兒出現癲癇發作、肌張力降低以及轉氨酶升高等情況。33例病例有14例在5歲前死亡,包括2例宮內死亡和1例流產胎兒,可見該病死亡率高。目前文獻報道年齡最大的DPAGT1-CDG患者,是來自同一非近親家族的2例成年人,分別是32、34歲,這2例患者出生時正常,母孕期間和分娩過程均無異常。該2例患者在1.5~2歲時開始說話,1.5歲時開始走路,這2例的表型相對較輕,主要包括中度智障、癲癇、肌張力低下、攻擊行為和平衡問題。外顯子組測序提示復合雜合錯義突變,即c.85A>T(p.I29F)和c.503T>C (p.L168P)[11]。另有2例胎兒宮內死亡,為同一個健康產婦,分別于妊娠27、31周出現胎兒宮內死亡,死胎表現為小頜畸形、足內收和足上翻,基因檢測DPAGT1復合雜合錯義變異:c.324G>A:p.Met108Ile遺傳自父親;c.122T>C:p.Leu41Pro遺傳自母親[16]。33例DPAGT1變異位點總結詳見表1。
討論 糖基化包括各種基于糖的共修飾或翻譯后修飾,主要發生在內質網(Endoplasmic reticulum,ER)和高爾基體[17]。主要有兩種途徑:N-糖基化和O-糖基化。在真核細胞中,N-糖基化始于脂質連接的低聚糖在一個稱為多羥基循環的循環途徑中的合成,N-糖基化對蛋白質的正常折疊和功能至關重要[17,18]。N-糖基化抑制或異常,會導致錯誤折疊的蛋白質在ER腔內堆積、ER應激和誘導未折疊蛋白反應(Unfolded protein response,UPR)[18,19]。UPR是一種轉錄反應,旨在恢復細胞內的平衡狀態,若應激狀態無法得到緩解,可能導致細胞凋亡。N-糖基化的初始代謝產物是GlcNAc-PP-dolichol,其由DPAGT1酶合成。DPAGT1是N-糖基化途徑中的關鍵蛋白,參與寡糖生物合成,在早期胚胎發育中扮演著重要角色。該基因位于染色體11q23.3,其功能在于催化N-乙酰基葡萄糖氨酰-焦磷酸多立醇(GlcNAc-PP-dolichol)、磷酸多立醇以及UDP-GlcNAc的合成,這是多立醇循環中N-糖基化的首要步驟[16,20,21]。目前糖基化不足導致智力障礙和畸形的病理機制尚不清楚。越來越多的文獻認為DPAGT1是Wnt/β-catenin信號通路的靶點,而Wnt/β-catenin信號通路在發育過程中起著多種關鍵作用,為腦發育過程中形態發生過程出現的腦結構異常提供了可能的機制[11,15,22]。
DPAGT基因變異相關疾病具有多種表型,包括先天性肌無力綜合征、Nonaka肌病、伴眼外肌麻痹的小核肌病、杜氏肌營養不良、貝氏肌營養不良、遺傳性淋巴水腫3型、先天性糖基化障礙等。DPAGT1缺陷主要的兩種亞型:DPAGT1-CDG和肢帶型先天性肌無力綜合征(Limb girdle congenital myasthenic syndrome-13,CMS13),前者往往是一種嚴重的多系統疾病,可以表現為:發育不良、神經系統癥狀(震顫、精神運動遲滯、癲癇發作、過度興奮、共濟失調和小頭畸形)和肝功能異常(凝血異常和低蛋白血癥)。有些患者可能具有面部異常、乳頭內陷和皮下脂肪墊。據報道,80% 以上的患者在 5 歲前死亡[13,16]。后者臨床表現為肌無力,可能伴有或不伴有輕微的顱腦癥狀。與DPAGT1-CDG相比,CMS13的病程較輕,主要表現為肌肉無力,尤其是肢帶型肌無力,以及可能的肌肉活檢中出現的管狀聚集體。
33例DPAGT1-CDG患兒中,23例出現肌張力低下、20例出現發育遲緩,以及癲癇或眼部異常等表現,部分存在小頭畸形、關節畸形或者攣縮、肝臟腫大或者轉氨酶升高、脊柱側彎等,還有少部分存在特殊面容(顳狹窄、鼻翼模糊、耳垂薄、口小、牙齒中線融合)以及骨質疏松等情況。與文獻報道一致,本例患兒也出現發育遲緩、肌無力、肝功能異常,但嬰兒痙攣可能為一種新的表型。
DPAGT1-CDG患兒80% 以上在 5 歲前死亡[13,16],目前尚無確切有效的特定治療方式,主要為對癥治療,如抗癲癇、膽堿酯酶抑制劑對抗肌無力、康復訓練等,以提高生活質量為主[23]。本例患兒使用溴比斯的明口服,肌無力癥狀較前有所改善。在特殊情況下,飲食補充或器官移植可能會有所幫助[24]。然而,實現更有效的治療方法的關鍵在于開發細胞和動物模型,以深入了解疾病的發病機制,并促進藥物研發[25]。
為了確定潛在的治療靶點,有學者在果蠅模型中進行了CRISPR基因敲除篩選,以尋找與更好的存活率和糖蛋白水平相關的基因,發現了數百個可能有助于治療的候選基因。有意思的是,抑制甘露基轉移酶Dpm1或其下游的糖基化途徑可以挽救DPAGT1抑制和內質網應激兩個體內模型,盡管這些途徑的損傷通常會導致CDG。雖然這兩種體內模型都表現出細胞應激(通過DPAGT1抑制或錯誤折疊的蛋白質),但發現了果糖代謝的新差異,表明糖酵解可能是DPAGT1-CDG的調節因子。相關研究結果為DPAGT1-CDG提供了新的治療靶點,包括Dpm1相關通路挽救DPAGT1抑制的獨特發現,并揭示了果糖代謝與內質網應激之間的新型相互作用[26]。也有相關研究表明,某些家系存在著DPAGT1基因的新型錯義突變(NM_001382,c.1217 A>G),在這些家系利用單基因遺傳病胚胎植入前基因檢測(PGT-M)的診斷技術,揭示了單基因遺傳病胚胎的遺傳特征。通過不同的在線生物信息學工具,包括MutationTaster、I-Mutant v2.0、T-Coffee和CADD v1.0,對這一突變的致病性進行了預測。最終,在進行了PGT-M之后成功妊娠的家庭中,誕生一例正常嬰兒,證明了利用PGT-M技術防止有害變異體遺傳到下一代的重要性[14]。
綜上所述,DPAGT1基因變異相關CDG較罕見,以多系統異常為特征,包括發育遲緩、認知及運動發育障礙、癲癇發作、眼部異常、肌張力低下、關節畸形、肝功能異常等,臨床診斷存在一定困難。因此在臨床上對于嬰兒早期出現生長發育遲緩、癲癇發作、肌張力低下、肝功能異常、眼部異常等表現,需警惕該病,建議完善遺傳病全外顯子組測序以輔助診斷。目前在治療上,尚無確切有效的治療方式,主要為對癥治療,如抗癲癇發作、膽堿酯酶抑制劑對抗肌無力、康復訓練等,基因修飾治療或將成為該病治療的突破口,未來仍需進一步深入探究。
利益沖突 所有作者聲明無利益沖突。
先天性糖基化紊亂(Congenital disorders of glycosylation ,CDG)是一組由蛋白質和脂質糖基化缺陷引起的罕見遺傳病,涉及160多種亞型,與103余種不同的基因相關[1,2]。在這些病癥中,多乙酰磷酸N-乙酰葡萄糖胺-1-磷酸轉移酶(Dolichylphosphate N-acetylglucosamine phosphotransferase 1,DPAGT1)基因的變異會引起相關的先天性糖基化障礙(DPAGT1-CDG)。該病以多系統異常為特征,包括精神運動遲滯、癲癇發作、共濟失調和視網膜病變,其中部分出現肝功能異常和凝血功能障礙。發育畸形特征包括斜視、面部異常、乳頭內陷和皮下脂肪墊[3-5]。文章報道一例福建省兒童醫院2023年11月6日入院治療的DPAGT1-CDG患兒的臨床特點,并文獻復習,以提高對該病的臨床認識。本研究經我院倫理委員會批準(2023ETKLR11006)及患兒監護人知情同意。
病例資料 患者 女,5月齡23天齡。因“反復抽搐12天”于2023年11月至福建省兒童醫院神經內科就診。患兒入院前12天無明顯誘因出現反復抽搐,具體表現為點頭擁抱樣動作,成串發作,每串發作8~9下,持續1~2 min自行緩解,緩解后精神倦怠,每天發作8~9串,偶伴吐奶,無發熱、腹瀉,無大小便失禁,為進一步診治來我院。自發病以來,精神欠佳,食納、睡眠一般。患兒系其母第 2胎第 2 產,足月剖宮產娩出,出生體重2 800 g,否認出生時缺氧窒息史。目前尚不會抬頭。父母健康,非近親婚配,姐姐18歲,體健。既往患兒生后發育遲緩,營養不良,肌無力,目前不能抬頭,平素吃奶差,容易呼吸道感染,既往4月齡3天齡時,因“嗆奶”后導致“重癥肺炎、呼吸衰竭”于我院兒科重癥監護病房(Pediatric intensive care unit,PICU)治療,治療上予氣管插管、呼吸機輔助通氣、抗感染等處理,病情好轉后出院,平素予雞尾酒療法口服改善肌無力癥狀。此次入院體格檢查:神志清楚,精神差,表情淡漠,哭聲低弱,營養不良,自主體位,消瘦外觀,皮下脂肪消失,皮膚彈性稍差。追視不佳,眼球水平震顫,直接對光反射遲鈍,間接對光反射尚可。口唇紅,咽充血,頸軟,呼吸節律不規則,吸凹征陰性,雙肺呼吸音粗,聞及少許痰鳴。心音有力律齊,腹平軟。四肢自主活動少,頸軟無力,四肢肌力Ⅲ級,肌張力低下。跟腱、膝腱反射可引出。腦膜刺激征陰性,病理征陰性。入院輔檢:血常規、腦脊液常規、生化、培養、甲功、凝血等均大致正常。血生化提示:丙氨酸氨基轉移酶149 U/L、天門冬氨酸氨基轉移酶81 U/L,均偏高。長程腦電圖提示異常嬰兒腦電圖,出現高度失律(圖1a),后頭部顯著,監測到清醒期3次成串痙攣發作(圖1b)。頭顱核磁共振成像3.0T薄層平掃提示雙側額部腦外間隙略增寬。診斷嬰兒癲癇性痙攣綜合征,入院后予托吡酯、左乙拉西坦口服抗癲癇治療,患兒抽搐好轉后出院。同時行全外顯子基因測序,出院后結果回報DPAGT1復合雜合變異:DPAGT1 c.764G>T:p.Cys255Phe雜合變異,變異來自母親,父親為野生型;DPAGT1c.315C>G:p.lle105Met雜合變異,為新發變異,父母均為野生型(圖2);根據ACMG評級、腦表達、基因多態性、基因功能、基因保守性、等位基因頻率等評估致病性,考慮為可能致病性變異,結合患兒有肌無力、癲癇發作、發育遲緩、呼吸功能不全、肝功能異常等表現,最終診斷為DPAGT1基因變異相關先天性糖基化障礙。出院后1個月至我院神經內科門診隨診,未再抽搐。但患兒肌無力癥狀未見明顯改善,考慮DPAGT1基因變異相關先天性糖基化障礙部分患兒口服溴比斯的明有效,遂加用溴比斯的明口服,逐漸加量至5 mg/(kg·d),4個月后隨診患兒肌無力癥狀較前改善,未見抽搐。
圖1
患兒腦電圖
a. 睡眠期高度失律,后頭部顯著;b. 腦電監測到成串痙攣發作
圖2
DPAGT1復合雜合變異
文獻復習 分別以“先天性糖基化、DPAGT1、DPAGT1-CDG、Congenital disorders of glycosylation” 為檢索詞檢索 PubMed、中國知網、萬方醫學網等數據庫(建庫至2024年5月),發現檢索符合條件的中文文獻 0 篇、英文文獻 13篇共32例DPAGT1-CDG患兒[4-16]。結合本組 1 例,共 33例DPAGT1-CDG患兒的臨床資料進行總結。
33例患兒中6例患兒父母親近結婚,14例在5歲前死亡,包括2例宮內死亡和1例流產胎兒。患兒從出生即可出現異常表現,如呼吸衰竭、肌張力下降、白內障、多關節攣縮等。除宮內死亡胎兒,均出現不同程度的運動發育遲緩或語言發育遲緩、智力低下。33例患兒中有24例出現肌張力低下,1例出現肌張力增高;20例出現發育遲緩;17例出現眼部異常(包括8例白內障以及眼震顫、斜視、夜盲癥、視神經萎縮、散光、視網膜色素變性、乳突萎縮等);8例出現癲癇發作,其中3例為藥物難治性癲癇,大部分未明確發作類型;8例出現肝臟腫大或者轉氨酶升高;6例出現關節攣縮,3例出現關節畸形(第2和第3趾聯合畸形等)。與文獻類似,本例患兒出現癲癇發作、肌張力降低以及轉氨酶升高等情況。33例病例有14例在5歲前死亡,包括2例宮內死亡和1例流產胎兒,可見該病死亡率高。目前文獻報道年齡最大的DPAGT1-CDG患者,是來自同一非近親家族的2例成年人,分別是32、34歲,這2例患者出生時正常,母孕期間和分娩過程均無異常。該2例患者在1.5~2歲時開始說話,1.5歲時開始走路,這2例的表型相對較輕,主要包括中度智障、癲癇、肌張力低下、攻擊行為和平衡問題。外顯子組測序提示復合雜合錯義突變,即c.85A>T(p.I29F)和c.503T>C (p.L168P)[11]。另有2例胎兒宮內死亡,為同一個健康產婦,分別于妊娠27、31周出現胎兒宮內死亡,死胎表現為小頜畸形、足內收和足上翻,基因檢測DPAGT1復合雜合錯義變異:c.324G>A:p.Met108Ile遺傳自父親;c.122T>C:p.Leu41Pro遺傳自母親[16]。33例DPAGT1變異位點總結詳見表1。
討論 糖基化包括各種基于糖的共修飾或翻譯后修飾,主要發生在內質網(Endoplasmic reticulum,ER)和高爾基體[17]。主要有兩種途徑:N-糖基化和O-糖基化。在真核細胞中,N-糖基化始于脂質連接的低聚糖在一個稱為多羥基循環的循環途徑中的合成,N-糖基化對蛋白質的正常折疊和功能至關重要[17,18]。N-糖基化抑制或異常,會導致錯誤折疊的蛋白質在ER腔內堆積、ER應激和誘導未折疊蛋白反應(Unfolded protein response,UPR)[18,19]。UPR是一種轉錄反應,旨在恢復細胞內的平衡狀態,若應激狀態無法得到緩解,可能導致細胞凋亡。N-糖基化的初始代謝產物是GlcNAc-PP-dolichol,其由DPAGT1酶合成。DPAGT1是N-糖基化途徑中的關鍵蛋白,參與寡糖生物合成,在早期胚胎發育中扮演著重要角色。該基因位于染色體11q23.3,其功能在于催化N-乙酰基葡萄糖氨酰-焦磷酸多立醇(GlcNAc-PP-dolichol)、磷酸多立醇以及UDP-GlcNAc的合成,這是多立醇循環中N-糖基化的首要步驟[16,20,21]。目前糖基化不足導致智力障礙和畸形的病理機制尚不清楚。越來越多的文獻認為DPAGT1是Wnt/β-catenin信號通路的靶點,而Wnt/β-catenin信號通路在發育過程中起著多種關鍵作用,為腦發育過程中形態發生過程出現的腦結構異常提供了可能的機制[11,15,22]。
DPAGT基因變異相關疾病具有多種表型,包括先天性肌無力綜合征、Nonaka肌病、伴眼外肌麻痹的小核肌病、杜氏肌營養不良、貝氏肌營養不良、遺傳性淋巴水腫3型、先天性糖基化障礙等。DPAGT1缺陷主要的兩種亞型:DPAGT1-CDG和肢帶型先天性肌無力綜合征(Limb girdle congenital myasthenic syndrome-13,CMS13),前者往往是一種嚴重的多系統疾病,可以表現為:發育不良、神經系統癥狀(震顫、精神運動遲滯、癲癇發作、過度興奮、共濟失調和小頭畸形)和肝功能異常(凝血異常和低蛋白血癥)。有些患者可能具有面部異常、乳頭內陷和皮下脂肪墊。據報道,80% 以上的患者在 5 歲前死亡[13,16]。后者臨床表現為肌無力,可能伴有或不伴有輕微的顱腦癥狀。與DPAGT1-CDG相比,CMS13的病程較輕,主要表現為肌肉無力,尤其是肢帶型肌無力,以及可能的肌肉活檢中出現的管狀聚集體。
33例DPAGT1-CDG患兒中,23例出現肌張力低下、20例出現發育遲緩,以及癲癇或眼部異常等表現,部分存在小頭畸形、關節畸形或者攣縮、肝臟腫大或者轉氨酶升高、脊柱側彎等,還有少部分存在特殊面容(顳狹窄、鼻翼模糊、耳垂薄、口小、牙齒中線融合)以及骨質疏松等情況。與文獻報道一致,本例患兒也出現發育遲緩、肌無力、肝功能異常,但嬰兒痙攣可能為一種新的表型。
DPAGT1-CDG患兒80% 以上在 5 歲前死亡[13,16],目前尚無確切有效的特定治療方式,主要為對癥治療,如抗癲癇、膽堿酯酶抑制劑對抗肌無力、康復訓練等,以提高生活質量為主[23]。本例患兒使用溴比斯的明口服,肌無力癥狀較前有所改善。在特殊情況下,飲食補充或器官移植可能會有所幫助[24]。然而,實現更有效的治療方法的關鍵在于開發細胞和動物模型,以深入了解疾病的發病機制,并促進藥物研發[25]。
為了確定潛在的治療靶點,有學者在果蠅模型中進行了CRISPR基因敲除篩選,以尋找與更好的存活率和糖蛋白水平相關的基因,發現了數百個可能有助于治療的候選基因。有意思的是,抑制甘露基轉移酶Dpm1或其下游的糖基化途徑可以挽救DPAGT1抑制和內質網應激兩個體內模型,盡管這些途徑的損傷通常會導致CDG。雖然這兩種體內模型都表現出細胞應激(通過DPAGT1抑制或錯誤折疊的蛋白質),但發現了果糖代謝的新差異,表明糖酵解可能是DPAGT1-CDG的調節因子。相關研究結果為DPAGT1-CDG提供了新的治療靶點,包括Dpm1相關通路挽救DPAGT1抑制的獨特發現,并揭示了果糖代謝與內質網應激之間的新型相互作用[26]。也有相關研究表明,某些家系存在著DPAGT1基因的新型錯義突變(NM_001382,c.1217 A>G),在這些家系利用單基因遺傳病胚胎植入前基因檢測(PGT-M)的診斷技術,揭示了單基因遺傳病胚胎的遺傳特征。通過不同的在線生物信息學工具,包括MutationTaster、I-Mutant v2.0、T-Coffee和CADD v1.0,對這一突變的致病性進行了預測。最終,在進行了PGT-M之后成功妊娠的家庭中,誕生一例正常嬰兒,證明了利用PGT-M技術防止有害變異體遺傳到下一代的重要性[14]。
綜上所述,DPAGT1基因變異相關CDG較罕見,以多系統異常為特征,包括發育遲緩、認知及運動發育障礙、癲癇發作、眼部異常、肌張力低下、關節畸形、肝功能異常等,臨床診斷存在一定困難。因此在臨床上對于嬰兒早期出現生長發育遲緩、癲癇發作、肌張力低下、肝功能異常、眼部異常等表現,需警惕該病,建議完善遺傳病全外顯子組測序以輔助診斷。目前在治療上,尚無確切有效的治療方式,主要為對癥治療,如抗癲癇發作、膽堿酯酶抑制劑對抗肌無力、康復訓練等,基因修飾治療或將成為該病治療的突破口,未來仍需進一步深入探究。
利益沖突 所有作者聲明無利益沖突。