引用本文: 黃玲玲, 吳正玉, 劉程桃, 孟麗萍, 涂松濟, 儲金華. 拉考沙胺聯合吡侖帕奈添加治療兒童難治性局灶性癲癇合并骨髓增生異常綜合征一例并文獻復習. 癲癇雜志, 2024, 10(5): 449-453. doi: 10.7507/2096-0247.202406003 復制
版權信息: ?四川大學華西醫院華西期刊社《癲癇雜志》版權所有,未經授權不得轉載、改編
難治性癲癇(Drug resistant epilepsy,DRE)指應用選擇正確且能耐受的兩種抗癲癇發作藥物(單藥或聯合用藥)仍未能達到持續無發作[1]。DRE反復發作病程長,對患兒危害大,藥物治療多采用聯合用藥,目前尚無標準推薦方案。血液腫瘤是一類起源于造血系統的惡性腫瘤,包括白血病、淋巴瘤、骨髓增生異常綜合征(Myelodysplastic syndrome,MDS)等疾病,發病率及死亡率均位居腫瘤前列[2],藥物化療是主要治療手段。當DRE合并惡性腫瘤時,治療用藥選擇應更為慎重。文章通過報道1例安徽醫科大學第二附屬醫院診治的難治性局灶性癲癇患兒治療過程合并MDS且演變為急性髓細胞白血病(Acute myelocytic leukemia,AML)的臨床資料,總結患兒疾病演變特點、血液學、骨髓、腦電圖、全外顯子測序結果及診治用藥經過,分析在血液腫瘤疾病基礎上,拉考沙胺(Lacosamide,LCM)聯合吡侖帕奈(Perampanel,PER)作為添加治療局灶性癲癇的療效和安全性,同時檢索LCM、PER添加治療相關文獻資料進行復習,旨在為血液疾病合并癲癇的診治提供一定參考。該研究獲得安徽醫科大學第二附屬醫院醫學倫理委員會審核批準(YX2024-076)及所有患兒監護人知情同意。
病例資料 患兒 女,7歲4月齡。因“反復抽搐發作5年余,血三系下降2年余”于2023年6月至安徽醫科大學第二附屬醫院兒科住院治療。現病史:8月齡呼吸道感染后出現抽搐,表現為四肢抽動、雙眼凝視,持續2~3 min緩解,時測體溫38℃。9月齡呼吸道感染后出現抽搐發作持續1 h,時測體溫超過39℃,外院住院對癥處理后緩解,出院后仍有反復局灶性發作,表現為口唇抽動、雙眼斜視或凝視、雙上肢或單側上肢僵直抖動,持續1 min緩解,不足半數發作伴有口唇紫紺及意識不清,無流涎及大小便失禁,發作頻率6~7次/d,1歲時就診外院,視頻腦電圖檢查見癇樣放電,頭顱核磁共振(Magnetic resonance imaging,MRI)及血尿串聯質譜檢查正常,診斷“癲癇”,給予左乙拉西坦(Levetiracetam,LEV)口服半年,未能控制癲癇發作,遂聯合丙戊酸鈉(Sodium valproate,VPA)治療3年余,發作頻率減少至4~5次/年,發作形式為頭眼一側偏轉、口歪斜、四肢僵直抖動,無口唇紫紺,持續1min,病程期間數次高熱時發作后繼發雙側強直陣攣發作。2年前檢查發現血三系下降,血小板最低值為20×1012/L[參考值(167~453)×1012/L],外院骨髓細胞學檢查示骨髓增生活躍,全外顯子二代測序基因分析報告提示未發現與患者表型吻合的致病性或疑似致病性變異。腦電圖提示背景活動慢、睡眠期右側顳區慢波發放。給與“甲強龍”沖擊及靜脈用“丙種球蛋白”治療,同時減停“LEV”及“VPA”,考慮到“卡馬西平”和“拉莫三嗪”存在特異體質不良反應“再生障礙性貧血”,外院與家屬溝通后予“托吡酯(Topiramate,TPM)”口服抗癲癇發作治療。近2年來監測血三系一直未能恢復正常,白細胞輕度下降,貧血呈重度,血小板波動在20~60×1012/L,外院給與“潑尼松、蛋白琥珀酸鐵劑、地榆生白片”口服及輸血等治療,無效。近半年來癲癇發作頻率增多,2~3次/月,發作形式較前無變化,多數癲癇發作系呼吸道感染高熱時出現,腦電圖提示背景活動偏慢、睡眠期右側中央、頂、中線區棘波、棘慢波發放,監測到睡眠期1次局灶性發作(右側中央、頂區起始)(同期表現為覺醒后軀體右偏,雙目凝視,反應減低)。起病前患兒生長發育正常,8月齡起病后生長發育落后,智力低下,語言發育落后,目前僅會發少數雙音節。
既往史及家族史:8月齡前生長發育正常,健康,否認疾病史。否認家族遺傳性疾病史,否認癲癇家族史。
入院體檢:身高118cm,體重24.6 kg,發簡單雙音節詞,無法交流,神清,反應可,面容無異常,頸軟,心肺聽診陰性,腹軟,肝脾肋下未及,脊柱四肢無畸形,深淺反射可引出,腦膜刺激征陰性。韋氏智力量表評估智力商數(Intelligence quotient,IQ)值<40,存在智力低下。
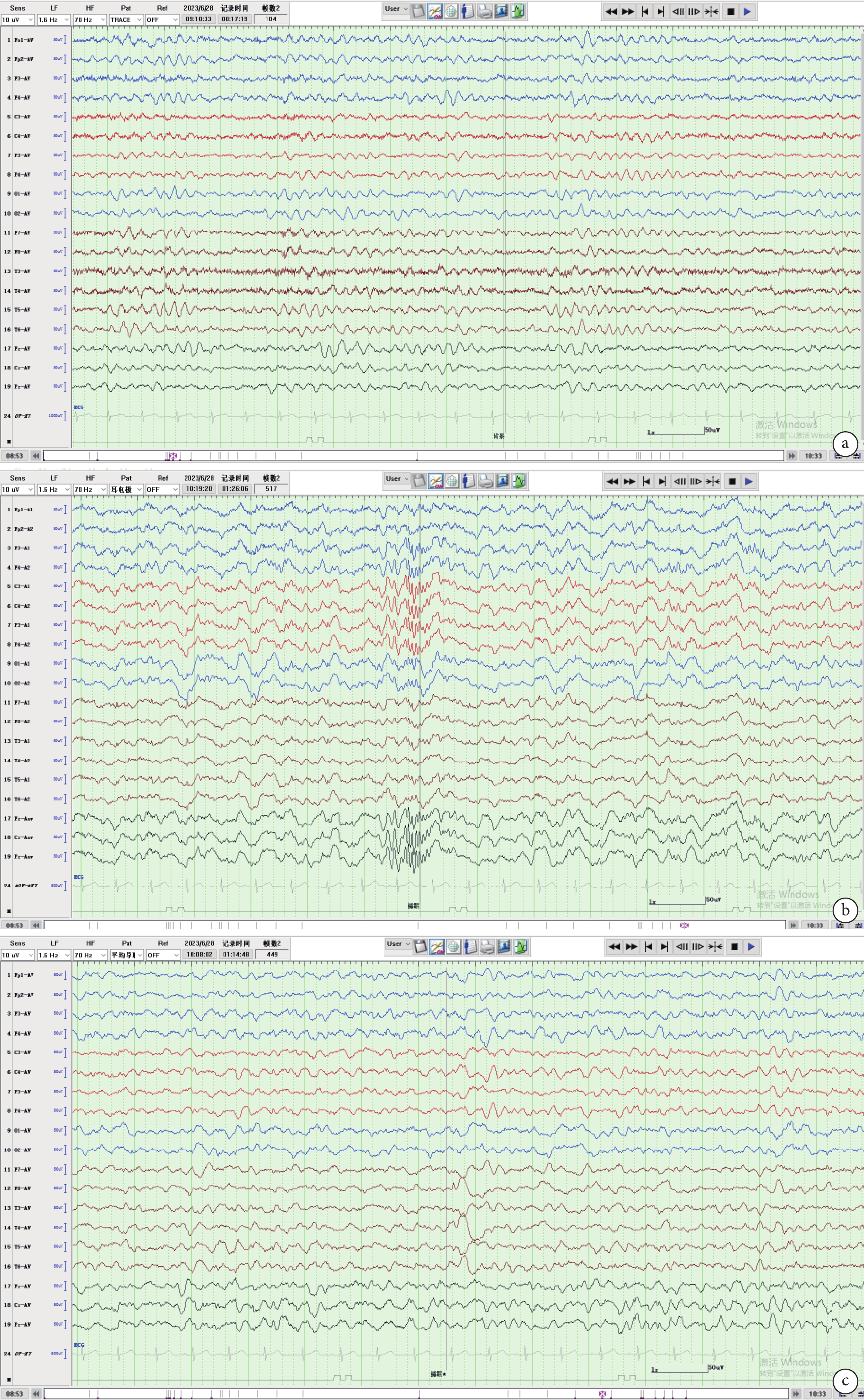
輔助檢查:實驗室檢查: 血常規白細胞3.7×109/L,血紅蛋白 68 g/L,血小板82×1012/L。血生化、電解質、血糖、血脂未見明顯異常。骨髓涂片檢查:骨髓細胞學提示粒、紅系可見病態改變。骨髓病理考慮MDS。骨髓MDS基因芯片顯示該標本染色體為女性核型,且含有12處染色體變異,其中5處強臨床意義的染色體變異。骨髓染色體檢查結果示:46,XX,add(1)(q21),der(1,5)(q10,p10),del(7)(q22),del(12)(p13),18,del(20)(q13),?20.inc[cp20]。頭顱影像學及腦電圖檢查:頭顱MRI平掃腦實質未見明顯異常。腦電圖提示背景活動慢、睡眠期右側前中顳區為主的限局或右側半球廣泛性多位相慢波、不規則慢波、尖型慢波發放(圖1a、1b、1c)。
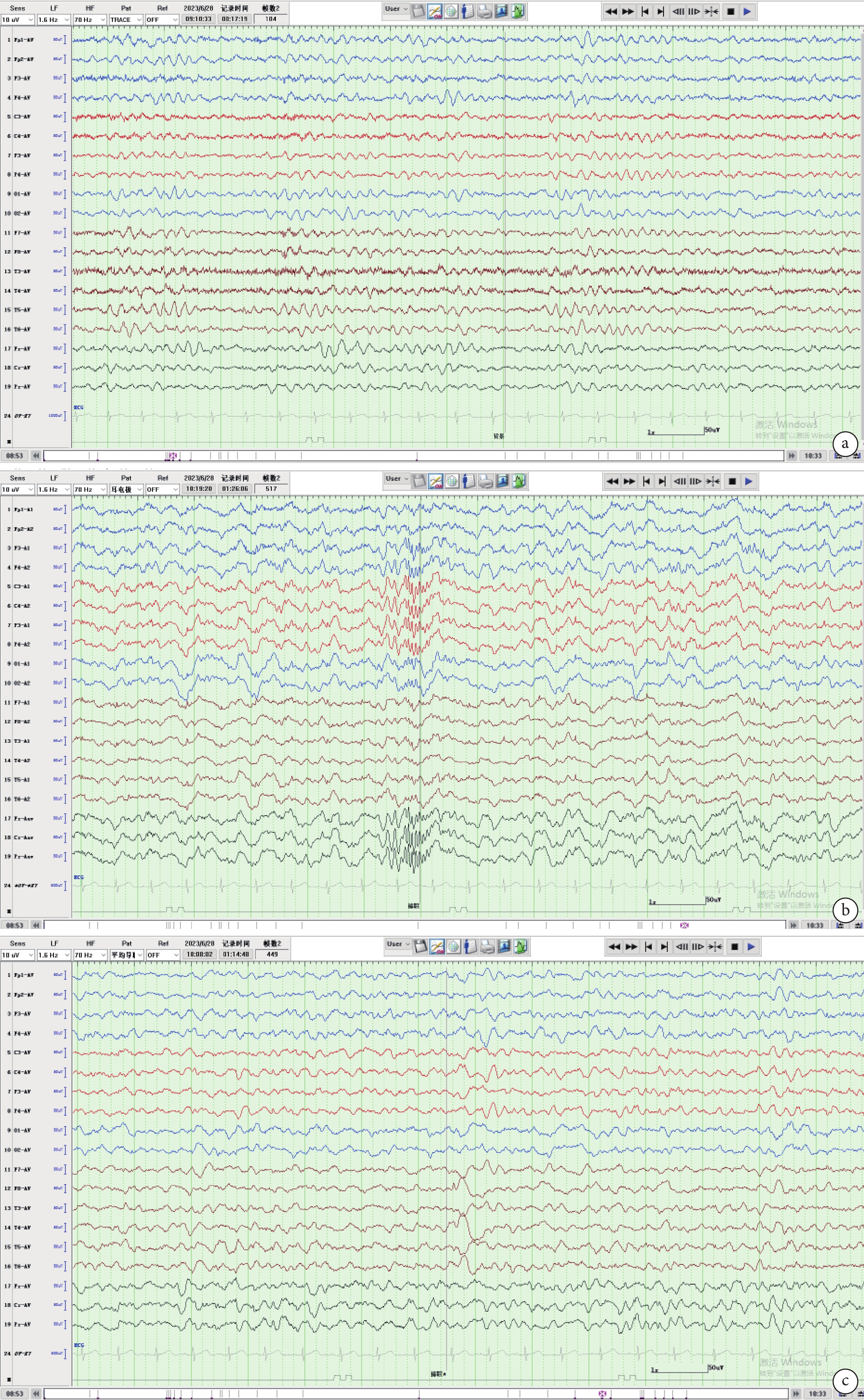 圖1
患兒6歲3月齡腦電圖
圖1
患兒6歲3月齡腦電圖
a. 彌漫性中波幅4~6 Hz θ節律,背景活動慢;b. 睡眠波及睡眠分期正常,可見睡眠期紡錘波;c. 監測期間記錄到睡眠期右側前、中顳區為主的限局或廣泛性多位相慢波、尖形慢波,可見右側顳區異常放電
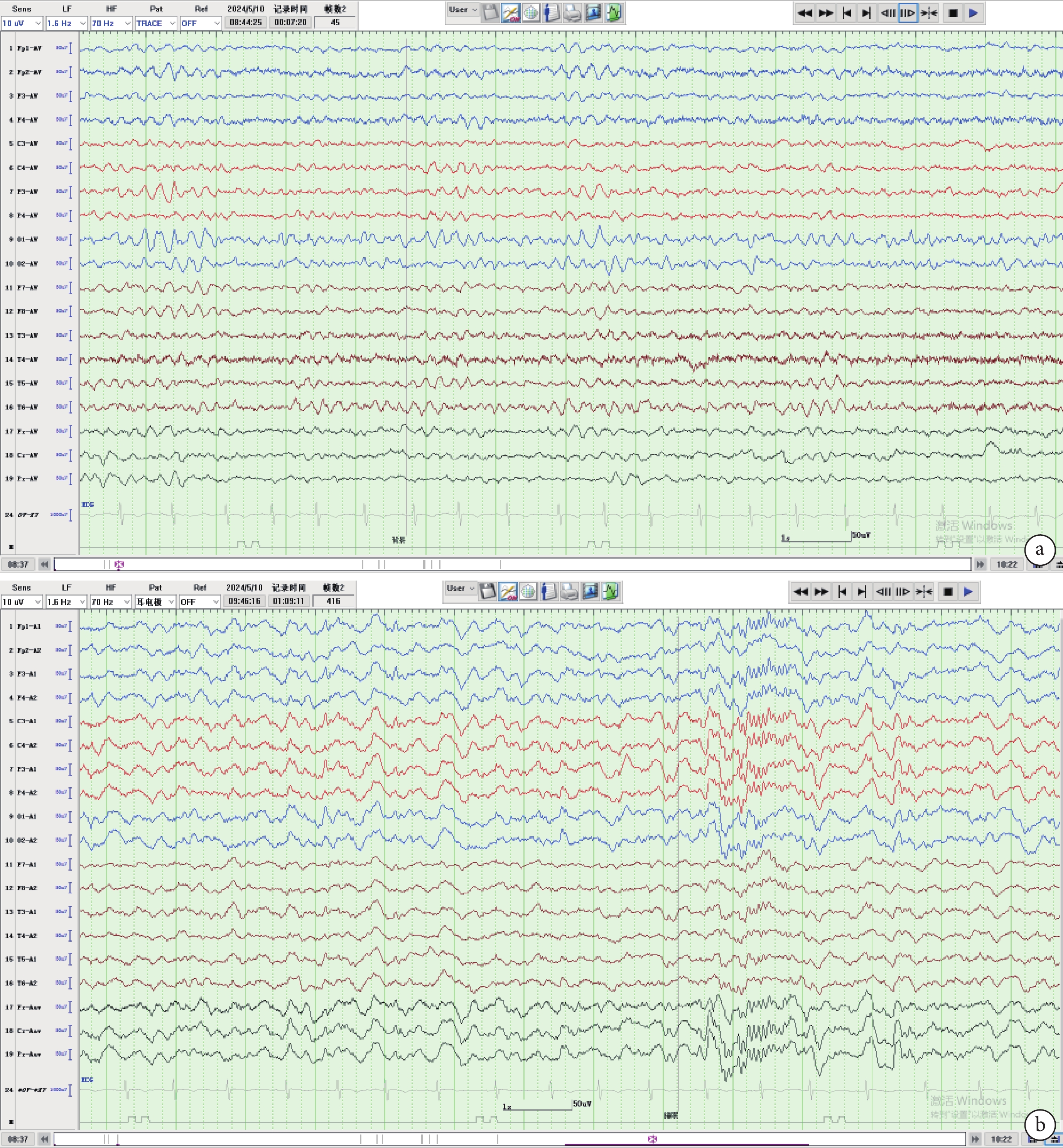
治療經過:入院后綜合病史、體檢、腦電圖、頭顱MRI及全外顯子二代測序基因結果等,診斷“癲癇(局灶性發作、局灶性發作繼發雙側強直陣攣發作)”,不符合目前兒童癲癇綜合征分類標準,病因不明。結合骨髓相關檢查,符合MDS診斷標準,分組系高危組,治療需要造血干細胞移植治療,但存在移植反指征(IQ<40),家屬選擇對癥支持治療,多次輸注懸浮紅細胞糾正貧血,監測中性粒細胞水平呈下降趨勢,近半年多來一直呈粒細胞缺乏狀態,呼吸道感染頻率增加,約2周出現一次發熱感染。轉入我院后抗癲癇發作治療調整為TPM(早62.5 mg,晚50 mg)聯合PER治療,PER劑量增加至6 mg/d頭暈不能耐受,降至5 mg/d可耐受,TPM聯合PER治療半年,發作頻率無明顯下降,且在一次感染高熱40℃時出現局灶性繼發雙側強直陣攣發作并呈持續狀態,添加LCM(50 mg,每日兩次)治療,目前LCM聯合PER及TPM治療半年多,3個月前監測血常規發現異常波動,復查骨髓細胞學,結果提示急性白血病骨髓象傾向AML,骨髓免疫分型提示為AML伴白細胞分化抗原(Cluster of differentiation,CD)7表達可能,目前完成個體化化療方案2療程(第一療程藥物:阿扎胞苷、維奈克拉、高三尖杉酯、阿糖胞苷;第二療程藥物:地西他濱),評估未緩解。LCM、PER、TPM聯合抗發作治療1個月時發熱期間出現局灶性癲癇發作1次,后至今近半年未再出現癲癇發作,近期復查腦電圖提示背景活動慢,未見癇樣放電(圖2a、2b)。
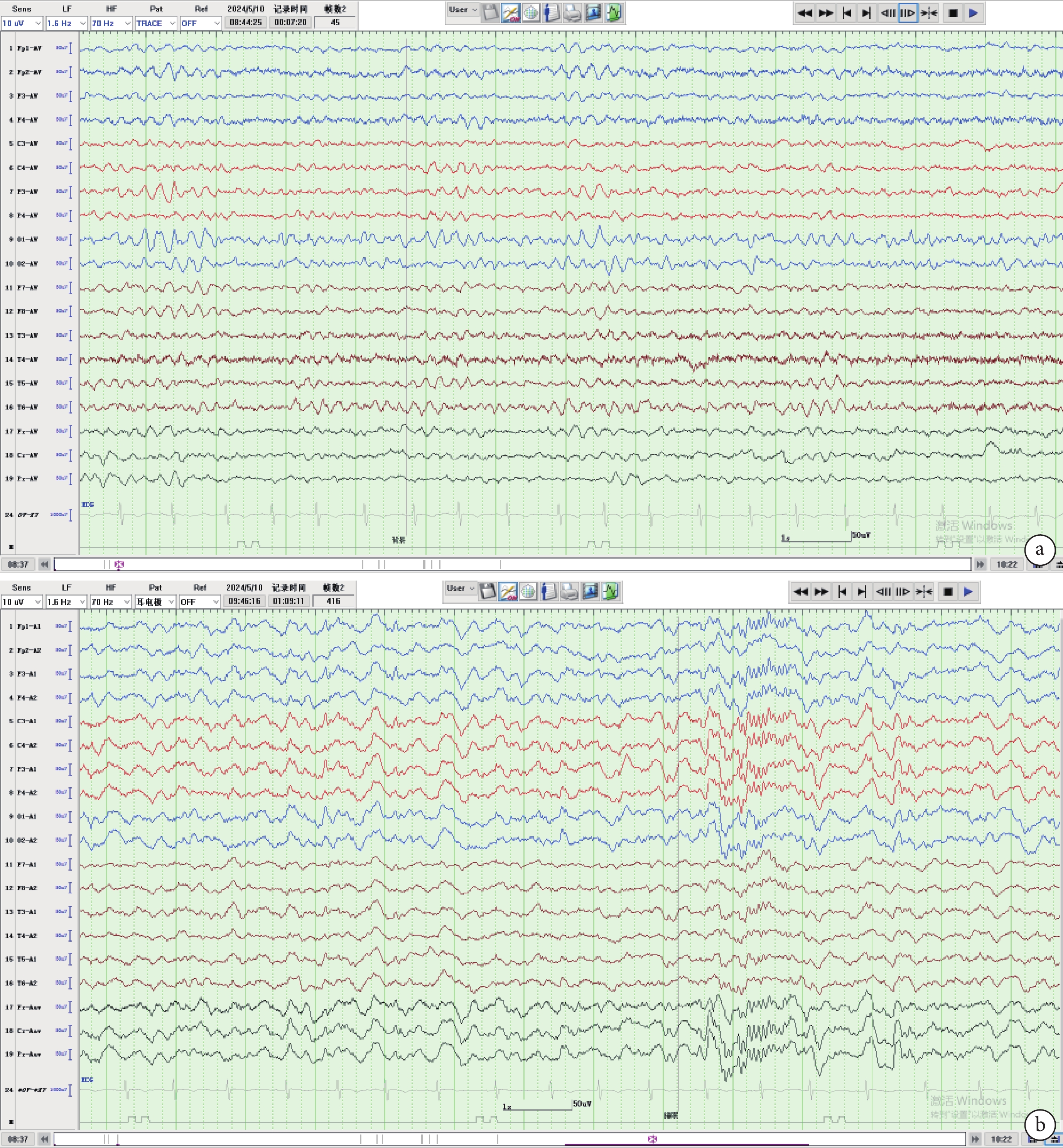 圖2
患兒7歲2月齡腦電圖
圖2
患兒7歲2月齡腦電圖
a. 彌漫性中波幅4~6 Hz θ節律,背景活動慢;b. 睡眠波及睡眠分期正常,睡眠期各導未見明顯異常放電,可見睡眠期紡錘波
討論 根據國際抗癲癇聯盟的定義,DRE指應用選擇正確且能耐受的兩種抗癲癇發作藥物(單藥或聯合用藥)仍未能達到持續無發作[1]。DRE反復發作病程長,對患兒危害大,藥物治療多采用聯合用藥,目前尚無標準推薦方案,若合并惡性疾病,治療用藥更為棘手。血液腫瘤是一類起源于造血系統的惡性腫瘤,包括白血病、淋巴瘤、MDS等疾病,發病率及死亡率均位居腫瘤前列[2],藥物化療是主要治療手段。當兩病合并發生時,治療用藥選擇需更為慎重。本研究病例在VPA和LEV聯合治療過程中發現血液學改變,停藥后不能恢復,最終診斷血液系統腫瘤MDS,后演變為AML。VPA和LEV分別為第一代和第二代抗癲癇發作藥物,VPA可通過多種機制發揮抗癲癇發作作用,LEV抗癲癇作用可能與神經突觸囊泡蛋白有關,兩者在血液腫瘤患者中均有使用報道[3-6]。VPA和LEV血液系統不良反應有所差別, VPA血液系統不良反應更為常見及多樣,血小板減少、纖維蛋白原減少、白細胞減少、凝血因子缺乏等均有報道[7],減停藥后預后良好。LEV血液系統不良反應少見,發生率約0.01%~0.13%[8],主要表現為血小板減少及全血細胞減少,停藥后短時間內可恢復[9]。本研究病例在減停VPA和LEV后血三系不能恢復,激素治療無效,評估骨髓細胞學提示骨髓增生活躍,并無骨髓抑制表現,提示并非藥物所致血液系統不良反應。停藥約1年半后監測骨髓符合MDS診斷,MDS特點為高風險向AML轉化[10],本研究病例后續MDS演變為AML系疾病本身演變過程。
PER和LCM均為第三代抗癲癇發作藥物,PER是第一個選擇性ɑ-氨基-3-羥基-5甲基-4-異噁唑受體拮抗劑,LCM是新型N-甲基-D-天門冬氨酸受體甘氨酸位點結合拮抗劑,兩者均有口服及注射制劑,口服給藥時吸收迅速,可與食物同服[11,12]。在藥物合用相互作用方面,PER在體外既不是細胞色素P450氧化酶(Cytochrome P450,CYP)或尿苷二磷酸葡萄糖醛基轉移酶(Uridine diphosphate glucuronyl transferases,UGT)同功酶的有效抑制劑,也不是誘導劑,并未發現PER對托吡酯、氯硝西泮、LEV、苯巴比妥或唑尼沙胺的清除率有顯著影響[13]。LCM不誘導或抑制CYP或已知的藥物轉運系統,蛋白質結合率低于15%,由于它具有多種消除途徑,因此與常用處方藥物之間并無臨床相關的相互作用[12]。
PER和LCM已在多個國家被應用于局灶性和全面性癲癇患者,新靶點及新機制可以降低藥物不良反應發生率并提高藥物耐受性。年齡在兒童患者中是影響PER療效的重要因素,一項納入62例6~18歲兒童藥物難治性癲癇患兒的研究顯示PER添加治療總體有效率為50.0%,不同年齡段療效存在差異,12歲以下年齡組有效率25.0%,12歲以上年齡組有效率53.7%[14]。國內一項研究顯示類似結果,年齡越大PER療效呈越好趨勢[15]。PER不良反應主要包括精神不良事件和非精神不良事件,在精神不良事件中,抑郁癥和雙相情感障礙最為常見,分別占4.5%和10.4%;在非精神不良事件中,輕度頭暈、嗜睡和惡心/嘔吐最為常見,分別占28.4%、19.4%和6.0%[16]。LCM同樣是一種有效的添加治療,一項納入60例4~17歲局灶性癲癇患兒的研究顯示,LCM添加治療患兒自基線至末次隨訪的50%應答率為40.0%,75%應答率為28.3%,結果提示在≥4歲中國患兒中LCM添加治療可有效降低癲癇發作頻率[17]。國外LCM添加治療兒童癲癇的研究也有類似結果[18,19]。有數項研究顯示LCM口服制劑及靜脈制劑在癲癇患兒中具有良好的安全性和耐受性[20,21]。LCM最常見的不良反應為上呼吸道感染(33.3%)、嗜睡(26.7%)、頭暈(25.0%)及嘔吐(21.7%),未發現具有臨床意義的心電圖異常變化[17]。對比兩者療效及安全性,有Meta分析結果顯示成人患者中PER 4mg/d癲癇發作反應率低于LCM 400mg/d,另外LCM耐受性更優[22]。PER和LCM聯合添加治療相關研究較少,目前多為個案報道,有報道顯示在熱性感染相關性癲癇綜合征中兩者聯合添加治療表現出良好的治療效果[23]。
本研究病例年齡<12歲,在PER治療過程中出現頭暈,減量后不良反應消失,耐受良好,但添加治療6個月效果不理想, LCM聯合PER添加治療后癲癇發作控制。聯合治療期間患兒接受多次輸血、抗感染治療及多藥化療,未發現異常不良反應。對比LCM聯合PER治療前后腦電圖結果,背景變化差別不大,間期電變化比較明顯,治療前腦電圖右側睡眠期有少量放電,以右側前中顳區明顯,治療后腦電圖無明顯異常波,提示對于兒童難治性局灶性癲癇,LCM聯合PER添加治療為有效的治療選擇之一,且與其它藥物間相互影響小,但目前樣本量小,后續需開展多中心、大樣本長期隨訪研究,進一步評估LCM和PRE聯合添加治療療效及長期耐受性。
利益沖突聲明 所有作者無利益沖突。
難治性癲癇(Drug resistant epilepsy,DRE)指應用選擇正確且能耐受的兩種抗癲癇發作藥物(單藥或聯合用藥)仍未能達到持續無發作[1]。DRE反復發作病程長,對患兒危害大,藥物治療多采用聯合用藥,目前尚無標準推薦方案。血液腫瘤是一類起源于造血系統的惡性腫瘤,包括白血病、淋巴瘤、骨髓增生異常綜合征(Myelodysplastic syndrome,MDS)等疾病,發病率及死亡率均位居腫瘤前列[2],藥物化療是主要治療手段。當DRE合并惡性腫瘤時,治療用藥選擇應更為慎重。文章通過報道1例安徽醫科大學第二附屬醫院診治的難治性局灶性癲癇患兒治療過程合并MDS且演變為急性髓細胞白血病(Acute myelocytic leukemia,AML)的臨床資料,總結患兒疾病演變特點、血液學、骨髓、腦電圖、全外顯子測序結果及診治用藥經過,分析在血液腫瘤疾病基礎上,拉考沙胺(Lacosamide,LCM)聯合吡侖帕奈(Perampanel,PER)作為添加治療局灶性癲癇的療效和安全性,同時檢索LCM、PER添加治療相關文獻資料進行復習,旨在為血液疾病合并癲癇的診治提供一定參考。該研究獲得安徽醫科大學第二附屬醫院醫學倫理委員會審核批準(YX2024-076)及所有患兒監護人知情同意。
病例資料 患兒 女,7歲4月齡。因“反復抽搐發作5年余,血三系下降2年余”于2023年6月至安徽醫科大學第二附屬醫院兒科住院治療。現病史:8月齡呼吸道感染后出現抽搐,表現為四肢抽動、雙眼凝視,持續2~3 min緩解,時測體溫38℃。9月齡呼吸道感染后出現抽搐發作持續1 h,時測體溫超過39℃,外院住院對癥處理后緩解,出院后仍有反復局灶性發作,表現為口唇抽動、雙眼斜視或凝視、雙上肢或單側上肢僵直抖動,持續1 min緩解,不足半數發作伴有口唇紫紺及意識不清,無流涎及大小便失禁,發作頻率6~7次/d,1歲時就診外院,視頻腦電圖檢查見癇樣放電,頭顱核磁共振(Magnetic resonance imaging,MRI)及血尿串聯質譜檢查正常,診斷“癲癇”,給予左乙拉西坦(Levetiracetam,LEV)口服半年,未能控制癲癇發作,遂聯合丙戊酸鈉(Sodium valproate,VPA)治療3年余,發作頻率減少至4~5次/年,發作形式為頭眼一側偏轉、口歪斜、四肢僵直抖動,無口唇紫紺,持續1min,病程期間數次高熱時發作后繼發雙側強直陣攣發作。2年前檢查發現血三系下降,血小板最低值為20×1012/L[參考值(167~453)×1012/L],外院骨髓細胞學檢查示骨髓增生活躍,全外顯子二代測序基因分析報告提示未發現與患者表型吻合的致病性或疑似致病性變異。腦電圖提示背景活動慢、睡眠期右側顳區慢波發放。給與“甲強龍”沖擊及靜脈用“丙種球蛋白”治療,同時減停“LEV”及“VPA”,考慮到“卡馬西平”和“拉莫三嗪”存在特異體質不良反應“再生障礙性貧血”,外院與家屬溝通后予“托吡酯(Topiramate,TPM)”口服抗癲癇發作治療。近2年來監測血三系一直未能恢復正常,白細胞輕度下降,貧血呈重度,血小板波動在20~60×1012/L,外院給與“潑尼松、蛋白琥珀酸鐵劑、地榆生白片”口服及輸血等治療,無效。近半年來癲癇發作頻率增多,2~3次/月,發作形式較前無變化,多數癲癇發作系呼吸道感染高熱時出現,腦電圖提示背景活動偏慢、睡眠期右側中央、頂、中線區棘波、棘慢波發放,監測到睡眠期1次局灶性發作(右側中央、頂區起始)(同期表現為覺醒后軀體右偏,雙目凝視,反應減低)。起病前患兒生長發育正常,8月齡起病后生長發育落后,智力低下,語言發育落后,目前僅會發少數雙音節。
既往史及家族史:8月齡前生長發育正常,健康,否認疾病史。否認家族遺傳性疾病史,否認癲癇家族史。
入院體檢:身高118cm,體重24.6 kg,發簡單雙音節詞,無法交流,神清,反應可,面容無異常,頸軟,心肺聽診陰性,腹軟,肝脾肋下未及,脊柱四肢無畸形,深淺反射可引出,腦膜刺激征陰性。韋氏智力量表評估智力商數(Intelligence quotient,IQ)值<40,存在智力低下。
輔助檢查:實驗室檢查: 血常規白細胞3.7×109/L,血紅蛋白 68 g/L,血小板82×1012/L。血生化、電解質、血糖、血脂未見明顯異常。骨髓涂片檢查:骨髓細胞學提示粒、紅系可見病態改變。骨髓病理考慮MDS。骨髓MDS基因芯片顯示該標本染色體為女性核型,且含有12處染色體變異,其中5處強臨床意義的染色體變異。骨髓染色體檢查結果示:46,XX,add(1)(q21),der(1,5)(q10,p10),del(7)(q22),del(12)(p13),18,del(20)(q13),?20.inc[cp20]。頭顱影像學及腦電圖檢查:頭顱MRI平掃腦實質未見明顯異常。腦電圖提示背景活動慢、睡眠期右側前中顳區為主的限局或右側半球廣泛性多位相慢波、不規則慢波、尖型慢波發放(圖1a、1b、1c)。
圖1
患兒6歲3月齡腦電圖
a. 彌漫性中波幅4~6 Hz θ節律,背景活動慢;b. 睡眠波及睡眠分期正常,可見睡眠期紡錘波;c. 監測期間記錄到睡眠期右側前、中顳區為主的限局或廣泛性多位相慢波、尖形慢波,可見右側顳區異常放電
治療經過:入院后綜合病史、體檢、腦電圖、頭顱MRI及全外顯子二代測序基因結果等,診斷“癲癇(局灶性發作、局灶性發作繼發雙側強直陣攣發作)”,不符合目前兒童癲癇綜合征分類標準,病因不明。結合骨髓相關檢查,符合MDS診斷標準,分組系高危組,治療需要造血干細胞移植治療,但存在移植反指征(IQ<40),家屬選擇對癥支持治療,多次輸注懸浮紅細胞糾正貧血,監測中性粒細胞水平呈下降趨勢,近半年多來一直呈粒細胞缺乏狀態,呼吸道感染頻率增加,約2周出現一次發熱感染。轉入我院后抗癲癇發作治療調整為TPM(早62.5 mg,晚50 mg)聯合PER治療,PER劑量增加至6 mg/d頭暈不能耐受,降至5 mg/d可耐受,TPM聯合PER治療半年,發作頻率無明顯下降,且在一次感染高熱40℃時出現局灶性繼發雙側強直陣攣發作并呈持續狀態,添加LCM(50 mg,每日兩次)治療,目前LCM聯合PER及TPM治療半年多,3個月前監測血常規發現異常波動,復查骨髓細胞學,結果提示急性白血病骨髓象傾向AML,骨髓免疫分型提示為AML伴白細胞分化抗原(Cluster of differentiation,CD)7表達可能,目前完成個體化化療方案2療程(第一療程藥物:阿扎胞苷、維奈克拉、高三尖杉酯、阿糖胞苷;第二療程藥物:地西他濱),評估未緩解。LCM、PER、TPM聯合抗發作治療1個月時發熱期間出現局灶性癲癇發作1次,后至今近半年未再出現癲癇發作,近期復查腦電圖提示背景活動慢,未見癇樣放電(圖2a、2b)。
圖2
患兒7歲2月齡腦電圖
a. 彌漫性中波幅4~6 Hz θ節律,背景活動慢;b. 睡眠波及睡眠分期正常,睡眠期各導未見明顯異常放電,可見睡眠期紡錘波
討論 根據國際抗癲癇聯盟的定義,DRE指應用選擇正確且能耐受的兩種抗癲癇發作藥物(單藥或聯合用藥)仍未能達到持續無發作[1]。DRE反復發作病程長,對患兒危害大,藥物治療多采用聯合用藥,目前尚無標準推薦方案,若合并惡性疾病,治療用藥更為棘手。血液腫瘤是一類起源于造血系統的惡性腫瘤,包括白血病、淋巴瘤、MDS等疾病,發病率及死亡率均位居腫瘤前列[2],藥物化療是主要治療手段。當兩病合并發生時,治療用藥選擇需更為慎重。本研究病例在VPA和LEV聯合治療過程中發現血液學改變,停藥后不能恢復,最終診斷血液系統腫瘤MDS,后演變為AML。VPA和LEV分別為第一代和第二代抗癲癇發作藥物,VPA可通過多種機制發揮抗癲癇發作作用,LEV抗癲癇作用可能與神經突觸囊泡蛋白有關,兩者在血液腫瘤患者中均有使用報道[3-6]。VPA和LEV血液系統不良反應有所差別, VPA血液系統不良反應更為常見及多樣,血小板減少、纖維蛋白原減少、白細胞減少、凝血因子缺乏等均有報道[7],減停藥后預后良好。LEV血液系統不良反應少見,發生率約0.01%~0.13%[8],主要表現為血小板減少及全血細胞減少,停藥后短時間內可恢復[9]。本研究病例在減停VPA和LEV后血三系不能恢復,激素治療無效,評估骨髓細胞學提示骨髓增生活躍,并無骨髓抑制表現,提示并非藥物所致血液系統不良反應。停藥約1年半后監測骨髓符合MDS診斷,MDS特點為高風險向AML轉化[10],本研究病例后續MDS演變為AML系疾病本身演變過程。
PER和LCM均為第三代抗癲癇發作藥物,PER是第一個選擇性ɑ-氨基-3-羥基-5甲基-4-異噁唑受體拮抗劑,LCM是新型N-甲基-D-天門冬氨酸受體甘氨酸位點結合拮抗劑,兩者均有口服及注射制劑,口服給藥時吸收迅速,可與食物同服[11,12]。在藥物合用相互作用方面,PER在體外既不是細胞色素P450氧化酶(Cytochrome P450,CYP)或尿苷二磷酸葡萄糖醛基轉移酶(Uridine diphosphate glucuronyl transferases,UGT)同功酶的有效抑制劑,也不是誘導劑,并未發現PER對托吡酯、氯硝西泮、LEV、苯巴比妥或唑尼沙胺的清除率有顯著影響[13]。LCM不誘導或抑制CYP或已知的藥物轉運系統,蛋白質結合率低于15%,由于它具有多種消除途徑,因此與常用處方藥物之間并無臨床相關的相互作用[12]。
PER和LCM已在多個國家被應用于局灶性和全面性癲癇患者,新靶點及新機制可以降低藥物不良反應發生率并提高藥物耐受性。年齡在兒童患者中是影響PER療效的重要因素,一項納入62例6~18歲兒童藥物難治性癲癇患兒的研究顯示PER添加治療總體有效率為50.0%,不同年齡段療效存在差異,12歲以下年齡組有效率25.0%,12歲以上年齡組有效率53.7%[14]。國內一項研究顯示類似結果,年齡越大PER療效呈越好趨勢[15]。PER不良反應主要包括精神不良事件和非精神不良事件,在精神不良事件中,抑郁癥和雙相情感障礙最為常見,分別占4.5%和10.4%;在非精神不良事件中,輕度頭暈、嗜睡和惡心/嘔吐最為常見,分別占28.4%、19.4%和6.0%[16]。LCM同樣是一種有效的添加治療,一項納入60例4~17歲局灶性癲癇患兒的研究顯示,LCM添加治療患兒自基線至末次隨訪的50%應答率為40.0%,75%應答率為28.3%,結果提示在≥4歲中國患兒中LCM添加治療可有效降低癲癇發作頻率[17]。國外LCM添加治療兒童癲癇的研究也有類似結果[18,19]。有數項研究顯示LCM口服制劑及靜脈制劑在癲癇患兒中具有良好的安全性和耐受性[20,21]。LCM最常見的不良反應為上呼吸道感染(33.3%)、嗜睡(26.7%)、頭暈(25.0%)及嘔吐(21.7%),未發現具有臨床意義的心電圖異常變化[17]。對比兩者療效及安全性,有Meta分析結果顯示成人患者中PER 4mg/d癲癇發作反應率低于LCM 400mg/d,另外LCM耐受性更優[22]。PER和LCM聯合添加治療相關研究較少,目前多為個案報道,有報道顯示在熱性感染相關性癲癇綜合征中兩者聯合添加治療表現出良好的治療效果[23]。
本研究病例年齡<12歲,在PER治療過程中出現頭暈,減量后不良反應消失,耐受良好,但添加治療6個月效果不理想, LCM聯合PER添加治療后癲癇發作控制。聯合治療期間患兒接受多次輸血、抗感染治療及多藥化療,未發現異常不良反應。對比LCM聯合PER治療前后腦電圖結果,背景變化差別不大,間期電變化比較明顯,治療前腦電圖右側睡眠期有少量放電,以右側前中顳區明顯,治療后腦電圖無明顯異常波,提示對于兒童難治性局灶性癲癇,LCM聯合PER添加治療為有效的治療選擇之一,且與其它藥物間相互影響小,但目前樣本量小,后續需開展多中心、大樣本長期隨訪研究,進一步評估LCM和PRE聯合添加治療療效及長期耐受性。
利益沖突聲明 所有作者無利益沖突。