引用本文: 賈晨露, 范淑慧, 楊海坡, 姜玉武, 王爽. 細胞周期蛋白依賴性激酶樣-5基因所致發育性癲癇性腦病的臨床電生理特點. 癲癇雜志, 2024, 10(5): 384-392. doi: 10.7507/2096-0247.202407007 復制
版權信息: ?四川大學華西醫院華西期刊社《癲癇雜志》版權所有,未經授權不得轉載、改編
細胞周期蛋白依賴性激酶樣-5基因(Cyclin-dependent kinase-like 5,CDKL5)位于Xp22上,其編碼的絲氨酸-蘇氨酸蛋白激酶在大腦中廣泛分布,參與神經功能的各個環節,與神經發育障礙相關[1-3]。該基因于2003年首次在2例嬰兒痙攣伴嚴重發育落后的女性患兒中發現[4],2004年被確定為疾病病因[5-6]。CDKL5基因變異可引起發育性癲癇性腦病(Developmental and epileptic encephalopathies, DEE),即CDKL5相關發育性癲癇性腦病(CDKL5-DEE)。2022年國際抗癲癇聯盟(International League Against Epilepsy,ILAE)癲癇綜合征分類和標準將其列為新生兒及嬰兒期起病的特異性病因的癲癇綜合征之一[7]。CDKL5-DEE的臨床特征為早發性難治性癲癇發作及全面性發育遲緩。本文通過回顧分析16例CDKL5-DEE患兒的臨床及4h系列視頻腦電圖(Video electroencephalography ,VEEG)等資料,以期提高對該疾病癲癇發作及腦電圖特點的認識。
1 資料與方法
1.1 納入標準
納入2016年6月—2024年5月北京大學第一醫院兒童醫學中心腦電圖監測單元資料庫CDKL5基因變異的患兒。納入標準:基因變異位點明確的致病性變異,臨床表現為運動和認知發育遲緩,藥物難治性癲癇發作[8]。對入組病例臨床資料、系列4h VEEG、視覺誘發電位(Visual evoked potentials,VEP)及頭顱核磁共振(Magnetic Resonance Imaging,MRI)檢查進行回顧分析。該研究獲得北京大學第一醫院醫學倫理委員會審核批準(2005-004)及所有患兒監護人知情同意。
1.2 視頻腦電圖
所有患兒均在我院兒科腦電圖監測單元進行至少1次4h VEEG監測,使用日本光電32導視頻腦電圖儀(EEG-1200C)采集腦電信號,按照國際標準10 - 20系統安放 19 導記錄電極,同時安放雙側三角肌肌電及心電電極,部分患兒根據發作情況額外添加雙側股四頭肌、眼瞼或口角肌電。腦電濾波設置為:高頻15~120 Hz,低頻0.016~160 Hz。采樣率為500 Hz。監測過程包括至少1個清醒-睡眠-覺醒周期,根據患兒配合情況進行誘發實驗,包括睜閉眼測試、間斷閃光刺激(Intermittent photic stimulation,IPS)、過度換氣(Hyperventilation,HV)。腦電圖結果由至少1名技術員及1名腦電圖醫師進行雙重審核判讀。
1.3 視覺誘發電位
使用丹麥維迪肌電圖誘發電位儀(Keypoint4)采集檢測數據。閃光刺激視覺誘發電位采用紅色LED作為刺激光源,刺激頻率為1.9 Hz。采用Fpz-Oz單通道記錄,電極阻抗≤5 kΩ。記錄參數設置為:掃描速度50 ms/D,靈敏度5 μV/D,低頻濾波5 Hz,高頻濾波200 Hz,疊加平均次數50~100次。嬰幼兒患者在水合氯醛誘導睡眠狀態下監測。
2 結果
2.1 一般情況
共納入16例患兒,其中女13例、男3例。所有患兒均為CDKL5基因新生變異(表1),其中錯義變異6例(均為女性),移碼變異5例(女3例、男2例),無義變異4例(女3例、男1例),大片段缺失1例(均為女性)。所有患兒均存在發育落后或遲滯。家族史均無特殊。出生史/既往史:10例無特殊;1例出生后半小時出現低血糖,其母親孕中晚期有手足搐搦史;1例亦于生后出現低血糖,為異卵雙胎之小,36周(w)+3天(d)剖宮產;1例生后4 h出現頻繁嘔吐;1例其母親肝炎于36 w剖宮產;2例有先天性心臟病。頭顱MRI: 5例未見明顯異常、9例表現為額顳為主腦外間隙或蛛網膜下腔增寬、1例左側腦室增寬、1例具體不詳(表2)。
2.2 癲癇發作及治療
所有患兒首次癲癇發作年齡為8天齡(d)~1歲(y)10月齡(m),中位年齡85.94±95.76 d。起病時癲癇發作類型包括:強直發作(Tonic seizure,TS) 4例[起病年齡10~52 d,中位年齡(25.5±15.84)d];局灶性發作(Focal seizure,FS)5例[起病年齡8 d~8 m,中位年齡(77.76±85.97)d];癲癇性痙攣發作(Epileptic spasms,ES)4例[起病年齡3 m~1 y 10 m,中位年齡(6.25±3.49)m];雙側強直-陣攣(Bilateral tonic-clonic seizure,BTCS)發作2例[起病年齡1 m~40 d,中位年齡(35.00±5.00)d];FS并發ES 1例(起病年齡2 m)。詳見表2。
16例患兒起病后均嘗試3種及以上抗癲癇發作藥物(Anti-seizure medications ,ASMs),其中5例曾用維生素B6(VitB6),7例曾接受生酮治療(Ketogenic diet,KD),11例患兒接受促腎上腺皮質激素(Adrenocorticotropic hormone,ACTH)治療。其中1例患兒ACTH后40余天無發作;1例患兒起病后左乙拉西坦(Levetiracetam,LEV)單藥控制4年余無發作,5歲時復發。詳見表2。
2.3 視頻腦電圖
2016年6月—2024年5月期間16例患兒行4 h VEEG監測共59次。所有1歲以上且配合的患兒進行睜眼IPS(共33次),僅1例患兒完成睜閉眼、HV及睜眼、閉眼及合眼IPS全部測試。59次結果均為異常,其中背景正常26次、背景慢節律差25次、無背景8次;間期為后頭部或局灶性放電16次、多灶放電19次、廣泛性或伴局灶/多灶放電17次、高度失律7次;發作期監測到ES 33次、肌陣攣發作(Myoclonic seizure,MS)6次、FS 4次、BTCS發作2次,不典型失神發作(Atypical absence seizure,AS)2次、MS序貫FS 1次、FS序貫成串ES發作1次、過度運動-TS-ES序貫性發作1次、TS 2次。將VEEG結果按監測時年齡分為3組,分別為<6 m組、6 m~2 y組及>2 y組(表3)。背景: <6 m組背景均正常,隨年齡增長,背景正常明顯減少,背景異常明顯增多。發作間期:<6 m組多灶性放電最常見,6 m~2 y組后頭部及局灶性放電最常見,>2 y組廣泛性放電最常見,>2 y組未見高度失律。發作期:三組記錄到發作期的比例均超過60%,<6 m組比另兩組更易記錄到發作期。任何年齡組ES均為最常見的發作類型;FS及BTCS隨年齡增長減少,且BTCS僅出現在<6 m組;MS、AS及TS隨年齡增長增加,AS及TS出現在>2 y組。
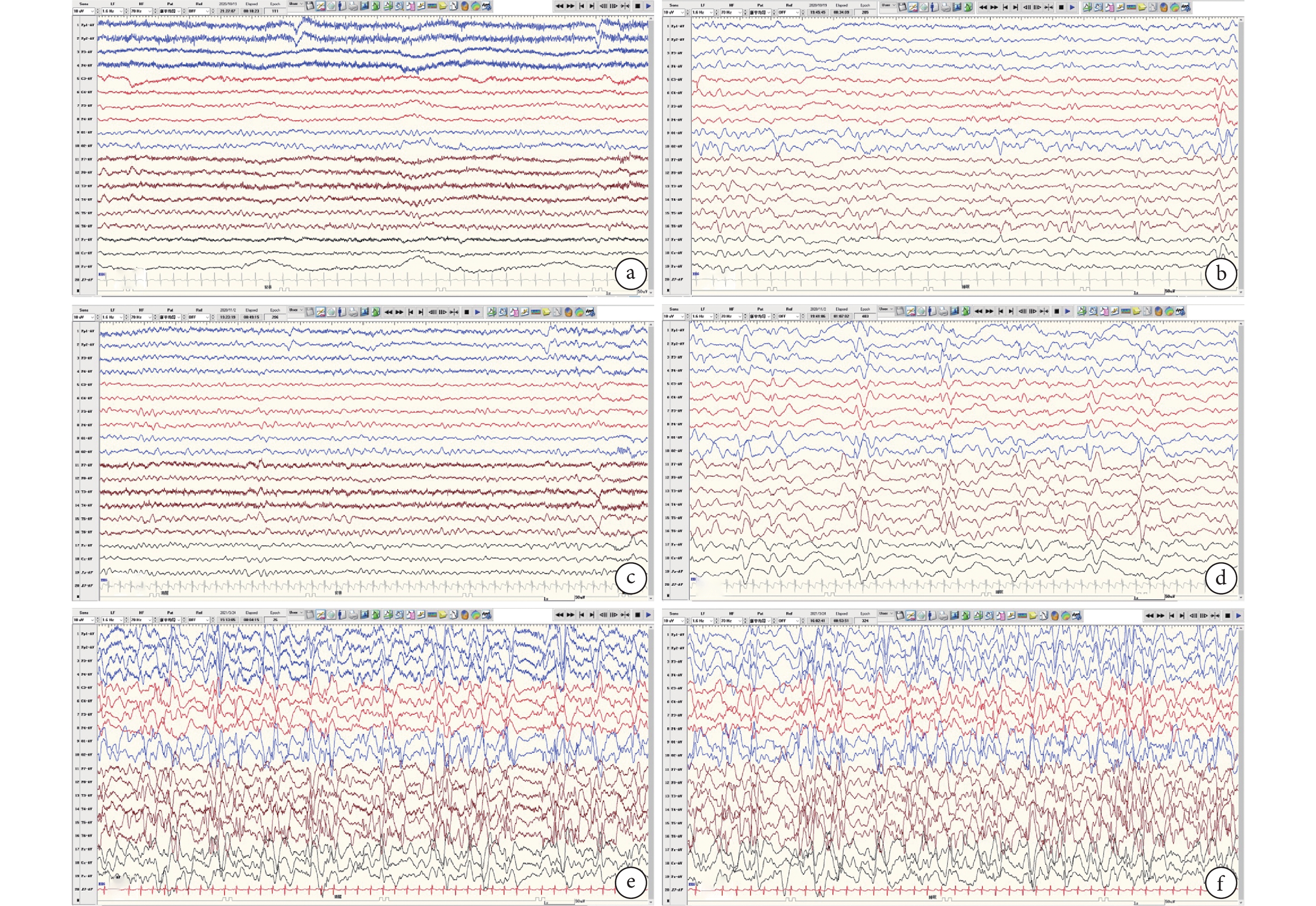
8例患兒進行2次以上系列VEEG監測共計51次(表4)。背景:病例1~4背景由正常→無背景,病例5~6背景由正常→背景慢,病例7~8背景由異常→正常→背景慢。發作間期:病例1多灶放電→高度失律,病例3~5后頭部放電→數量增多或廣泛,病例2、6~7高度失律或多灶放電→后頭部局灶性放電→多灶/廣泛性放電或高度失律。病例8持續多灶及廣泛性放電。發作期:所有患兒均經歷ES,其中病例4~5及7~8 ES持續存在,病例1、2、6由其他發作類型演變為ES,且病例2、6均為BTCS→無發作→ES演變過程,病例3為FS-ES→FS。詳見圖1。
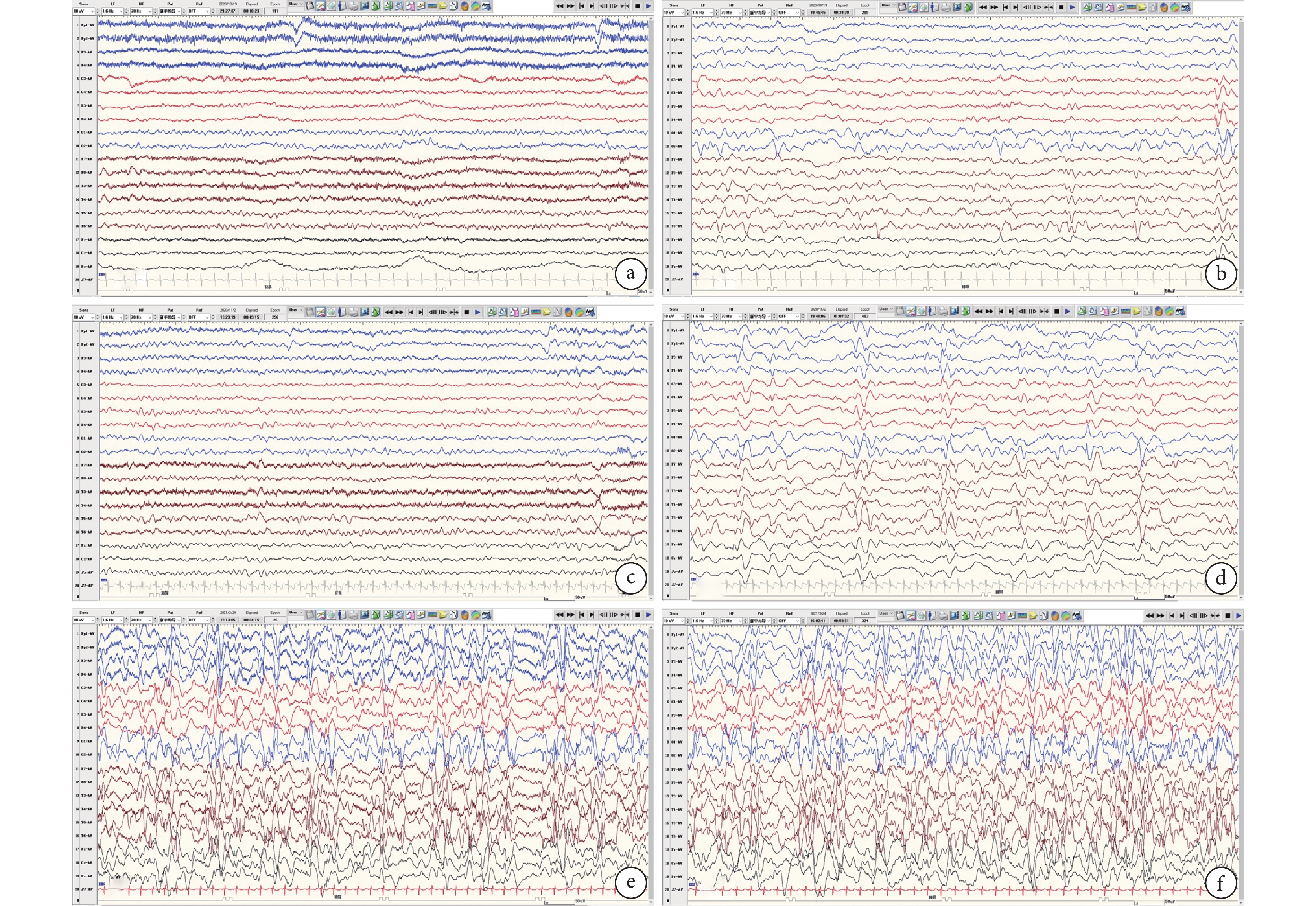 圖1
患兒腦電圖
圖1
患兒腦電圖
ab、cd、ef分別為同一患兒5 m~6 m~10 m背景及間期放電演變;背景:正常-失節律-無背景;間期:后頭部-多灶-高度失律
2.4 視覺誘發電位
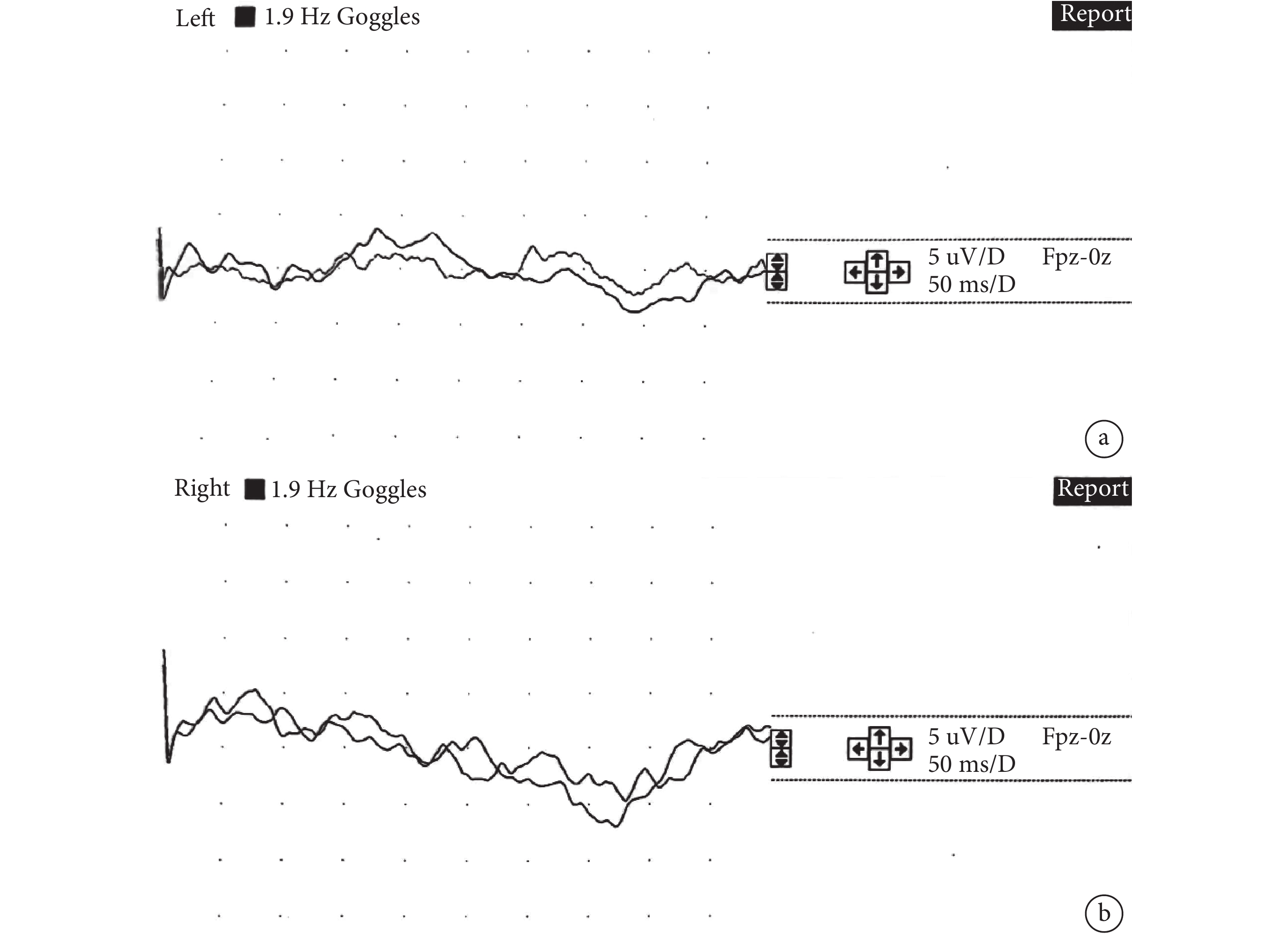
16例患兒僅有3例在我院行視覺誘發電位檢查,均為女性,年齡為3~7 m。2例雙側P1潛伏期正常、1例雙側經多次刺激誘發均未見可重復有效波形引出。詳見圖2。
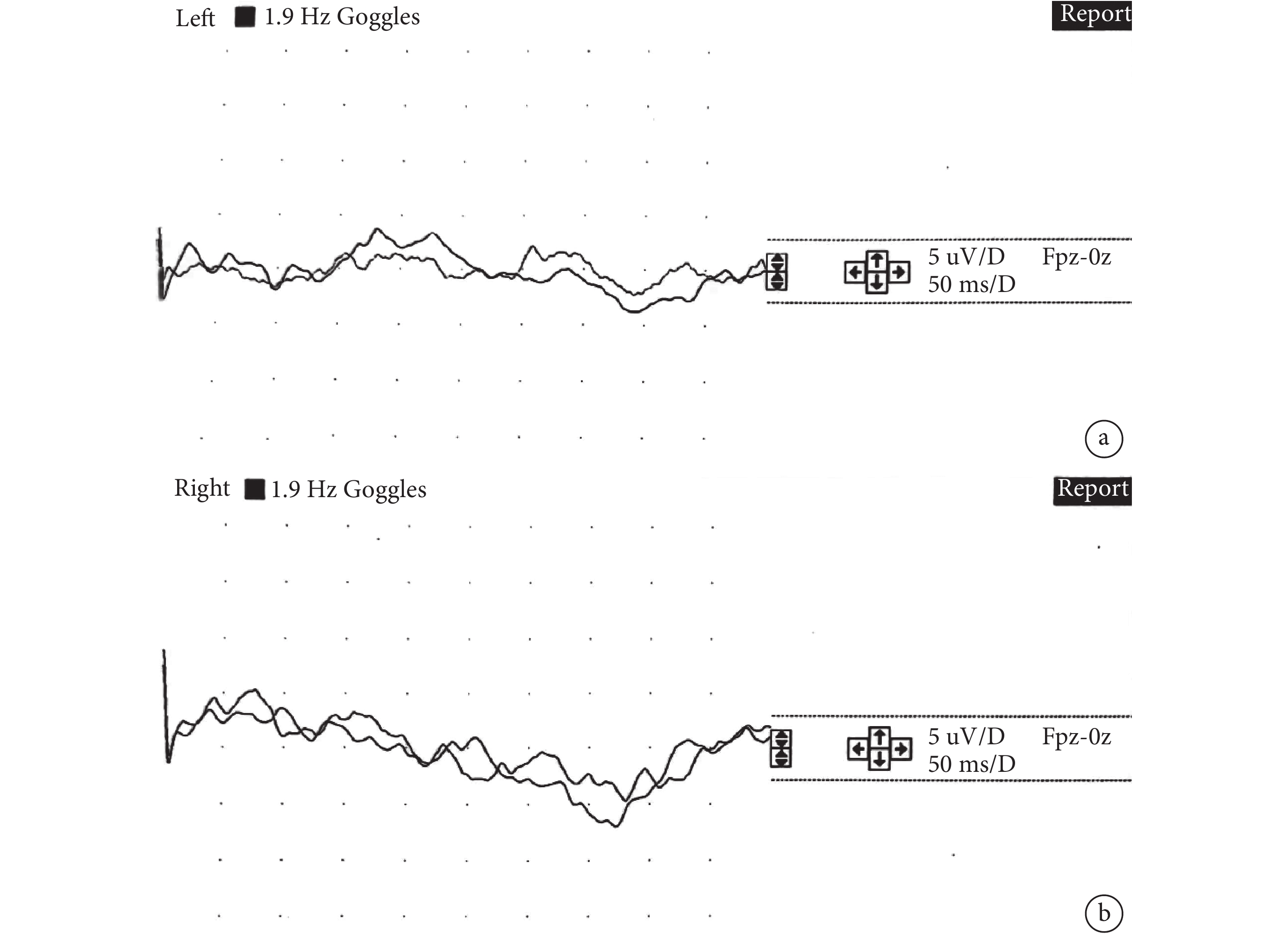 圖2
患兒VEP
圖2
患兒VEP
女,7 m,VEP雙側經多次刺激誘發均未見可重復有效波形引出
3 討論
CDKL5-DEE是X 連鎖遺傳,女∶男為 4∶1[8-9]。本組16例中女13例、男3例,女性占比更高。一項CDKL5-DEE患者的顱腦MRI研究表明進行性腦萎縮可能是該綜合征的一部分,此研究中患者行MRI檢查的年齡為2.5~23 y(中位年齡13.4 y)[10]。本組患兒行MRI時,1例年齡為4 y,其余均為2 y以內,頭顱MRI所見額顳腦外間隙增寬最常見,未發現明顯特異性改變。國內多個報道 CDKL5-DEE患者頭部MRI多表現為額顳腦外間隙增寬 [11,12],與本組病例結果一致。本組患兒仍需進一步隨訪頭顱MRI隨年齡的變化。
癲癇發作是的CDKL5-DEE的重要癥狀,文獻報道其癲癇發作起病中位年齡為6 w[7,13],多在3 m內。本組病例首次癲癇發作年齡為8 d~1 y 10 m,中位年齡46 d,75%的患兒3 m內起病。本組病例起病時發作類型TS 4例、FS 5例、ES 4例,既往有文獻報道首發癲癇發作類型以TS最多見[14],本組病例與該報道不一致。但本組病例多數起病時未能及時行VEEG監測,僅憑癥狀描述無法判斷起源。由于歷史局限性,既往文獻中對新生兒期癲癇發作分類十分混亂,且常常缺乏同期VEEG印證。2020年ILAE新生兒癲癇發作分類中提出,新生兒期不存在全面性癲癇發作[15]。本組病例以TS起病的患兒多在新生兒期,因此CDKL5-DEE起病時TS為局灶性起始可能性很大。國內也有多個研究報道CDKL5-DEE患者以局灶性發作為首發癲癇發作類型最常見 [16-19]。因此CDKL5-DEE新生兒及早嬰期起病時癲癇發作多為FS。本組病例以ES起病共5例,較其他發作類型起病晚。本組病例系列VEEG可見各年齡段均有明顯的癲癇發作,但發作類型有所演變。各年齡段ES均占比最高,隨年齡增長ES持續存在,2 y后進行的VEEG監測中,超過半數記錄到了ES。FS發作隨年齡增長減少,2 y后進行的VEEG監測中,未記錄到FS。而全面性發作隨年齡增長明顯增多。
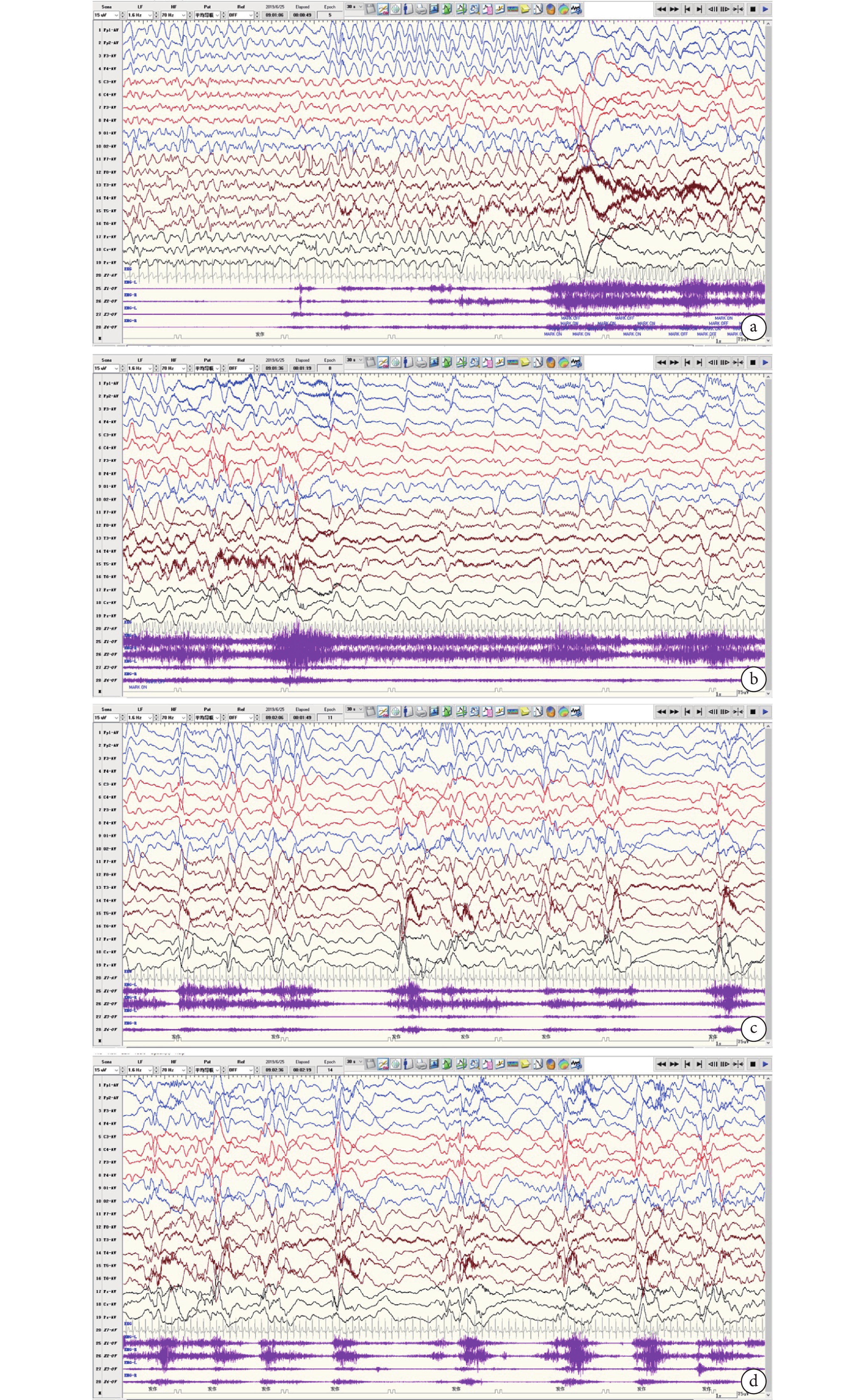
個別文獻報道過度運動-TS-ES序貫性發作為CDKL5-DEE特征性發作,且常見于3.5~13 m(中位年齡7.5 m) [20]。本組2 y以內的VEEG監測共38次,僅在1次監測中發現1次類似發作,患兒表現為睡眠中覺醒,表情驚恐伴輕微挪胯→四肢伸直上抬用力維持伴哭鬧→成串四肢上抬(圖3)。本組2Y以內病例中監測到的最常見的發作類型是ES,其次是FS,此2種發作類型缺乏病因特異性。CDKL5-DEE患者是否存在特征性發作類型有待商榷。
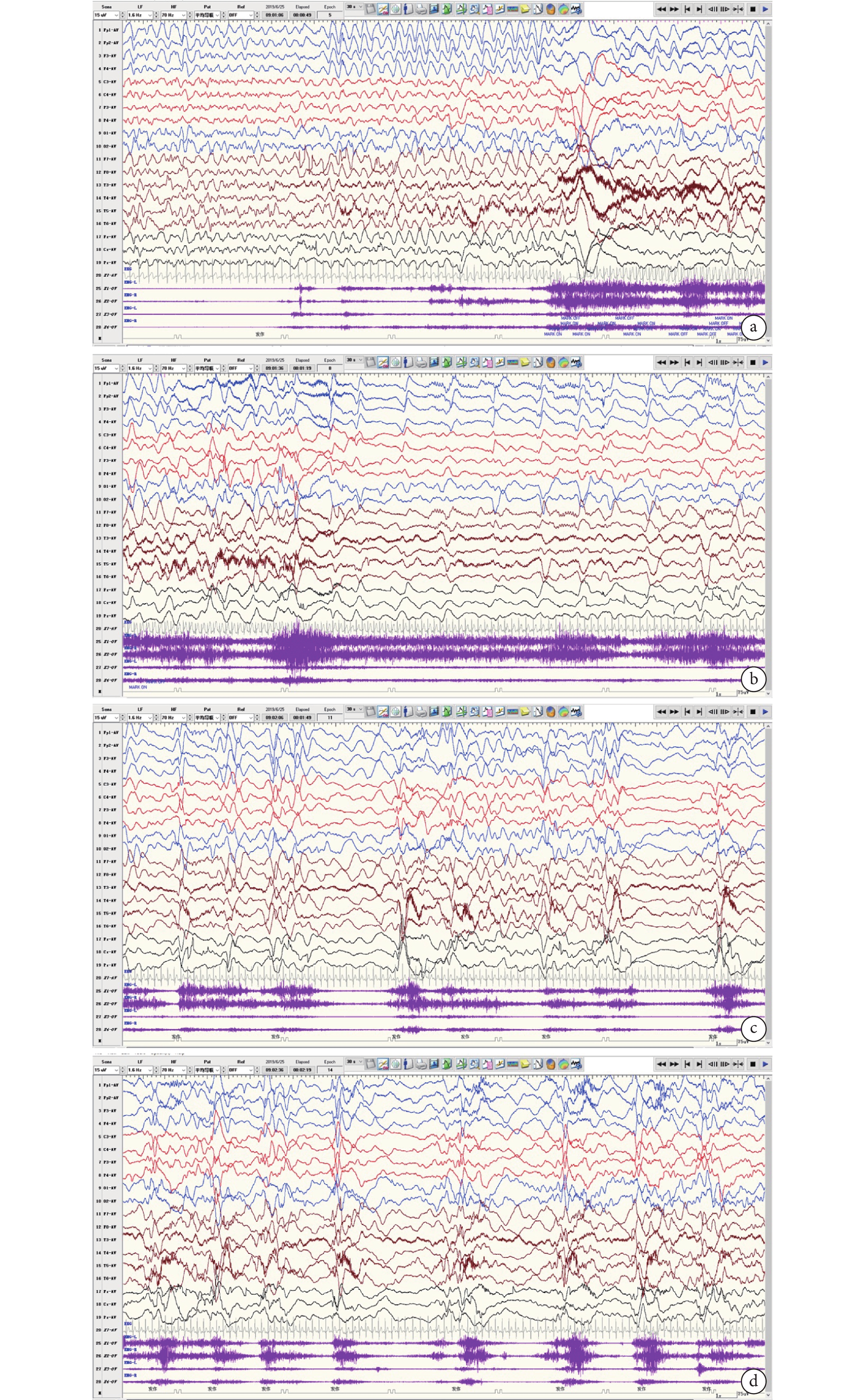 圖3
患兒腦電圖
圖3
患兒腦電圖
男,9 m,睡眠中1次過度運動-強直-成串痙攣發作
與既往文獻報道[7-8]一致,本組所有病例均嘗試3種以上ASMs,符合CDKL5-DEE癲癇發作藥物難治性的特點。Müller等[21]研究CDKL5-DEE患者隨年齡增長對藥物反應性逐漸減低,39例中有2例ASMs或KD治療超過3年無癲癇發作。本組有1例患兒在起病后LEV單藥控制4年,5歲時復發,但也有報道稱LEV在某些情況下可加重CDKL5-DEE患者的癲癇發作[22]。
本組患兒不同年齡的VEEG監測及8例患兒的系列VEEG監測可見一些趨勢。盡管CDKL5-DEE是嚴重的DEE,但在起病初期EEG不一定符合癲癇性腦病。本組所有6m以內的VEEG監測均顯示背景正常,同期可見良好的睡眠紡錘波,符合嬰幼兒期正常生理節律[23]。但此時患兒已可能存在發育落后,本組8例在癲癇發作前即有發育落后,說明CDKL5-DEE在起病的初期發育性腦病更為突出。6 m后背景異常比例逐漸增加。所有VEEG監測發作間期均有明顯異常放電,隨年齡增長放電由多灶放電逐漸同步化,2 y以內放電更多灶,可限局在后頭部,2 y以后廣泛性放電比例增加。這可能與腦發育及髓鞘化逐漸完善有關[24]。系列VEEG可見所有患兒背景及間期放電均伴惡化趨勢。文獻報道部分患兒可出現短暫“蜜月期”,但癲癇性腦病及難治性癲癇發作在晚期很常見[22]。本組有3例患兒臨床干預可出現短暫背景好轉及間期放電減少,同期發作也明顯減輕,但隨后仍會惡化,發作增多。
相比其他的癲癇性腦病,CDKL5-DEE視覺受損較顯著[8,25],Demarest 等[14]研究稱皮質視力障礙(Cortical visual impairment,CVI)存在于75%的CDKL5-DEE患者中。本組病例僅3例行VEP,1例雙側經多次刺激誘發均未見可重復有效波形引出。文獻報道CVI的腦電圖可表現為枕區節律受損、放電或閃光陽性[26],后頭部放電可能與視覺受損有關[27]。本組病例VEEG顯示間期放電部位后頭部或枕區最突出,56%背景異常。
既往文獻關于CDKL5變異基因型與表型關系的研究結果存在不一致,有報道認為錯義變異臨床表型較輕[8],也有報道稱p.A178T和 p.A178G兩種錯義變異臨床表型較嚴重,相比之下,p.Arg134*和 p.Arg550* 表型較輕[28]。本組患兒樣本量小,無法進行基因型與表型之間的相關性研究。僅發現本組患兒中所有錯義變異均為女性,3例男性患兒均為非錯義變異。由于CDKL5為 X連鎖疾病,因此CDKL5-DEE的基因型與表型,以及性別與表型之間存在著復雜的關系,至今未見明確的結論。
綜上所述,CDKL5-DEE是X連鎖遺傳,女性多見,臨床特點為明顯發育落后及藥物難治性癲癇發作。ES是每個患者均會經歷的發作類型,且持續時間長,隨年齡增長局灶性發作減少,全面性發作明顯增多。腦電圖特點為6月齡以內背景正常,隨年齡增長背景及間期放電均具有惡化趨勢。CDKL5-DEE后頭部放電常見,可能與其臨床顯著視覺受損相關。
利益沖突聲明 所有作者無利益沖突。
細胞周期蛋白依賴性激酶樣-5基因(Cyclin-dependent kinase-like 5,CDKL5)位于Xp22上,其編碼的絲氨酸-蘇氨酸蛋白激酶在大腦中廣泛分布,參與神經功能的各個環節,與神經發育障礙相關[1-3]。該基因于2003年首次在2例嬰兒痙攣伴嚴重發育落后的女性患兒中發現[4],2004年被確定為疾病病因[5-6]。CDKL5基因變異可引起發育性癲癇性腦病(Developmental and epileptic encephalopathies, DEE),即CDKL5相關發育性癲癇性腦病(CDKL5-DEE)。2022年國際抗癲癇聯盟(International League Against Epilepsy,ILAE)癲癇綜合征分類和標準將其列為新生兒及嬰兒期起病的特異性病因的癲癇綜合征之一[7]。CDKL5-DEE的臨床特征為早發性難治性癲癇發作及全面性發育遲緩。本文通過回顧分析16例CDKL5-DEE患兒的臨床及4h系列視頻腦電圖(Video electroencephalography ,VEEG)等資料,以期提高對該疾病癲癇發作及腦電圖特點的認識。
1 資料與方法
1.1 納入標準
納入2016年6月—2024年5月北京大學第一醫院兒童醫學中心腦電圖監測單元資料庫CDKL5基因變異的患兒。納入標準:基因變異位點明確的致病性變異,臨床表現為運動和認知發育遲緩,藥物難治性癲癇發作[8]。對入組病例臨床資料、系列4h VEEG、視覺誘發電位(Visual evoked potentials,VEP)及頭顱核磁共振(Magnetic Resonance Imaging,MRI)檢查進行回顧分析。該研究獲得北京大學第一醫院醫學倫理委員會審核批準(2005-004)及所有患兒監護人知情同意。
1.2 視頻腦電圖
所有患兒均在我院兒科腦電圖監測單元進行至少1次4h VEEG監測,使用日本光電32導視頻腦電圖儀(EEG-1200C)采集腦電信號,按照國際標準10 - 20系統安放 19 導記錄電極,同時安放雙側三角肌肌電及心電電極,部分患兒根據發作情況額外添加雙側股四頭肌、眼瞼或口角肌電。腦電濾波設置為:高頻15~120 Hz,低頻0.016~160 Hz。采樣率為500 Hz。監測過程包括至少1個清醒-睡眠-覺醒周期,根據患兒配合情況進行誘發實驗,包括睜閉眼測試、間斷閃光刺激(Intermittent photic stimulation,IPS)、過度換氣(Hyperventilation,HV)。腦電圖結果由至少1名技術員及1名腦電圖醫師進行雙重審核判讀。
1.3 視覺誘發電位
使用丹麥維迪肌電圖誘發電位儀(Keypoint4)采集檢測數據。閃光刺激視覺誘發電位采用紅色LED作為刺激光源,刺激頻率為1.9 Hz。采用Fpz-Oz單通道記錄,電極阻抗≤5 kΩ。記錄參數設置為:掃描速度50 ms/D,靈敏度5 μV/D,低頻濾波5 Hz,高頻濾波200 Hz,疊加平均次數50~100次。嬰幼兒患者在水合氯醛誘導睡眠狀態下監測。
2 結果
2.1 一般情況
共納入16例患兒,其中女13例、男3例。所有患兒均為CDKL5基因新生變異(表1),其中錯義變異6例(均為女性),移碼變異5例(女3例、男2例),無義變異4例(女3例、男1例),大片段缺失1例(均為女性)。所有患兒均存在發育落后或遲滯。家族史均無特殊。出生史/既往史:10例無特殊;1例出生后半小時出現低血糖,其母親孕中晚期有手足搐搦史;1例亦于生后出現低血糖,為異卵雙胎之小,36周(w)+3天(d)剖宮產;1例生后4 h出現頻繁嘔吐;1例其母親肝炎于36 w剖宮產;2例有先天性心臟病。頭顱MRI: 5例未見明顯異常、9例表現為額顳為主腦外間隙或蛛網膜下腔增寬、1例左側腦室增寬、1例具體不詳(表2)。
2.2 癲癇發作及治療
所有患兒首次癲癇發作年齡為8天齡(d)~1歲(y)10月齡(m),中位年齡85.94±95.76 d。起病時癲癇發作類型包括:強直發作(Tonic seizure,TS) 4例[起病年齡10~52 d,中位年齡(25.5±15.84)d];局灶性發作(Focal seizure,FS)5例[起病年齡8 d~8 m,中位年齡(77.76±85.97)d];癲癇性痙攣發作(Epileptic spasms,ES)4例[起病年齡3 m~1 y 10 m,中位年齡(6.25±3.49)m];雙側強直-陣攣(Bilateral tonic-clonic seizure,BTCS)發作2例[起病年齡1 m~40 d,中位年齡(35.00±5.00)d];FS并發ES 1例(起病年齡2 m)。詳見表2。
16例患兒起病后均嘗試3種及以上抗癲癇發作藥物(Anti-seizure medications ,ASMs),其中5例曾用維生素B6(VitB6),7例曾接受生酮治療(Ketogenic diet,KD),11例患兒接受促腎上腺皮質激素(Adrenocorticotropic hormone,ACTH)治療。其中1例患兒ACTH后40余天無發作;1例患兒起病后左乙拉西坦(Levetiracetam,LEV)單藥控制4年余無發作,5歲時復發。詳見表2。
2.3 視頻腦電圖
2016年6月—2024年5月期間16例患兒行4 h VEEG監測共59次。所有1歲以上且配合的患兒進行睜眼IPS(共33次),僅1例患兒完成睜閉眼、HV及睜眼、閉眼及合眼IPS全部測試。59次結果均為異常,其中背景正常26次、背景慢節律差25次、無背景8次;間期為后頭部或局灶性放電16次、多灶放電19次、廣泛性或伴局灶/多灶放電17次、高度失律7次;發作期監測到ES 33次、肌陣攣發作(Myoclonic seizure,MS)6次、FS 4次、BTCS發作2次,不典型失神發作(Atypical absence seizure,AS)2次、MS序貫FS 1次、FS序貫成串ES發作1次、過度運動-TS-ES序貫性發作1次、TS 2次。將VEEG結果按監測時年齡分為3組,分別為<6 m組、6 m~2 y組及>2 y組(表3)。背景: <6 m組背景均正常,隨年齡增長,背景正常明顯減少,背景異常明顯增多。發作間期:<6 m組多灶性放電最常見,6 m~2 y組后頭部及局灶性放電最常見,>2 y組廣泛性放電最常見,>2 y組未見高度失律。發作期:三組記錄到發作期的比例均超過60%,<6 m組比另兩組更易記錄到發作期。任何年齡組ES均為最常見的發作類型;FS及BTCS隨年齡增長減少,且BTCS僅出現在<6 m組;MS、AS及TS隨年齡增長增加,AS及TS出現在>2 y組。
8例患兒進行2次以上系列VEEG監測共計51次(表4)。背景:病例1~4背景由正常→無背景,病例5~6背景由正常→背景慢,病例7~8背景由異常→正常→背景慢。發作間期:病例1多灶放電→高度失律,病例3~5后頭部放電→數量增多或廣泛,病例2、6~7高度失律或多灶放電→后頭部局灶性放電→多灶/廣泛性放電或高度失律。病例8持續多灶及廣泛性放電。發作期:所有患兒均經歷ES,其中病例4~5及7~8 ES持續存在,病例1、2、6由其他發作類型演變為ES,且病例2、6均為BTCS→無發作→ES演變過程,病例3為FS-ES→FS。詳見圖1。
圖1
患兒腦電圖
ab、cd、ef分別為同一患兒5 m~6 m~10 m背景及間期放電演變;背景:正常-失節律-無背景;間期:后頭部-多灶-高度失律
2.4 視覺誘發電位
16例患兒僅有3例在我院行視覺誘發電位檢查,均為女性,年齡為3~7 m。2例雙側P1潛伏期正常、1例雙側經多次刺激誘發均未見可重復有效波形引出。詳見圖2。
圖2
患兒VEP
女,7 m,VEP雙側經多次刺激誘發均未見可重復有效波形引出
3 討論
CDKL5-DEE是X 連鎖遺傳,女∶男為 4∶1[8-9]。本組16例中女13例、男3例,女性占比更高。一項CDKL5-DEE患者的顱腦MRI研究表明進行性腦萎縮可能是該綜合征的一部分,此研究中患者行MRI檢查的年齡為2.5~23 y(中位年齡13.4 y)[10]。本組患兒行MRI時,1例年齡為4 y,其余均為2 y以內,頭顱MRI所見額顳腦外間隙增寬最常見,未發現明顯特異性改變。國內多個報道 CDKL5-DEE患者頭部MRI多表現為額顳腦外間隙增寬 [11,12],與本組病例結果一致。本組患兒仍需進一步隨訪頭顱MRI隨年齡的變化。
癲癇發作是的CDKL5-DEE的重要癥狀,文獻報道其癲癇發作起病中位年齡為6 w[7,13],多在3 m內。本組病例首次癲癇發作年齡為8 d~1 y 10 m,中位年齡46 d,75%的患兒3 m內起病。本組病例起病時發作類型TS 4例、FS 5例、ES 4例,既往有文獻報道首發癲癇發作類型以TS最多見[14],本組病例與該報道不一致。但本組病例多數起病時未能及時行VEEG監測,僅憑癥狀描述無法判斷起源。由于歷史局限性,既往文獻中對新生兒期癲癇發作分類十分混亂,且常常缺乏同期VEEG印證。2020年ILAE新生兒癲癇發作分類中提出,新生兒期不存在全面性癲癇發作[15]。本組病例以TS起病的患兒多在新生兒期,因此CDKL5-DEE起病時TS為局灶性起始可能性很大。國內也有多個研究報道CDKL5-DEE患者以局灶性發作為首發癲癇發作類型最常見 [16-19]。因此CDKL5-DEE新生兒及早嬰期起病時癲癇發作多為FS。本組病例以ES起病共5例,較其他發作類型起病晚。本組病例系列VEEG可見各年齡段均有明顯的癲癇發作,但發作類型有所演變。各年齡段ES均占比最高,隨年齡增長ES持續存在,2 y后進行的VEEG監測中,超過半數記錄到了ES。FS發作隨年齡增長減少,2 y后進行的VEEG監測中,未記錄到FS。而全面性發作隨年齡增長明顯增多。
個別文獻報道過度運動-TS-ES序貫性發作為CDKL5-DEE特征性發作,且常見于3.5~13 m(中位年齡7.5 m) [20]。本組2 y以內的VEEG監測共38次,僅在1次監測中發現1次類似發作,患兒表現為睡眠中覺醒,表情驚恐伴輕微挪胯→四肢伸直上抬用力維持伴哭鬧→成串四肢上抬(圖3)。本組2Y以內病例中監測到的最常見的發作類型是ES,其次是FS,此2種發作類型缺乏病因特異性。CDKL5-DEE患者是否存在特征性發作類型有待商榷。
圖3
患兒腦電圖
男,9 m,睡眠中1次過度運動-強直-成串痙攣發作
與既往文獻報道[7-8]一致,本組所有病例均嘗試3種以上ASMs,符合CDKL5-DEE癲癇發作藥物難治性的特點。Müller等[21]研究CDKL5-DEE患者隨年齡增長對藥物反應性逐漸減低,39例中有2例ASMs或KD治療超過3年無癲癇發作。本組有1例患兒在起病后LEV單藥控制4年,5歲時復發,但也有報道稱LEV在某些情況下可加重CDKL5-DEE患者的癲癇發作[22]。
本組患兒不同年齡的VEEG監測及8例患兒的系列VEEG監測可見一些趨勢。盡管CDKL5-DEE是嚴重的DEE,但在起病初期EEG不一定符合癲癇性腦病。本組所有6m以內的VEEG監測均顯示背景正常,同期可見良好的睡眠紡錘波,符合嬰幼兒期正常生理節律[23]。但此時患兒已可能存在發育落后,本組8例在癲癇發作前即有發育落后,說明CDKL5-DEE在起病的初期發育性腦病更為突出。6 m后背景異常比例逐漸增加。所有VEEG監測發作間期均有明顯異常放電,隨年齡增長放電由多灶放電逐漸同步化,2 y以內放電更多灶,可限局在后頭部,2 y以后廣泛性放電比例增加。這可能與腦發育及髓鞘化逐漸完善有關[24]。系列VEEG可見所有患兒背景及間期放電均伴惡化趨勢。文獻報道部分患兒可出現短暫“蜜月期”,但癲癇性腦病及難治性癲癇發作在晚期很常見[22]。本組有3例患兒臨床干預可出現短暫背景好轉及間期放電減少,同期發作也明顯減輕,但隨后仍會惡化,發作增多。
相比其他的癲癇性腦病,CDKL5-DEE視覺受損較顯著[8,25],Demarest 等[14]研究稱皮質視力障礙(Cortical visual impairment,CVI)存在于75%的CDKL5-DEE患者中。本組病例僅3例行VEP,1例雙側經多次刺激誘發均未見可重復有效波形引出。文獻報道CVI的腦電圖可表現為枕區節律受損、放電或閃光陽性[26],后頭部放電可能與視覺受損有關[27]。本組病例VEEG顯示間期放電部位后頭部或枕區最突出,56%背景異常。
既往文獻關于CDKL5變異基因型與表型關系的研究結果存在不一致,有報道認為錯義變異臨床表型較輕[8],也有報道稱p.A178T和 p.A178G兩種錯義變異臨床表型較嚴重,相比之下,p.Arg134*和 p.Arg550* 表型較輕[28]。本組患兒樣本量小,無法進行基因型與表型之間的相關性研究。僅發現本組患兒中所有錯義變異均為女性,3例男性患兒均為非錯義變異。由于CDKL5為 X連鎖疾病,因此CDKL5-DEE的基因型與表型,以及性別與表型之間存在著復雜的關系,至今未見明確的結論。
綜上所述,CDKL5-DEE是X連鎖遺傳,女性多見,臨床特點為明顯發育落后及藥物難治性癲癇發作。ES是每個患者均會經歷的發作類型,且持續時間長,隨年齡增長局灶性發作減少,全面性發作明顯增多。腦電圖特點為6月齡以內背景正常,隨年齡增長背景及間期放電均具有惡化趨勢。CDKL5-DEE后頭部放電常見,可能與其臨床顯著視覺受損相關。
利益沖突聲明 所有作者無利益沖突。